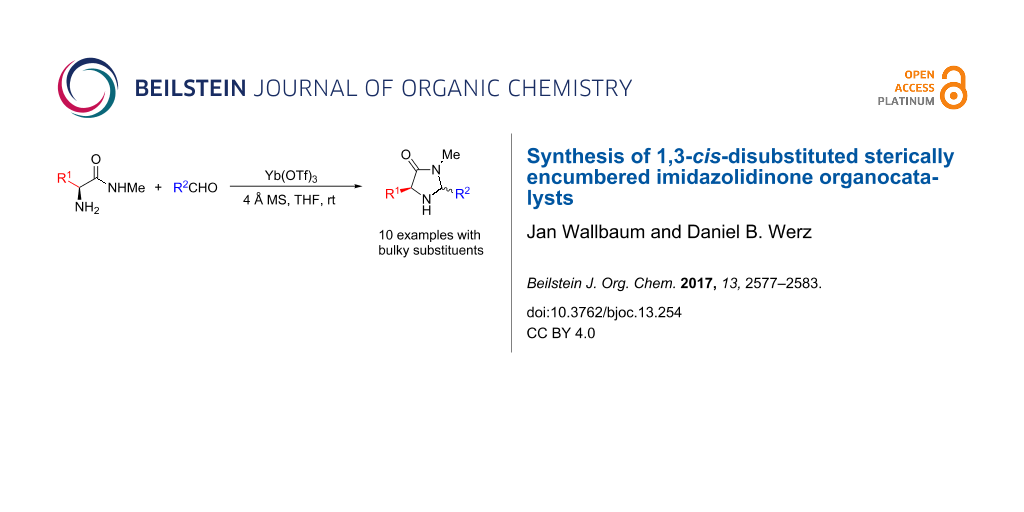
Abstract
A variety of novel imidazolidinone-based organocatalysts with bulky substituents were synthesized under mild reaction conditions starting from easily accessible substrates. Different natural and unnatural amino acid methyl amides were cyclized with aromatic carbaldehydes to yield two diastereomeric MacMillan-type catalysts. Special emphasis was put on bulky residues such as mesityl and pyrene moieties.

Graphical Abstract
Introduction
Organocatalytic iminium and enamine activation has attracted organic chemists for more than one century [1]. Until today a wide and constantly increasing number of different organocatalytic transformations with various substrates have been accomplished [2-7]. Initially, proline and proline-derived catalysts have been widely used in asymmetric iminium and enamine organocatalysis [8-12]. Since the beginning of the 21th century imidazolidinone-based organocatalysts developed by MacMillan and co-workers, which are easily accessible from amino acids, are widely used in these kinds of reactions [13-18]. More recent examples demonstrate the applicability in various reactions like diastereoselective α-fluorination [19], total syntheses [20,21], cross-dehydrogenative couplings [22], selectivity-reversed Friedel–Crafts alkylation [23] and in combination with photoredox catalysis (Scheme 1a) [24].
![[1860-5397-13-254-i1]](/bjoc/content/inline/1860-5397-13-254-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: a) MacMillan’s enantioselective α-alkylation of aldehydes. b) Our enantioselective 1,3-chlorosulfenation of meso-cyclopropyl carbaldehydes.
Scheme 1: a) MacMillan’s enantioselective α-alkylation of aldehydes. b) Our enantioselective 1,3-chlorosulfen...
The enantioselective α-alkylation was achieved by merging the common photoredox catalyst Ru(bpy)3Cl2 with imidazolidinone catalyst 3a·TfOH, controlling the stereochemistry of the radical addition via an intermediate enamine complex.
Scheme 1b shows our regio-, diastereo- and enantioselective 1,3-chlorosulfenation of meso-cyclopropyl carbaldehydes employing a newly designed organocatalyst 7a·DCA for chiral induction [25]. In the course of these studies we prepared a variety of imidazolidinone organocatalysts with rather bulky substituents. In this paper we report on these MacMillan-type catalysts which will be of great value for the screening of further transformations based on iminium–enamine mechanisms.
Results and Discussion
In order to screen a certain transformation by using a variety of organocatalysts with subtle differences in the substitution pattern a modular approach for their preparation is highly desirable. A simple retrosynthetic cut of the five-membered ring delivers an amino acid methyl amide and a carbaldehyde as starting materials. Chirality is commonly introduced by the use of derivatives of naturally occurring L-amino acid derivatives. During the condensation process two diastereomers can be formed with the substituents in either a 1,3-cis or 1,3-trans arrangement. Since our reaction design of the above-mentioned reaction (depicted in Scheme 1b) showed a strong preference to use the 1,3-cis-disubstituted derivatives we concentrated our efforts on the isolation of these isomers.
Methyl amides 9 were produced from the corresponding methyl or ethyl esters via reaction with ethanolic methyl amide solution [26]. Solid methyl amides were further recrystallized to achieve a higher purity. Protected methyl amides 9a and 9c were synthesized in five or three literature-known steps from commercially available substrates, respectively [27-29].
The different methyl amides were subjected to the reaction with 1-naphthyl carbaldehyde employing 4 Å molecular sieves as dehydrating agent and 10 mol % Yb(OTf)3 in THF. This Lewis acid proved to be the Lewis acid of choice since the reaction proceeds without loss of optical purity [30]. The naphthyl residue was chosen since it revealed a high selectivity in the desired organocatalytic transformation. Table 1 depicts the scope with respect to 1-naphthyl carbaldehyde. Crude 1H NMR data of the reaction mixture showed in every case approximately 90% product formation of both diastereomers together, using an internal standard. The corresponding 1,3-trans diastereomers were not isolated in pure form. As reaction partners different methyl amides 9a–f were employed in the reaction. The use of methyl-protected L-histidine-derived methyl amide 9a (Table 1, entry 1) gave the desired imidazolidinone 7b in 42% yield, whereas the unprotected derivative showed no conversion, probably because of its insolubility. The unprotected as well as the benzyl-protected L-tryptophan-derived methyl amides 9b and 9c were converted in good yields to the corresponding imidazolidinones (Table 1, entries 2 and 3). In the case of 7c it is important to use not more than 1.0 equivalent of the aldehyde, otherwise a further reaction with the indole moiety is observed.
Table 1: Scope of the reaction using 1-naphthyl carbaldehyde (10a).a
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-254-i2.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Methyl amide 9 | Imidazolidinone 7 | Yield [%] (ratio)b | ||
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-254-i3.svg?max-width=637&scale=1.0)
|
9a |
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-254-i4.svg?max-width=637&scale=1.0)
|
7b |
42
(1:1) |
| 2 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-254-i5.svg?max-width=637&scale=1.0)
|
9b |
![[Graphic 5]](/bjoc/content/inline/1860-5397-13-254-i6.svg?max-width=637&scale=1.0)
|
7c |
45
(1:1) |
| 3 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-13-254-i7.svg?max-width=637&scale=1.0)
|
9c |
![[Graphic 7]](/bjoc/content/inline/1860-5397-13-254-i8.svg?max-width=637&scale=1.0)
|
7d |
42
(1:1) |
| 4 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-13-254-i9.svg?max-width=637&scale=1.0)
|
9d |
![[Graphic 9]](/bjoc/content/inline/1860-5397-13-254-i10.svg?max-width=637&scale=1.0)
|
7e |
12
(1:5) |
| 5 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-13-254-i11.svg?max-width=637&scale=1.0)
|
9e |
![[Graphic 11]](/bjoc/content/inline/1860-5397-13-254-i12.svg?max-width=637&scale=1.0)
|
7f |
46
(3:2) |
| 6 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-13-254-i13.svg?max-width=637&scale=1.0)
|
9f |
![[Graphic 13]](/bjoc/content/inline/1860-5397-13-254-i14.svg?max-width=637&scale=1.0)
|
7g |
32
(2:3) |
aReaction conditions: 9 (1.0 equiv), 10a (0.9–1.1 equiv), Yb(OTf)3 (10 mol %), THF (4–12 mL/mmol), 4 Å MS (40 mg/mL), rt, 24 h. bIsolated yield after column chromatography; the yield of both diastereomers together is in every case about 80–90%; the ratio of the two diastereomers is given in brackets, the desired cis-isomer is underlined (determined via 1H NMR spectroscopy of the crude reaction mixture).
A significant change in selectivity was observed while using electron-withdrawing methyl amide 9d; the influence of the para-nitro substituent decreases the yield of the desired cis-diastereomer to only 12%. Utilization of non-aromatic methyl amides 9e and 9f, synthesized from L-norvaline and L-methionine methyl esters, gave rise to the desired imidazolidinones 7f and 7g (Table 1, entries 5 and 6) in 46% and 32%, respectively.
After evaluating the scope of imidazolidinones 7 we were keen to investigate which sterically more demanding aldehydes could be employed. Since the anticipated organocatalytic transformation (shown in Scheme 1b) delivered relatively high selectivity with imidazolidinones derived from L-phenylalanine this amino acid derivative was used as coupling partner. 1-Pyrene carbaldehyde 10b gave only a minor excess of the cis-diastereomer (S,S)-3b. For mesityl carbaldehyde 10c the desired isomer was obtained in 52% yield whereas the more sterically demanding aldehyde 10d led to a switch in the selectivity, giving the desired (S,S)-diastereomer in only 23% yield (Table 2).
Table 2: Scope of the imidazolidinone formation with respect to aldehyde 10.a
![[Graphic 14]](/bjoc/content/inline/1860-5397-13-254-i15.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Methyl amide 9 | Aldehyde 10 | Imidazolidinone 3 | Yield [%] (ratio)b | ||
| 1 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-13-254-i16.svg?max-width=637&scale=1.0)
|
9g |
![[Graphic 16]](/bjoc/content/inline/1860-5397-13-254-i17.svg?max-width=637&scale=1.0)
|
10b |
![[Graphic 17]](/bjoc/content/inline/1860-5397-13-254-i18.svg?max-width=637&scale=1.0)
|
|
| (R,S)-3b, 41 | ||||||
| (S,S)-3b, 49 | ||||||
| 2c |
![[Graphic 18]](/bjoc/content/inline/1860-5397-13-254-i19.svg?max-width=637&scale=1.0)
|
9g |
![[Graphic 19]](/bjoc/content/inline/1860-5397-13-254-i20.svg?max-width=637&scale=1.0)
|
10c |
![[Graphic 20]](/bjoc/content/inline/1860-5397-13-254-i21.svg?max-width=637&scale=1.0)
|
3c, 52
(3:2) |
| 3c |
![[Graphic 21]](/bjoc/content/inline/1860-5397-13-254-i22.svg?max-width=637&scale=1.0)
|
9g |
![[Graphic 22]](/bjoc/content/inline/1860-5397-13-254-i23.svg?max-width=637&scale=1.0)
|
10d |
![[Graphic 23]](/bjoc/content/inline/1860-5397-13-254-i24.svg?max-width=637&scale=1.0)
|
3d, 23
(1:2) |
aReaction conditions: 9 (1.0 equiv), 10 (1.1–1.5 equiv), Yb(OTf)3 (10 mol %), THF (4–5 mL/mmol), 4 Å MS (40 mg/mL), rt, 24 h. bIsolated yield after column chromatography. The ratio of the two diastereomers is given in brackets, the desired cis-isomer is underlined (determined via 1H NMR spectroscopy of the crude reaction mixture). cCombined yield of both diastereomers is 89% (entry 2) and 78% (entry 3), respectively.
Conclusion
We were able to synthesize 10 different imidazolidone-based organocatalysts with yields of the desired 1,3-cis-disubstituted diastereomers of up to 52%. Different methyl amides derived from α-amino acids and bulky aromatic carbaldehydes were employed to access the MacMillan-type catalysts. The key to the success is the activation with ytterbium triflate as Lewis acid and the use of dehydrating molecular sieves. The prepared organocatalysts will be a useful contribution for the screening of a multitude of different organocatalytic transformations.
Experimental
General. All solvents were distilled before use unless otherwise stated. Tetrahydrofuran (THF) was distilled over sodium and benzophenone under an argon atmosphere. Air- and moisture-sensitive reactions were carried out in oven-dried or flame-dried glassware, septum-capped under atmospheric pressure of argon. Commercially available compounds were used without further purification unless otherwise stated.
Proton (1H) and carbon (13C) NMR spectra were recorded on a 300, 400 or 600 MHz instrument using the residual signals from CHCl3, δ = 7.26 ppm and δ = 77.0 ppm, or MeOH, δ = 3.31 ppm and δ = 49.2 ppm, using TMS as internal reference for 1H and 13C chemical shifts, respectively. Assignments of the respective signals and the stereochemistry were made by combination of H,H-COSY, HSQC, HMBC and NOESY experiments. In some cases signals in the 13C NMR spectrum are missing because of bad relaxation of the respective signals. ESI high-resolution mass spectrometry was carried out on a FTICR instrument. IR spectra were measured on an ATR spectrometer. Optical rotation was measured on a common polarimeter.
General procedure (GP) for the synthesis of imidazolidinones of type 3 and 7. A Schlenk flask was charged with a magnetic stirring bar and powdered 4 Å molecular sieves (20 mg/mL) and flame-dried for 10 minutes under high vacuum. After cooling to ambient temperature methyl amide (1.0 equiv) was added and the flask subjected to the glovebox. After addition of Yb(OTf)3 (10 mol %) the flask was removed from the glovebox and THF (4.0 to 12.0 mL/mmol) and the corresponding aldehyde (0.9 to 1.2 equiv) were added subsequently. The resulting mixture was stirred at ambient temperature for 24 h. The suspension was filtered over Celite and the solvent was removed in vacuo.
(2S,5S)-5-((N-Methyl-1H-imidazol-4-yl)methyl)-3-methyl-2-(naphthalen-1-yl)imidazolidin-4-one (7b). (S)-N-Methylhistidine methyl amide (190 mg, 1.04 mmol, 1.0 equiv), 1-naphthyl carbaldehyde (180 mg, 156 µL, 1.15 mmol, 1.1 equiv), 4 Å molecular sieves (240 mg) and Yb(OTf)3 (64.5 mg, 104 µmol, 10 mol %) in THF (12 mL) were reacted according to the GP. Silica gel column chromatography (DCM/MeOH 1:0 → 50:1) afforded the desired (S,S)-diastereomer 7b (140 mg, 437 µmol, 42%) as pale yellow oil: 1H NMR (200 MHz, MeOD) δ 2.65 (s, 3H), 3.05 (dd, J = 9.4, 5.1 Hz, 2H), 3.70 (s, 3H), 3.91 (t, J = 5.1 Hz, 1H), 6.09 (br s, 1H), 6.87–6.98 (m, 1H), 6.97–7.12 (m, 1H), 7.43–7.58 (m, 4H), 7.84–8.00 (m, 2H), 8.02–8.16 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 28.7, 33.3, 60.4, 60.4, 118.3, 122.7, 125.1, 125.1, 126.0, 126.6, 126.7, 128.8, 137.2, 137.3, 137.8 (the carbonyl signal as well as one aliphatic, one aromatic CH and one aromatic Cq-signal are missing); ![[Graphic 24]](/bjoc/content/inline/1860-5397-13-254-i25.svg?max-width=637&scale=1.18182) (c 1.0, CHCl3) +38.0°; IR (ATR)
(c 1.0, CHCl3) +38.0°; IR (ATR) ![[Graphic 25]](/bjoc/content/inline/1860-5397-13-254-i26.svg?max-width=637&scale=1.18182) (cm−1): 3349, 2922, 1683, 1261, 1159; HRMS (ESI) m/z: calcd for C19H20N4O, 343.1529; found, 343.1529.
(cm−1): 3349, 2922, 1683, 1261, 1159; HRMS (ESI) m/z: calcd for C19H20N4O, 343.1529; found, 343.1529.
(2S,5S)-5-((Indol-3-yl)methyl)-3-methyl-2-(naphthalen-1-yl)imidazolidin-4-one (7c). (S)-Tryptophan methyl amide (340 mg, 1.56 mmol, 1.0 equiv), 1-naphthyl carbaldehyde (243 mg, 212 µL, 1.56 mmol, 1.0 equiv), 4 Å molecular sieves (160 mg) and Yb(OTf)3 (97.0 mg, 156 µmol, 10 mol %) in THF (8.0 mL) were reacted according to the GP. Silica gel column chromatography (n-pentane/EtOAc 1:1 → 2:3 → 0:1) gave the desired (S,S)-diastereomer 7c (250 mg, 203 µmol, 45%) as pale yellow solid: mp 145 °C; 1H NMR (300 MHz, MeOD) δ 2.52 (s, 3H), 3.23 (dd, J = 14.8, 5.3 Hz, 1H), 3.49 (dd, J = 14.8, 3.9 Hz, 1H), 3.85–3.96 (m, 1H), 5.98 (br s, 1H), 6.86–6.98 (m, 1H), 6.99–7.17 (m, 4H), 7.33–7.47 (m, 3H), 7.53 (d, J = 7.9 Hz, 1H), 7.69–7.87 (m, 3H); 13C NMR (75 MHz, MeOD) δ 30.6, 30.6, 64.3, 112.2, 115.0, 122.3, 122.7, 125.3, 125.6, 128.2, 128.9, 129.6, 130.3, 131.6, 132.5, 133.3, 133.4, 134.8, 137.9, 140.9, 180.3 (one aliphatic CH and one aromatic CH are missing); ![[Graphic 26]](/bjoc/content/inline/1860-5397-13-254-i27.svg?max-width=637&scale=1.18182) (c 1.0, CHCl3) −6.0°; IR (ATR)
(c 1.0, CHCl3) −6.0°; IR (ATR) ![[Graphic 27]](/bjoc/content/inline/1860-5397-13-254-i28.svg?max-width=637&scale=1.18182) (cm−1): 3404, 3280, 1681, 1435, 1097; HRMS (ESI) m/z: calcd for C23H21N3O, 378.1577; found, 378.1578.
(cm−1): 3404, 3280, 1681, 1435, 1097; HRMS (ESI) m/z: calcd for C23H21N3O, 378.1577; found, 378.1578.
(2S,5S)-5-((N-Benzylindol-3-yl)methyl)-3-methyl-2-(naphthalen1-yl)imidazolidin-4-one (7d). (S)-N-Benzyltryptophan methyl amide (307 mg, 1.00 mmol, 1.0 equiv), 1-naphthyl carbaldehyde (156 mg, 136 µL, 1.10 mmol, 1.1 equiv), 4 Å molecular sieves (120 mg) and Yb(OTf)3 (62.0 mg, 100 µmol, 10 mol %) in THF (6.0 mL) were reacted according to the GP. Silica gel column chromatography (n-pentane/EtOAc 1:0 → 50:1) the desired (S,S)-diastereomer 7d (188 mg, 422 µmol, 42%) as pale yellow solid: mp 120 °C (dec); 1H NMR (200 MHz, MeOD) δ 2.54 (s, 3H), 3.11–3.32 (dd, J = 14.9, 5.5 Hz, 1H), 3.51 (dd, J = 14.9, 3.6 Hz, 1H), 3.88–3.96 (m, 1H), 5.23 (s, 2H), 6.01 (br s, 1H), 6.87–7.22 (m, 11H), 7.32–7.48 (m, 3H), 7.55 (d, J = 7.9 Hz, 1H), 7.71 (d, J = 8.3 Hz, 1H), 7.83 (d, J = 8.4 Hz, 1H); 13C NMR (150 MHz, CDCl3) δ 26.3, 27.9, 49.9, 60.3, 109.3, 109.6, 119.6, 122.0, 122.3, 125.3, 126.0, 126.7, 126.7, 127.5, 127.8, 128.3, 128.6, 128.8, 131.0, 133.7, 136.8, 137.2, 176.1 (one aliphatic, two aromatic CH and one aromatic Cq-signals are missing); ![[Graphic 28]](/bjoc/content/inline/1860-5397-13-254-i29.svg?max-width=637&scale=1.18182) (c 1.0, CHCl3) −6.0°; IR (ATR)
(c 1.0, CHCl3) −6.0°; IR (ATR) ![[Graphic 29]](/bjoc/content/inline/1860-5397-13-254-i30.svg?max-width=637&scale=1.18182) (cm−1): 3491, 1684, 1259, 1170, 1035; HRMS (ESI) m/z: calcd for C30H27N3O, 468.2046; found, 468.2050.
(cm−1): 3491, 1684, 1259, 1170, 1035; HRMS (ESI) m/z: calcd for C30H27N3O, 468.2046; found, 468.2050.
(2S,5S)-5-(p-Nitrobenzyl)-3-methyl-2-(naphthalen-1-yl)imidazolidin-4-one (7e). (S)-p-Nitrophenylalanine methyl amide (892 mg, 4.00 mmol, 1.0 equiv), 1-naphthyl carbaldehyde (625 mg, 543 µL, 4.00 mmol, 1.0 equiv), 4 Å molecular sieves (480 mg) and Yb(OTf)3 (248 mg, 400 µmol, 10 mol %) in THF (24 mL) were reacted according to the GP. Silica gel column chromatography (n-pentane/EtOAc 2:1 → 1:1 → 0:1) gave the desired (S,S)-diastereomer 7e (180 mg, 498 µmol, 12%) as pale yellow solid: mp 75 °C; 1H NMR (300 MHz, CDCl3) δ 2.71 (s, 3H), 3.16–3.37 (m, 2H), 4.02 (dd, J = 5.1, 5.1 Hz, 1H), 5.99 (br s, 1H), 7.02 (br s, 1H), 7.28–7.41 (m, 3H), 7.41– 7.57 (m, 2H), 7.79–7.94 (m, 2H), 7.94–8.06 (m, 1H), 8.05–8.13 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 28.0, 37.3, 60.4, 77.2, 122.3, 123.7, 125.2, 126.2, 126.8, 129.1, 130.0, 130.6, 131.0, 134.0, 144.9, 147.0 (the carbonyl and two aromatic CH signals are missing); ![[Graphic 30]](/bjoc/content/inline/1860-5397-13-254-i31.svg?max-width=637&scale=1.18182) (c 1.0, CHCl3) −26.0°; IR (ATR)
(c 1.0, CHCl3) −26.0°; IR (ATR) ![[Graphic 31]](/bjoc/content/inline/1860-5397-13-254-i32.svg?max-width=637&scale=1.18182) (cm−1): 3324, 1690, 1513, 1342, 1107; HRMS (ESI) m/z: calcd for C21H19N3O3, 384.1319; found, 384.1319.
(cm−1): 3324, 1690, 1513, 1342, 1107; HRMS (ESI) m/z: calcd for C21H19N3O3, 384.1319; found, 384.1319.
(2S,5S)-3-Methyl-2-(naphthalen-1-yl)-5-propylimidazolidin-4-one (7f). (S)-Norvaline methyl amide (521 mg, 4.00 µmol, 1.0 equiv), 1-naphthyl carbaldehyde (562 mg, 489 µL, 3.60 mmol, 0.9 equiv), 4 Å molecular sieves (480 mg) and Yb(OTf)3 (248 mg, 400 µmol, 10 mol %) in THF (24 mL) were reacted according to GP. Silica gel column chromatography (n-pentane/EtOAc 4:1 → 1:1) gave the desired (S,S)-diastereomer 7f (440 mg, 1.64 mmol, 46%) as pale yellow oil. 1H NMR (200 MHz, CDCl3) δ 0.94 (t, J = 7.2 Hz, 3H), 1.34–1.71 (m, 3H), 1.87 (br s, 1H, NH), 1.89–2.13 (m, 1H), 3.57–3.70 (m, 1H), 6.01 (br s, 1H), 7.40–7.61 (m, 4H), 7.83–8.00 (m, 2H), 8.15–8.29 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 13.9, 19.2, 28.1, 34.4, 59.8, 77.2, 122.6, 125.4, 126.2, 126.9, 129.0, 129.8, 131.2, 131.4, 133.7, 134.1, 176.5; ![[Graphic 32]](/bjoc/content/inline/1860-5397-13-254-i33.svg?max-width=637&scale=1.18182) (c 1.0, CHCl3) +89.0°; IR (ATR)
(c 1.0, CHCl3) +89.0°; IR (ATR) ![[Graphic 33]](/bjoc/content/inline/1860-5397-13-254-i34.svg?max-width=637&scale=1.18182) (cm−1): 3321, 2957, 1689, 1396, 1322; HRMS (ESI) m/z: calcd for C17H20N2O, 291.1468; found, 291.1468.
(cm−1): 3321, 2957, 1689, 1396, 1322; HRMS (ESI) m/z: calcd for C17H20N2O, 291.1468; found, 291.1468.
(2S,5S)-3-Methyl-5-(2-(methylthio)ethyl)-2-(naphthalen-1-yl)imidazolidin-4-one (7g). (S)-Methionine methyl amide (649 mg, 4.00 µmol, 1.0 equiv), 1-naphthyl carbaldehyde (562 mg, 489 µL, 3.60 mmol, 0.9 equiv), 4 Å molecular sieves (480 mg) and Yb(OTf)3 (248 mg, 400 µmol, 10 mol %) in THF (24 mL) were reacted according to the GP. Silica gel column chromatography (n-pentane/EtOAc 2:1 → 1:2) gave the desired (S,S)-diastereomer 7g (350 mg, 1.17 mmol, 32%) as yellow oil. 1H NMR (300 MHz, CDCl3) δ 1.83–1.98 (m, 1H), 2.07 (s, 3H), 2.19 (br s, 1H, NH), 2.24–2.38 (m, 1H), 2.58–2.72 (m, 2H), 2.75 (s, 3H), 3.75–3.89 (m, 1H), 6.03 (br s, 1H), 7.45–7.60 (m, 4H), 7.85–7.97 (m, 3H), 8.20–8.25 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 15.2, 28.0, 30.6, 31.5, 58.7, 77.2, 122.6, 125.4, 126.2, 126.9, 129.0, 129.9, 131.2, 133.6, 134.1, 175.5 (one aromatic CH-signal is missing); ![[Graphic 34]](/bjoc/content/inline/1860-5397-13-254-i35.svg?max-width=637&scale=1.18182) (c 1.0, CHCl3) +66.0°; IR (ATR)
(c 1.0, CHCl3) +66.0°; IR (ATR) ![[Graphic 35]](/bjoc/content/inline/1860-5397-13-254-i36.svg?max-width=637&scale=1.18182) (cm−1): 3321, 2957, 1689, 1396, 1322; HRMS (ESI) m/z: calcd for C17H20N2OS, 323.1189; found, 323.1191.
(cm−1): 3321, 2957, 1689, 1396, 1322; HRMS (ESI) m/z: calcd for C17H20N2OS, 323.1189; found, 323.1191.
(2R,5S)-5-Benzyl-3-methyl-2-(pyren-1-yl)imidazolidin-4-one ((R,S)-3b) and (2S,5S)-5-benzyl-3-methyl-2-(pyren-1-yl)imidazolidin-4-one ((S,S)-3b). (S)-Phenylalanine methyl amide (0.89 g, 5.0 mmol, 1.0 equiv), pyrene-1-carbaldehyde (1.73g, 7.50 mmol, 1.5 equiv), 4 Å molecular sieves (400 mg) and Yb(OTf)3 (310 mg, 500 µmol, 10 mol %) in THF (20 mL) were reacted according to the GP. Silica gel column chromatography (n-pentane/EtOAc 4:1 → 2:1 → 1:2) gave the faster eluting (R,S)-diastereomer (R,S)-3b (801 mg, 2.05 mmol, 41%) and the desired (S,S)-diastereomer (S,S)-3b (952 mg, 2.44 mmol, 49%) as yellow solids. Analytical data of (R,S)-3b: mp 70 °C; 1H NMR (600 MHz, CDCl3) δ 2.36 (br s, 1H), 2.70 (s, 3H), 3.10–3.24 (m, 2H), 4.15 (dd, J = 5.2, 5.2 Hz, 1H), 5.74 (s, 1H), 7.29 (ddt, J = 9.2, 7.4, 1.2 Hz, 1H), 7.34–7.39 (m, 2H), 7.41–7.45 (m, 2H), 7.82 (d, J = 7.4 Hz, 1H), 8.02–8.07 (m, 2H), 8.10 (d, J = 9.2 Hz, 1H), 8.15–8.24 (m, 5H); 13C NMR (150 MHz, CDCl3) δ 27.9, 38.8, 60.2, 121.6, 122.8, 124.6, 125.1, 125.2, 125.5, 125.8, 126.3, 126.8, 127.2, 128.1, 128.4, 128.6, 129.1, 129.9, 130.5, 131.2, 131.2, 131.7, 137.7, 175.4 (one aliphatic CH-signal is missing); ![[Graphic 36]](/bjoc/content/inline/1860-5397-13-254-i37.svg?max-width=637&scale=1.18182) (c 1.0, CHCl3) −62.0°; IR (ATR)
(c 1.0, CHCl3) −62.0°; IR (ATR) ![[Graphic 37]](/bjoc/content/inline/1860-5397-13-254-i38.svg?max-width=637&scale=1.18182) (cm−1): 3310, 3029, 1688, 1395, 1086; HRMS (ESI) m/z: calcd for C27H22N2O, 413.1624; found, 413.1626. Analytical data of (S,S)-3b: mp 95 °C; 1H NMR (600 MHz, CDCl3) δ 2.68 (s, 3H), 3.18 (br d, J = 14.2 Hz, 1H), 3.36 (dd, J = 14.2, 5.4 Hz, 1H), 4.04 (t, J = 5.4 Hz, 1H), 6.32 (br s, 1H), 7.17–7.25 (m, 2H), 7.24–7.31 (m, 5H), 7.95–8.01 (m, 1H), 8.01–8.08 (m, 2H), 8.11 (d, J = 8.9 Hz, 1H), 8.19–8.25 (m, 2H), 8.28–8.39 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 27.9, 36.7, 60.9, 121.3, 124.6, 124.8, 125.3, 125.5, 125.8, 126.2, 127.0, 127.2, 128.2, 128.6, 128.9, 129.4, 129.8, 130.5, 131.2, 136.5, 175.0 (one aliphatic, two aromatic CH-signals as well as one aromatic Cq-signal are missing);
(cm−1): 3310, 3029, 1688, 1395, 1086; HRMS (ESI) m/z: calcd for C27H22N2O, 413.1624; found, 413.1626. Analytical data of (S,S)-3b: mp 95 °C; 1H NMR (600 MHz, CDCl3) δ 2.68 (s, 3H), 3.18 (br d, J = 14.2 Hz, 1H), 3.36 (dd, J = 14.2, 5.4 Hz, 1H), 4.04 (t, J = 5.4 Hz, 1H), 6.32 (br s, 1H), 7.17–7.25 (m, 2H), 7.24–7.31 (m, 5H), 7.95–8.01 (m, 1H), 8.01–8.08 (m, 2H), 8.11 (d, J = 8.9 Hz, 1H), 8.19–8.25 (m, 2H), 8.28–8.39 (m, 1H); 13C NMR (75 MHz, CDCl3) δ 27.9, 36.7, 60.9, 121.3, 124.6, 124.8, 125.3, 125.5, 125.8, 126.2, 127.0, 127.2, 128.2, 128.6, 128.9, 129.4, 129.8, 130.5, 131.2, 136.5, 175.0 (one aliphatic, two aromatic CH-signals as well as one aromatic Cq-signal are missing); ![[Graphic 38]](/bjoc/content/inline/1860-5397-13-254-i39.svg?max-width=637&scale=1.18182) (c 1.0, CHCl3) −10.0°; IR (ATR)
(c 1.0, CHCl3) −10.0°; IR (ATR) ![[Graphic 39]](/bjoc/content/inline/1860-5397-13-254-i40.svg?max-width=637&scale=1.18182) (cm−1): 3475, 2914, 1682, 1255, 1033; HRMS (ESI) m/z: calcd for C27H22N2O, 413.1624; found, 413.1627.
(cm−1): 3475, 2914, 1682, 1255, 1033; HRMS (ESI) m/z: calcd for C27H22N2O, 413.1624; found, 413.1627.
(2S,5S)-5-Benzyl-3-methyl-2-mesitylimidazolidin-4-one (3c). (S)-Phenylalanine methyl amide (71.3 mg, 400 µmol, 1.0 equiv), mesityl carbaldehyde (65.2 mg, 64.9 µL, 440 µmol, 1.1 equiv), 4 Å molecular sieves (40 mg) and Yb(OTf)3 (25 mg, 40 µmol, 10 mol %) in THF (2.0 mL) were reacted according to the GP. Silica gel column chromatography (n-pentane/EtOAc 3:1 → 1:1) gave the desired (S,S)-diastereomer 3c (64.1 mg, 208 µmol, 52%) as a white solid: mp 75 °C; 1H NMR (600 MHz, CDCl3) δ 1.80 (s, 3H), 2.24 (s, 3H), 2.37 (s, 3H), 2.58 (s, 3H), 3.18 (ddd, J = 14.0, 6.9, 0.9 Hz, 1H), 3.24 (ddd, J = 14.0, 3.8, 0.9 Hz, 1H), 3.83 (ddd, J = 6.9, 3.8, 1.7 Hz, 1H), 5.72 (d, J = 1.7 Hz, 1H), 6.75 (s, 1H), 6.83 (s, 1H), 7.20–7.24 (m, 1H), 7.25–7.28 (m, 4H); 13C NMR (150 MHz, CDCl3) δ 18.4, 20.3, 20.6, 26.8, 35.8, 60.6, 72.8, 126.7, 128.2, 128.5, 129.5, 129.6, 131.5, 137.1, 137.4, 137.9, 138.4, 173.8; ![[Graphic 40]](/bjoc/content/inline/1860-5397-13-254-i41.svg?max-width=637&scale=1.18182) (c 1.0, CHCl3) −62.0°; IR (ATR)
(c 1.0, CHCl3) −62.0°; IR (ATR) ![[Graphic 41]](/bjoc/content/inline/1860-5397-13-254-i42.svg?max-width=637&scale=1.18182) (cm−1): 3315, 2921, 1693, 1433, 1309; HRMS (ESI) m/z: calcd for C20H24N2O, 331.1781; found, 331.1781.
(cm−1): 3315, 2921, 1693, 1433, 1309; HRMS (ESI) m/z: calcd for C20H24N2O, 331.1781; found, 331.1781.
(2S,5S)-5-Benzyl-3-methyl-2-(2,4,6-triisopropylphen-1-yl)imidazolidin-4-one (3d). (S)-Phenylalanine methyl amide (217 mg, 1.00 µmol, 1.0 equiv), 2,4,6-triisopropylbenzaldehyde (255 mg, 273 µL, 1.10 mmol, 1.1 equiv), 4 Å molecular sieves (80 mg) and Yb(OTf)3 (62.1 mg, 100 µmol, 10 mol %) in THF (4.0 mL) were reacted according to the GP. Silica gel column chromatography (n-pentane/EtOAc 10:1 → 2:1) gave the desired (S,S)-diastereomer 3d (90.2 mg, 230 µmol, 23%) as a colorless oil: 1H NMR (600 MHz, CDCl3) δ 1.13 (d, J = 6.9 Hz, 3H), 1.15 (d, J = 6.9 Hz, 3H), 1.24 (d, J = 6.9 Hz, 12H), 2.69 (s, 3H), 2.83–2.90 (m, 2H), 3.30 (hept, J = 6.9 Hz, 1H), 3.41 (dd, J = 14.1, 3.4 Hz, 1H), 3.46 (hept, J = 6.9 Hz, 1H), 3.84 (d, J = 8.7 Hz, 1H), 5.92 (br s, 1H), 7.00 (d, J = 1.9 Hz, 1H), 7.07 (d, J = 1.9 Hz, 1H), 7.17–7.25 (m, 1H), 7.26–7.30 (m, 4H); 13C NMR (150 MHz, CDCl3) δ 23.8, 23.8, 24.2, 24.3, 24.7, 25.7, 27.5, 27.5, 29.1, 34.1, 37.7, 61.3, 71.5, 121.2, 123.6, 126.0, 126.6, 128.6, 129.2, 138.2, 148.4, 149.7, 150.1, 173.7; ![[Graphic 42]](/bjoc/content/inline/1860-5397-13-254-i43.svg?max-width=637&scale=1.18182) (c 1.0, CHCl3) −102°; IR (ATR)
(c 1.0, CHCl3) −102°; IR (ATR) ![[Graphic 43]](/bjoc/content/inline/1860-5397-13-254-i44.svg?max-width=637&scale=1.18182) (cm−1): 3548, 2957, 1683, 1397, 1098; HRMS (ESI) m/z: calcd for C26H36N2O, 415.2720; found, 415.2722.
(cm−1): 3548, 2957, 1683, 1397, 1098; HRMS (ESI) m/z: calcd for C26H36N2O, 415.2720; found, 415.2722.
Supporting Information
| Supporting Information File 1: Copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 698.3 KB | Download |
References
-
Knoevenagel, E. Ber. Dtsch. Chem. Ges. 1898, 31, 2596–2619. doi:10.1002/cber.18980310308
Return to citation in text: [1] -
Erkkilä, A.; Majander, I.; Pihko, P. M. Chem. Rev. 2007, 107, 5416–5470. doi:10.1021/cr068388p
Return to citation in text: [1] -
Mukherjee, S.; Yang, J. W.; Hoffmann, B.; List, B. Chem. Rev. 2007, 107, 5471–5569. doi:10.1021/cr0684016
Return to citation in text: [1] -
Moyano, A.; Rios, R. Chem. Rev. 2011, 111, 4703–4832. doi:10.1021/cr100348t
Return to citation in text: [1] -
Nielsen, M.; Worgull, D.; Zweifel, T.; Gschwend, B.; Bertelsen, S.; Jørgensen, K. A. Chem. Commun. 2011, 47, 632–649. doi:10.1039/C0CC02417A
Return to citation in text: [1] -
Torres, R. R., Ed. Stereoselective Organocatalysis: Bond Formation Methodologies and Activation Modes; Wiley-VCH: Weinheim, Germany, 2013. doi:10.1002/9781118604755
Return to citation in text: [1] -
Hayashi, Y. Chem. Sci. 2016, 7, 866–880. doi:10.1039/C5SC02913A
Return to citation in text: [1] -
Woodward, R. B.; Logusch, E.; Nambiar, K. P.; Sakan, K.; Ward, D. E.; Au-Yeung, B.-W.; Balaram, P.; Browne, L. J.; Card, P. J.; Chen, C. H.; Chênevert, R. B.; Fliri, A.; Frobel, K.; Gais, H.-J.; Garratt, D. G.; Hayakawa, K.; Heggie, W.; Hesson, D. P.; Hoppe, D.; Hoppe, I.; Hyatt, J. A.; Ikeda, D.; Jacobi, P. A.; Kim, K. S.; Kobuke, Y.; Kojima, K.; Krowicki, K.; Lee, V. J.; Leutert, T.; Malchenko, S.; Martens, J.; Matthews, R. S.; Ong, B. S.; Press, J. B.; Rajan Babu, T. V.; Rousseau, G.; Sauter, H. M.; Suzuki, M.; Tatsuta, K.; Tolbert, L. M.; Truesdale, E. A.; Uchida, I.; Ueda, Y.; Uyehara, T.; Vasella, A. T.; Vladuchick, W. C.; Wade, P. A.; Williams, R. M.; Wong, H. N.-C. J. Am. Chem. Soc. 1981, 103, 3210–3213. doi:10.1021/ja00401a049
Return to citation in text: [1] -
Yamaguchi, M.; Yokota, N.; Minami, T. J. Chem. Soc., Chem. Commun. 1991, 1088–1089. doi:10.1039/C39910001088
Return to citation in text: [1] -
Zhuang, W.; Marigo, M.; Jørgensen, K. A. Org. Biomol. Chem. 2005, 3, 3883–3885. doi:10.1039/b512542a
Return to citation in text: [1] -
Marigo, M.; Wabnitz, T. C.; Fielenbach, D.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2005, 44, 794–797. doi:10.1002/anie.200462101
Return to citation in text: [1] -
Hayashi, Y.; Gotoh, H.; Hayashi, T.; Shoji, M. Angew. Chem., Int. Ed. 2005, 44, 4212–4215. doi:10.1002/anie.200500599
Return to citation in text: [1] -
Ahrendt, K. A.; Borths, C. J.; MacMillan, D. W. C. J. Am. Chem. Soc. 2000, 122, 4243–4244. doi:10.1021/ja000092s
Return to citation in text: [1] -
Jen, W. S.; Wiener, J. J. M.; MacMillan, D. W. C. J. Am. Chem. Soc. 2000, 122, 9874–9875. doi:10.1021/ja005517p
Return to citation in text: [1] -
Paras, N. A.; MacMillan, D. W. C. J. Am. Chem. Soc. 2001, 123, 4370–4371. doi:10.1021/ja015717g
Return to citation in text: [1] -
Brown, S. P.; Goodwin, N. C.; MacMillan, D. W. C. J. Am. Chem. Soc. 2003, 125, 1192–1194. doi:10.1021/ja029095q
Return to citation in text: [1] -
Harmata, M.; Ghosh, S. K.; Hong, X.; Wacharasindhu, S.; Kirchhoefer, P. J. Am. Chem. Soc. 2003, 125, 2058–2059. doi:10.1021/ja029058z
Return to citation in text: [1] -
Mangion, I. K.; Northrup, A. B.; MacMillan, D. W. C. Angew. Chem., Int. Ed. 2004, 43, 6722–6724. doi:10.1002/anie.200461851
Return to citation in text: [1] -
Fjelbye, K.; Marigo, M.; Clausen, R. P.; Juhl, K. Org. Lett. 2016, 18, 1170–1173. doi:10.1021/acs.orglett.6b00293
Return to citation in text: [1] -
Yang, M.; Yang, X.; Sun, H.; Li, A. Angew. Chem., Int. Ed. 2016, 55, 2851–2855. doi:10.1002/anie.201510568
Return to citation in text: [1] -
Sun, W.-B.; Wang, X.; Sun, B.-F.; Zou, J.-P.; Lin, G.-Q. Org. Lett. 2016, 18, 1219–1221. doi:10.1021/acs.orglett.6b00150
Return to citation in text: [1] -
Xie, Z.; Zan, X.; Sun, S.; Pan, X.; Liu, L. Org. Lett. 2016, 18, 3944–3947. doi:10.1021/acs.orglett.6b01625
Return to citation in text: [1] -
Holland, M. C.; Metternich, J. B.; Daniliuc, C.; Schweizer, W. B.; Gilmour, R. Chem. – Eur. J. 2015, 21, 10031–10038. doi:10.1002/chem.201500270
Return to citation in text: [1] -
Welin, E. R.; Warkentin, A. A.; Conrad, J. C.; MacMillan, D. W. C. Angew. Chem., Int. Ed. 2015, 54, 9668–9672. doi:10.1002/anie.201503789
Return to citation in text: [1] -
Wallbaum, J.; Garve, L. K. B.; Jones, P. G.; Werz, D. B. Chem. – Eur. J. 2016, 22, 18756–18759. doi:10.1002/chem.201605265
Return to citation in text: [1] -
Paras, N. A.; MacMillan, D. W. C. J. Am. Chem. Soc. 2002, 124, 7894–7895. doi:10.1021/ja025981p
See as an example of an experimental procedure.
Return to citation in text: [1] -
Jain, R.; Cohen, L. A. Tetrahedron 1996, 52, 5363–5370. doi:10.1016/0040-4020(96)00187-1
Return to citation in text: [1] -
Daka, P.; Liu, A.; Karunaratne, C.; Csatary, E.; Williams, C.; Xiao, H.; Lin, J.; Xu, Z.; Page, R. C.; Wang, H. Bioorg. Med. Chem. 2015, 23, 1348–1355. doi:10.1016/j.bmc.2015.01.025
Return to citation in text: [1] -
Magnus, P.; Mugrage, B.; DeLuca, M. R.; Cain, G. A. J. Am. Chem. Soc. 1990, 112, 5220–5230. doi:10.1021/ja00169a033
Return to citation in text: [1] -
Samulis, L.; Tomkinson, N. C. O. Tetrahedron 2011, 67, 4263–4267. doi:10.1016/j.tet.2011.04.009
Return to citation in text: [1]
| 1. | Knoevenagel, E. Ber. Dtsch. Chem. Ges. 1898, 31, 2596–2619. doi:10.1002/cber.18980310308 |
| 19. | Fjelbye, K.; Marigo, M.; Clausen, R. P.; Juhl, K. Org. Lett. 2016, 18, 1170–1173. doi:10.1021/acs.orglett.6b00293 |
| 13. | Ahrendt, K. A.; Borths, C. J.; MacMillan, D. W. C. J. Am. Chem. Soc. 2000, 122, 4243–4244. doi:10.1021/ja000092s |
| 14. | Jen, W. S.; Wiener, J. J. M.; MacMillan, D. W. C. J. Am. Chem. Soc. 2000, 122, 9874–9875. doi:10.1021/ja005517p |
| 15. | Paras, N. A.; MacMillan, D. W. C. J. Am. Chem. Soc. 2001, 123, 4370–4371. doi:10.1021/ja015717g |
| 16. | Brown, S. P.; Goodwin, N. C.; MacMillan, D. W. C. J. Am. Chem. Soc. 2003, 125, 1192–1194. doi:10.1021/ja029095q |
| 17. | Harmata, M.; Ghosh, S. K.; Hong, X.; Wacharasindhu, S.; Kirchhoefer, P. J. Am. Chem. Soc. 2003, 125, 2058–2059. doi:10.1021/ja029058z |
| 18. | Mangion, I. K.; Northrup, A. B.; MacMillan, D. W. C. Angew. Chem., Int. Ed. 2004, 43, 6722–6724. doi:10.1002/anie.200461851 |
| 8. | Woodward, R. B.; Logusch, E.; Nambiar, K. P.; Sakan, K.; Ward, D. E.; Au-Yeung, B.-W.; Balaram, P.; Browne, L. J.; Card, P. J.; Chen, C. H.; Chênevert, R. B.; Fliri, A.; Frobel, K.; Gais, H.-J.; Garratt, D. G.; Hayakawa, K.; Heggie, W.; Hesson, D. P.; Hoppe, D.; Hoppe, I.; Hyatt, J. A.; Ikeda, D.; Jacobi, P. A.; Kim, K. S.; Kobuke, Y.; Kojima, K.; Krowicki, K.; Lee, V. J.; Leutert, T.; Malchenko, S.; Martens, J.; Matthews, R. S.; Ong, B. S.; Press, J. B.; Rajan Babu, T. V.; Rousseau, G.; Sauter, H. M.; Suzuki, M.; Tatsuta, K.; Tolbert, L. M.; Truesdale, E. A.; Uchida, I.; Ueda, Y.; Uyehara, T.; Vasella, A. T.; Vladuchick, W. C.; Wade, P. A.; Williams, R. M.; Wong, H. N.-C. J. Am. Chem. Soc. 1981, 103, 3210–3213. doi:10.1021/ja00401a049 |
| 9. | Yamaguchi, M.; Yokota, N.; Minami, T. J. Chem. Soc., Chem. Commun. 1991, 1088–1089. doi:10.1039/C39910001088 |
| 10. | Zhuang, W.; Marigo, M.; Jørgensen, K. A. Org. Biomol. Chem. 2005, 3, 3883–3885. doi:10.1039/b512542a |
| 11. | Marigo, M.; Wabnitz, T. C.; Fielenbach, D.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2005, 44, 794–797. doi:10.1002/anie.200462101 |
| 12. | Hayashi, Y.; Gotoh, H.; Hayashi, T.; Shoji, M. Angew. Chem., Int. Ed. 2005, 44, 4212–4215. doi:10.1002/anie.200500599 |
| 30. | Samulis, L.; Tomkinson, N. C. O. Tetrahedron 2011, 67, 4263–4267. doi:10.1016/j.tet.2011.04.009 |
| 2. | Erkkilä, A.; Majander, I.; Pihko, P. M. Chem. Rev. 2007, 107, 5416–5470. doi:10.1021/cr068388p |
| 3. | Mukherjee, S.; Yang, J. W.; Hoffmann, B.; List, B. Chem. Rev. 2007, 107, 5471–5569. doi:10.1021/cr0684016 |
| 4. | Moyano, A.; Rios, R. Chem. Rev. 2011, 111, 4703–4832. doi:10.1021/cr100348t |
| 5. | Nielsen, M.; Worgull, D.; Zweifel, T.; Gschwend, B.; Bertelsen, S.; Jørgensen, K. A. Chem. Commun. 2011, 47, 632–649. doi:10.1039/C0CC02417A |
| 6. | Torres, R. R., Ed. Stereoselective Organocatalysis: Bond Formation Methodologies and Activation Modes; Wiley-VCH: Weinheim, Germany, 2013. doi:10.1002/9781118604755 |
| 7. | Hayashi, Y. Chem. Sci. 2016, 7, 866–880. doi:10.1039/C5SC02913A |
| 24. | Welin, E. R.; Warkentin, A. A.; Conrad, J. C.; MacMillan, D. W. C. Angew. Chem., Int. Ed. 2015, 54, 9668–9672. doi:10.1002/anie.201503789 |
| 26. |
Paras, N. A.; MacMillan, D. W. C. J. Am. Chem. Soc. 2002, 124, 7894–7895. doi:10.1021/ja025981p
See as an example of an experimental procedure. |
| 23. | Holland, M. C.; Metternich, J. B.; Daniliuc, C.; Schweizer, W. B.; Gilmour, R. Chem. – Eur. J. 2015, 21, 10031–10038. doi:10.1002/chem.201500270 |
| 27. | Jain, R.; Cohen, L. A. Tetrahedron 1996, 52, 5363–5370. doi:10.1016/0040-4020(96)00187-1 |
| 28. | Daka, P.; Liu, A.; Karunaratne, C.; Csatary, E.; Williams, C.; Xiao, H.; Lin, J.; Xu, Z.; Page, R. C.; Wang, H. Bioorg. Med. Chem. 2015, 23, 1348–1355. doi:10.1016/j.bmc.2015.01.025 |
| 29. | Magnus, P.; Mugrage, B.; DeLuca, M. R.; Cain, G. A. J. Am. Chem. Soc. 1990, 112, 5220–5230. doi:10.1021/ja00169a033 |
| 22. | Xie, Z.; Zan, X.; Sun, S.; Pan, X.; Liu, L. Org. Lett. 2016, 18, 3944–3947. doi:10.1021/acs.orglett.6b01625 |
| 20. | Yang, M.; Yang, X.; Sun, H.; Li, A. Angew. Chem., Int. Ed. 2016, 55, 2851–2855. doi:10.1002/anie.201510568 |
| 21. | Sun, W.-B.; Wang, X.; Sun, B.-F.; Zou, J.-P.; Lin, G.-Q. Org. Lett. 2016, 18, 1219–1221. doi:10.1021/acs.orglett.6b00150 |
| 25. | Wallbaum, J.; Garve, L. K. B.; Jones, P. G.; Werz, D. B. Chem. – Eur. J. 2016, 22, 18756–18759. doi:10.1002/chem.201605265 |
© 2017 Wallbaum and Werz; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)