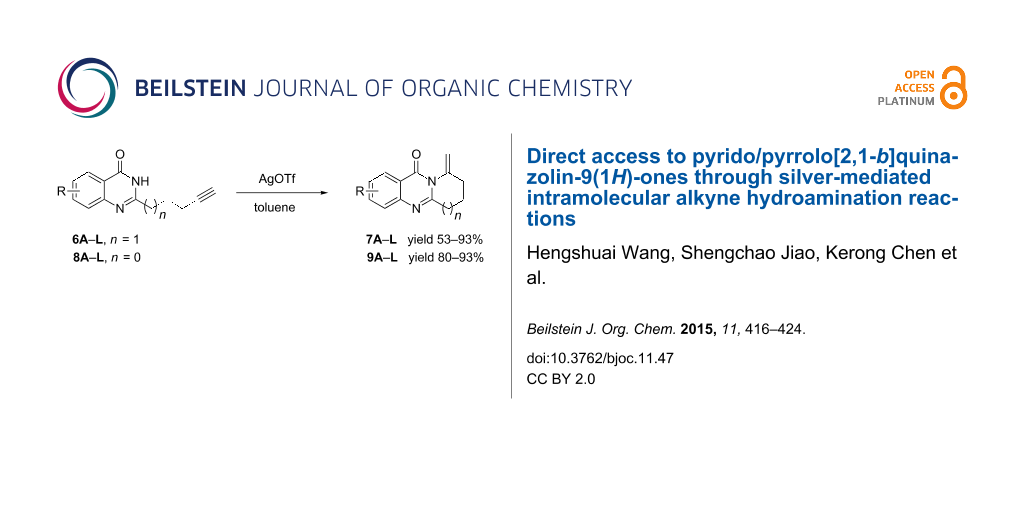
Abstract
We report a synthetic methodology for the construction of the fused heterocyclic compounds pyrido[2,1-b]quinazolin-9(1H)-ones and pyrrolo[2,1-b]quinazolin-9(1H)-ones through an AgOTf-catalyzed intramolecular alkyne hydroamination reaction. The methodology is applicable to a wide scope of substrates and produces a series of fused quinazolinone heterocycles in good to excellent yields.

Graphical Abstract
Introduction
Quinazolinone is a core skeleton for naturally existing phytochemicals. They were extracted from a variety of plant families. Among the quinazolinone derivatives, such as the pyrrolo[2,1-b]quinazolinone alkaloids, are a multitude of biomedically active substances [1,2]. For example, deoxyvasicione (1), 8-hydroxydeoxyvasicinone (2), compound 73/602 (3), mackinazolinone (4) and vasicinone (5) have been proven to act as bronchodilatory, anti-inflammatory, antimicrobial and antidepressant agents (Figure 1) [2-8].
![[1860-5397-11-47-1]](/bjoc/content/figures/1860-5397-11-47-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Selected structures of fused quinazolinones.
Figure 1: Selected structures of fused quinazolinones.
A variety of approaches have been employed to synthesize deoxyvasicione (1) and its derivatives, e.g., the Pd(OAc)2-catalyzed carbonyl-insertion reaction [7], the cycloaddition of anthranilic acid iminoketene to a methyl butyrolactam through a sulfinamide anhydride intermediate [9], the intramolecular aza-Wittig reaction with an azide substrate [10], and the cycloaddition of anthranilamide [11]. For the synthesis of vasicinone (5), deoxyvasicinone was subjected to a free-radical bromination using NBS and the subsequent treatment with NaOAc/AcOH as an acetoxylation reagent [12]. However, for most of these synthetic strategies harsh reaction conditions are a necessity, produce unstable sulfonamide anhydride intermediates [2,13], which are dangerous substrates bearing an azide group, and require a high reaction temperature and a long reaction time [2,10]. Recently, transition metal catalyzed hydroamination of alkynes [14-26], alkenes [15,27-31] and dienes [32,33] has been widely studied for the construction of heterocycles. We have reported on a highly efficient gold/silver-catalyzed intramolecular hydroamination of terminal alkynes in water for the synthesis of fused tricyclic xanthenes [34]. On the basis of this methodology, we have also afforded two fused benzimidazoles through silver-catalyzed intramolecular hydroamination from readily available starting materials with a long-chain alkyne [35,36]. Motivated by the unique structural properties and the biological activities characteristic of the vasicinone type alkaloids, we extended our work in this direction by elaborating the synthesis of fused quinazolinone derivatives. Herein, we present our recent findings of the synthesis of fused pyrrolo[2,1-b]quinazolin-9(1H)-ones by a silver-mediated chemoselective and regioselective intramolecular hydroamination cyclization (Scheme 1).
![[1860-5397-11-47-i1]](/bjoc/content/inline/1860-5397-11-47-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The intramolecular alkyne hydroamination of alkynes.
Scheme 1: The intramolecular alkyne hydroamination of alkynes.
Results and Discussion
To establish the overall best experimental conditions for the synthesis of pyrido/pyrrolo[2,1-b]quinazolin-9(1H)-ones, we chose 2-(4-pentynyl)-4(3H)-quinazolinone (6A) as a model substrate to prepare them by an intramolecular hydroamination cyclization. The results of these experiments are summarized in Table 1. Silver trifluoromethanesulfonate (AgOTf) seemed to be the most effective catalyst for this intramolecular hydroamination cyclization (Table 1, entries 1–6), whereas a product was not afforded in the absence of a catalyst (Table 1, entry 7). We also screened different solvents, and the results demonstrated that non-polar aprotic solvents could promote the reaction. Toluene was the most effective solvent for this cyclization (Table 1, entry 3 and entries 8–16). The concentration of the substrate in the reaction mixture also affected the product yield. When the concentration was changed from 0.1 M to 1 M, the yield dropped to 82% (Table 1, entry 17). Subsequently, we examined the influence of the reaction temperature, and no better yield could be obtained at a temperature either lower or higher than 80 °C (Table 1, entries 18 and 19). A prolongation of the reaction time to 12 h resulted in a slight decrease of the yield (Table 1, entry 20). Performing the reaction without inert gas (argon) atmosphere also led to a decrease of the yield (Table 1, entry 21). In summary, the optimum results were obtained when 2-(4-pentynyl)-4(3H)-quinazolinone (6A) in toluene was treated with 5 mol % of AgOTf in a sealed tube under argon protection at 80 °C for 3 h (Table 1, entry 3).
Table 1: Optimization of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-47-i3.svg?max-width=637&scale=1.0)
|
|||
| Entry | Catalyst | Solvent | Yield (%) |
|---|---|---|---|
| 1 | AgBF4 | toluene | 80 |
| 2 | AgSbF6 | toluene | 62 |
| 3 | AgOTf | toluene | 90 |
| 4 | AgNO3 | toluene | 39 |
| 5 | AgOCOCF3 | toluene | 85 |
| 6 | AgOAc | toluene | 41 |
| 7 | – | toluene | 0 |
| 8 | AgOTf | 1,2-dichloroethane | 82 |
| 9 | AgOTf | 1,4-dioxane | 70 |
| 10 | AgOTf | DME | 40 |
| 11 | AgOTf | THF | 47 |
| 12 | AgOTf | DMF | 37 |
| 13 | AgOTf | DMSO | 38 |
| 14 | AgOTf | MeCN | 73 |
| 15 | AgOTf | MeOH | 28 |
| 16 | AgOTf | EtOH | 28 |
| 17 | AgOTf | toluene | 82b |
| 18 | AgOTf | toluene | 70c |
| 19 | AgOTf | toluene | 83d |
| 20 | AgOTf | toluene | 85e |
| 21 | AgOTf | toluene | 71f |
a6A (0.2 mmol) and catalyst (5 mol %) in the specified solvent (2 mL) were heated in a sealed vial under argon protection at 80 °C for 3 h; bthe concentration of 6A is 1 M; cthe reaction temperature was 60 °C; dthe reaction temperature was 100 °C; ethe reaction time was 12 h; fthe reaction was performed without an argon inert gas atmosphere.
To evaluate the scope of the proposed silver-catalyzed intramolecular hydroamination cyclization reaction, we investigated its tolerance by probing changes in the substituted 2-(4-pentynyl)-4(3H)-quinazolinone (6A) under the optimum reaction conditions mentioned above (Table 2, entries 1–12). Various substituted 2-(4-pentynyl)-4(3H)-quinazolinones (6A–L) were tolerant of this transformation, and the desired products 7A–L were afforded with moderate to excellent yields (53–91%). It seems that the position and type of substituents on the 2-(4-pentynyl)-4(3H)-quinazolinones (6A) only slightly affected the yields of the target compounds (Table 2, entries 1–9). Higher yields could be obtained when the 6- and 7-positions of the 2-(4-pentynyl)-4(3H)-quinazolinone were substituted by methyl and methoxy groups (Table 2, entries 2–4). The introduction of a fluorine, a chlorine and a bromine atom at 5-, 6- and 7-positions resulted in a slight reduction of the yield of the products (Table 2, entries 5–9). However, a bulky phenyl group introduced at the 7-position led to a good yield (Table 2, entry 10). When the benzene ring of the skeleton of the substrate was replaced by a naphthalene ring, the product was obtained at a comparable yield of 87% (Table 2, entry 11). However, with 2-(4-pentynyl)-thieno[2,3-d]pyrimidin-4(1H)-one (6L) as a substrate, the reaction was significantly different compared to the other substituted 2-(4-pentynyl)-4(3H)-quinazolinones (Table 2, entry 12). Although the thieno analogue 6L was tolerated in the reaction, the cyclization required substantially longer (12 h), and the product was obtained in a relatively low yield (only 53%).
Table 2: Silver-mediated synthesis of target compounds 7A–L.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-47-i4.svg?max-width=637&scale=1.0)
|
|||
| Entry | Substrate | Product | Yield (%) |
|---|---|---|---|
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-47-i5.svg?max-width=637&scale=1.0)
|
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-47-i6.svg?max-width=637&scale=1.0)
7A |
90 |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-47-i7.svg?max-width=637&scale=1.0)
|
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-47-i8.svg?max-width=637&scale=1.0)
7B |
91 |
| 3 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-47-i9.svg?max-width=637&scale=1.0)
|
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-47-i10.svg?max-width=637&scale=1.0)
7C |
89 |
| 4 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-47-i11.svg?max-width=637&scale=1.0)
|
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-47-i12.svg?max-width=637&scale=1.0)
7D |
93 |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-47-i13.svg?max-width=637&scale=1.0)
|
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-47-i14.svg?max-width=637&scale=1.0)
7E |
82 |
| 6 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-47-i15.svg?max-width=637&scale=1.0)
|
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-47-i16.svg?max-width=637&scale=1.0)
7F |
83 |
| 7 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-47-i17.svg?max-width=637&scale=1.0)
|
![[Graphic 16]](/bjoc/content/inline/1860-5397-11-47-i18.svg?max-width=637&scale=1.0)
7G |
84 |
| 8 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-11-47-i19.svg?max-width=637&scale=1.0)
|
![[Graphic 18]](/bjoc/content/inline/1860-5397-11-47-i20.svg?max-width=637&scale=1.0)
7H |
85 |
| 9 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-11-47-i21.svg?max-width=637&scale=1.0)
|
![[Graphic 20]](/bjoc/content/inline/1860-5397-11-47-i22.svg?max-width=637&scale=1.0)
7I |
86 |
| 10 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-11-47-i23.svg?max-width=637&scale=1.0)
|
![[Graphic 22]](/bjoc/content/inline/1860-5397-11-47-i24.svg?max-width=637&scale=1.0)
7J |
89 |
| 11 |
![[Graphic 23]](/bjoc/content/inline/1860-5397-11-47-i25.svg?max-width=637&scale=1.0)
|
![[Graphic 24]](/bjoc/content/inline/1860-5397-11-47-i26.svg?max-width=637&scale=1.0)
7K |
87 |
| 12 |
![[Graphic 25]](/bjoc/content/inline/1860-5397-11-47-i27.svg?max-width=637&scale=1.0)
|
![[Graphic 26]](/bjoc/content/inline/1860-5397-11-47-i28.svg?max-width=637&scale=1.0)
7L |
53b |
aSubstrates 6A–L (0.4 mmol) and catalyst (5 mol %) in anhydrous toluene (4 mL) were heated in a sealed vial under argon atmosphere at 80 °C for 3 h; bthe reaction time was 12 h.
Further studies indicated that 2,3-dihydropyrrolo[2,1-b]quinazolin-9(1H)-ones 9A–L could be generated by the treatment of substituted 2-(3-butynyl)-4(3H)-quinazolinones 8A–L with AgOTf under the optimized reaction conditions. As illustrated in Table 3, 2-(3-butynyl)-4(3H)-quinazolinones 8A–L with different substituents were well-tolerated in this intramolecular cyclization reaction, and the expected products 9A–L were obtained in good to excellent yields (80–93%, Table 3, entries 1–12).
Table 3: Silver-mediated synthesis of target compounds 9A–L.a
![[Graphic 27]](/bjoc/content/inline/1860-5397-11-47-i29.svg?max-width=637&scale=1.0)
|
|||
| Entry | Substrate | Product | Yield (%) |
|---|---|---|---|
| 1 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-11-47-i30.svg?max-width=637&scale=1.0)
|
![[Graphic 29]](/bjoc/content/inline/1860-5397-11-47-i31.svg?max-width=637&scale=1.0)
9A |
93 |
| 2 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-11-47-i32.svg?max-width=637&scale=1.0)
|
![[Graphic 31]](/bjoc/content/inline/1860-5397-11-47-i33.svg?max-width=637&scale=1.0)
9B |
89 |
| 3 |
![[Graphic 32]](/bjoc/content/inline/1860-5397-11-47-i34.svg?max-width=637&scale=1.0)
|
![[Graphic 33]](/bjoc/content/inline/1860-5397-11-47-i35.svg?max-width=637&scale=1.0)
9C |
90 |
| 4 |
![[Graphic 34]](/bjoc/content/inline/1860-5397-11-47-i36.svg?max-width=637&scale=1.0)
|
![[Graphic 35]](/bjoc/content/inline/1860-5397-11-47-i37.svg?max-width=637&scale=1.0)
9D |
92 |
| 5 |
![[Graphic 36]](/bjoc/content/inline/1860-5397-11-47-i38.svg?max-width=637&scale=1.0)
|
![[Graphic 37]](/bjoc/content/inline/1860-5397-11-47-i39.svg?max-width=637&scale=1.0)
9E |
90 |
| 6 |
![[Graphic 38]](/bjoc/content/inline/1860-5397-11-47-i40.svg?max-width=637&scale=1.0)
|
![[Graphic 39]](/bjoc/content/inline/1860-5397-11-47-i41.svg?max-width=637&scale=1.0)
9F |
80 |
| 7 |
![[Graphic 40]](/bjoc/content/inline/1860-5397-11-47-i42.svg?max-width=637&scale=1.0)
|
![[Graphic 41]](/bjoc/content/inline/1860-5397-11-47-i43.svg?max-width=637&scale=1.0)
9G |
82 |
| 8 |
![[Graphic 42]](/bjoc/content/inline/1860-5397-11-47-i44.svg?max-width=637&scale=1.0)
|
![[Graphic 43]](/bjoc/content/inline/1860-5397-11-47-i45.svg?max-width=637&scale=1.0)
9H |
84 |
| 9 |
![[Graphic 44]](/bjoc/content/inline/1860-5397-11-47-i46.svg?max-width=637&scale=1.0)
|
![[Graphic 45]](/bjoc/content/inline/1860-5397-11-47-i47.svg?max-width=637&scale=1.0)
9I |
83 |
| 10 |
![[Graphic 46]](/bjoc/content/inline/1860-5397-11-47-i48.svg?max-width=637&scale=1.0)
|
![[Graphic 47]](/bjoc/content/inline/1860-5397-11-47-i49.svg?max-width=637&scale=1.0)
9J |
83 |
| 11 |
![[Graphic 48]](/bjoc/content/inline/1860-5397-11-47-i50.svg?max-width=637&scale=1.0)
|
![[Graphic 49]](/bjoc/content/inline/1860-5397-11-47-i51.svg?max-width=637&scale=1.0)
9K |
91 |
| 12 |
![[Graphic 50]](/bjoc/content/inline/1860-5397-11-47-i52.svg?max-width=637&scale=1.0)
|
![[Graphic 51]](/bjoc/content/inline/1860-5397-11-47-i53.svg?max-width=637&scale=1.0)
9L |
86 |
| 13 |
![[Graphic 52]](/bjoc/content/inline/1860-5397-11-47-i54.svg?max-width=637&scale=1.0)
|
![[Graphic 53]](/bjoc/content/inline/1860-5397-11-47-i55.svg?max-width=637&scale=1.0)
9M |
—b |
aSubstrates 8A–L (0.4 mmol) and catalyst (5 mol %) in anhydrous toluene (4 mL) were heated in a sealed vial under argon atmosphere at 80 °C for 3 h; bthe product could not be isolated due to large amounts of impurities formed during the reaction.
Based on the results of the present studies, we propose a plausible mechanism for the transformation. As depicted in Scheme 2, the intramolecular cyclization is initiated by the activation of the terminal alkyne moiety of the substrate with AgOTf to generate the Ag–alkyne π complex I (or its tautomer II). Subsequently, the Ag–alkyne π complex I or II is converted into complex III through a nucleophilic attack of the nitrogen atom of the amide, and then produces the final product. Products 7A and 9G were recrystallized and their structures were unambiguously confirmed by X-ray diffraction (XRD) studies (see Supporting Information File 1 for details).
![[1860-5397-11-47-i2]](/bjoc/content/inline/1860-5397-11-47-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In conclusion, we have developed a chemical methodology for the synthesis of pyrido/pyrrolo[2,1-b]quinazolin-9(1H)-ones through an AgOTf-catalyzed intramolecular alkyne hydroamination cyclization reaction. The methodology is applicable to a wide scope of substrates and generates a series of fused quinazolinone heterocycles in good to excellent yields. It lends itself an alternative method to the construction of innovative molecules with polycyclic architectures. It may be worthwhile to investigate the biological activity of the synthesized structures.
Experimental
Commercially available reagents and solvents were used without further purification. Column chromatography was performed on silica gel. TLC was performed on silica gel GF254 plates. 1H NMR and 13C NMR spectra were obtained on Varian 300, Bruker 400 and 500 spectrometers. The chemical shifts for 1H NMR were recorded in parts per million (ppm) downfield from tetramethylsilane (TMS) with the residual solvent resonance as the internal standard (7.26 ppm for CDCl3 or 2.50 ppm for DMSO-d6). The chemical shifts for 13C NMR were recorded in ppm by using the central peak of CDCl3 (77.23 ppm) or DMSO-d6 (39.52 ppm) as the internal standard. Coupling constants (J) are reported in Hz and refer to apparent peak multiplications. The abbreviations s, d, t, q, p and m stand for singlet, doublet, triplet, quartet, pentet and multiplet, respectively.
General procedure for the synthesis of substrates 6A–6L and 8A–8L: To a solution of 5-hexynoic acid (3.0 mmol) in dry CH2Cl2 (5 mL) was added EDCI (3.1 mmol) and HOBt (3.1 mmol). The resulting mixture was stirred at rt for 2 h. Then substituted or unsubstituted 2-aminobenzamide (3.0 mmol) was added, and the reaction mixture was stirred at rt for 12 h while being monitored by TLC. After the addition of H2O (10 mL) the mixture was extracted with ethyl acetate (3 × 20 mL). The organic layers were combined and concentrated under vacuum to give the amide intermediate.
The above intermediate was then dissolved in 95% EtOH (5 mL), and solid NaOH (6.0 mmol) was added. The mixture was heated under reflux for 2 h while being monitored by TLC. The solvent was evaporated under vacuum. Water (10 mL) was added, and the mixture was extracted with ethyl acetate (3 × 20 mL). The organic layers were combined and dried over anhydrous Na2SO4. After the removal of the solvent the crude product was purified by silica gel column chromatography with CH2Cl2/MeOH 50:1 (v/v) as an eluent to give the desired substrates 6A–6L.
For 8A–8L, the same procedure as described above was used, except that 4-pentynoic acid was used instead of 5-hexynoic acid. Compound 6A as an example: 1H NMR (300 MHz, CDCl3) δ 11.75 (s, 1H), 8.29 (dd, J = 8.0, 1.0 Hz, 1H), 7.82–7.73 (m, 1H), 7.73–7.66 (m, 1H), 7.52–7.43 (m, 1H), 2.98–2.88 (m, 2H), 2.41 (td, J = 6.9, 2.6 Hz, 2H), 2.21–2.08 (m, 2H), 2.01 (t, J = 2.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 164.3, 155.8, 149.4, 134.8, 127.3, 126.5, 128.2, 120.5, 83.2, 69.4, 34.4, 25.8, 18.0. LRMS (ESI) m/z: 213 [M + H]+; HRMS–ESI (m/z): [M + H]+ calcd for C13H13N2O, 213.1028; found, 213.1024.
General procedure for the synthesis of the target products 7A–7L and 9A–9L: A vial equipped with a magnetic stir bar was charged with the corresponding substrate 6A–6L or 8A–8L (0.4 mmol) and the catalyst AgOTf (5 mol %) and capped with a septum. The vial was evacuated and backfilled with argon, and this process was repeated three times. Under argon, anhydrous toluene (4 mL) was injected to the vial with a syringe, and the resulting mixture was stirred at rt for 10 min. Afterwards, the vial was kept in a preheated oil bath at 80 °C for the appropriate time. After the reaction was complete, the reaction mixture was cooled to rt and the solvent was evaporated under vacuum. The residue was purified by silica gel column chromatography with petroleum ether/EtOAc 20:1 (v/v) as an eluent to give the desired target compounds 7A–7L and 9A–9L. Compound 7A as an example: 1H NMR (400 MHz, CDCl3) δ 8.30 (dd, J = 8.0, 1.5 Hz, 1H), 7.82–7.66 (m, 1H), 7.61 (d, J = 8.1 Hz, 1H), 7.50–7.37 (m, 1H), 5.58 (s, 1H), 5.45 (s, 1H), 2.84 (t, J = 6.9 Hz, 2H), 2.76–2.56 (m, 2H), 2.00 (dt, J = 14.4, 7.1 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 160.3, 155.8, 146.9, 136.9, 134.4, 127.4, 126.6, 126.5, 121.3, 112.5, 31.8, 29.6, 18.3; LRMS (EI) m/z: 212 [M]+; HRMS–EI (m/z): [M]+ calcd for C13H12N2O, 212.0950; found, 212.0930.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures for all compounds and precursors, copies of 1H/13C NMR spectra for all compounds. | ||
| Format: PDF | Size: 5.2 MB | Download |
Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China Grants (91229204 and 21372235), Major Project of Chinese National Programs for Fundamental Research and Development (2015CB910304), National High Technology Research and Development Program of China (2012AA020302), National Basic Research Program of China (2012CB518005), National S&T Major Projects (2012ZX09103101-072, 2014ZX09507002-001 and 2013ZX09507-001).
References
-
Michael, J. P. Nat. Prod. Rep. 2004, 21, 650–668. doi:10.1039/b310691h
Return to citation in text: [1] -
Mhaske, S. B.; Argade, N. P. Tetrahedron 2006, 62, 9787–9826. doi:10.1016/j.tet.2006.07.098
Return to citation in text: [1] [2] [3] [4] -
Amin, A. H.; Mehta, D. R. Nature 1959, 184, 1317. doi:10.1038/1841317a0
Return to citation in text: [1] -
Al-Shamma, A.; Drake, S.; Flynn, D. L.; Mitscher, L. A.; Park, Y. H.; Rao, G. S. R.; Simpson, A.; Swayze, J. K.; Veysoglu, T.; Wu, S. T.-S. J. Nat. Prod. 1981, 44, 745–747. doi:10.1021/np50018a025
Return to citation in text: [1] -
Michael, J. P. Nat. Prod. Rep. 2008, 25, 166–187. doi:10.1039/b612168n
Return to citation in text: [1] -
Liu, J.-F.; Ye, P.; Sprague, K.; Sargent, K.; Yohannes, D.; Baldino, C. M.; Wilson, C. J.; Ng, S.-C. Org. Lett. 2005, 7, 3363–3366. doi:10.1021/ol0513084
Return to citation in text: [1] -
Mori, M.; Kobayashi, H.; Kimura, M.; Ban, Y. Heterocycles 1985, 23, 2803–2806. doi:10.3987/R-1985-11-2803
Return to citation in text: [1] [2] -
Johns, S. R.; Lamberton, J. A. Chem. Commun. 1965, 267a. doi:10.1039/c1965000267a
Return to citation in text: [1] -
Kametani, T.; Loc, C. V.; Higa, T.; Koizumi, M.; Ihara, M.; Fukumoto, K. J. Am. Chem. Soc. 1977, 99, 2306–2309. doi:10.1021/ja00449a047
Return to citation in text: [1] -
Eguchi, S.; Suzuki, T.; Okawa, T.; Matsushita, Y.; Yashima, E.; Okamoto, Y. J. Org. Chem. 1996, 61, 7316–7319. doi:10.1021/jo9609283
Return to citation in text: [1] [2] -
Mhaske, S. B.; Argade, N. P. J. Org. Chem. 2001, 66, 9038–9040. doi:10.1021/jo010727l
Return to citation in text: [1] -
Morris, R. C.; Hanford, W. E.; Adams, R. J. Am. Chem. Soc. 1935, 57, 951–954. doi:10.1021/ja01308a052
Return to citation in text: [1] -
Kametani, T.; Higa, T.; Loc, C. V.; Ihara, M.; Koizumi, M.; Fukumoto, K. J. Am. Chem. Soc. 1976, 98, 6186–6188. doi:10.1021/ja00436a019
Return to citation in text: [1] -
Severin, R.; Doye, S. Chem. Soc. Rev. 2007, 36, 1407–1420. doi:10.1039/b600981f
Return to citation in text: [1] -
Müller, T. E.; Hultzsch, K. C.; Yus, M.; Foubelo, F.; Tada, M. Chem. Rev. 2008, 108, 3795–3892. doi:10.1021/cr0306788
Return to citation in text: [1] [2] -
Alonso, F.; Beletskaya, I. P.; Yus, M. Chem. Rev. 2004, 104, 3079–3160. doi:10.1021/cr0201068
Return to citation in text: [1] -
Hesp, K. D.; Stradiotto, M. ChemCatChem 2010, 2, 1192–1207. doi:10.1002/cctc.201000102
Return to citation in text: [1] -
Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410–3449. doi:10.1002/anie.200604335
Return to citation in text: [1] -
Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395–3442. doi:10.1021/cr050041j
Return to citation in text: [1] -
Arcadi, A. Chem. Rev. 2008, 108, 3266–3325. doi:10.1021/cr068435d
Return to citation in text: [1] -
Wang, Y.; Rapakousiou, A.; Latouche, C.; Daran, J.-C.; Singh, A.; Ledoux-Rak, I.; Ruiz, J.; Saillard, J.-Y.; Astruc, D. Chem. Commun. 2013, 49, 5862–5864. doi:10.1039/c3cc42211a
Return to citation in text: [1] -
Wong, V. H. L.; Hor, T. S. A.; Hii, K. K. Chem. Commun. 2013, 49, 9272–9274. doi:10.1039/c3cc45500a
Return to citation in text: [1] -
Patil, N. T.; Singh, V. J. Organomet. Chem. 2011, 696, 419–432. doi:10.1016/j.jorganchem.2010.10.027
Return to citation in text: [1] -
Kinjo, R.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2011, 50, 5560–5563. doi:10.1002/anie.201100740
Return to citation in text: [1] -
McNulty, J.; Keskar, K. Eur. J. Org. Chem. 2014, 1622–1629. doi:10.1002/ejoc.201301368
Return to citation in text: [1] -
Müller, T. E.; Grosche, M.; Herdtweck, E.; Pleier, A.-K.; Walter, E.; Yan, Y.-K. Organometallics 1999, 19, 170–183. doi:10.1021/om9906013
Return to citation in text: [1] -
Sevov, C. S.; Zhou, J.; Hartwig, J. F. J. Am. Chem. Soc. 2014, 136, 3200–3207. doi:10.1021/ja412116d
Return to citation in text: [1] -
Hesp, K. D. Angew. Chem., Int. Ed. 2014, 53, 2034–2036. doi:10.1002/anie.201309262
Return to citation in text: [1] -
Nguyen, T. M.; Nicewicz, D. A. J. Am. Chem. Soc. 2013, 135, 9588–9591. doi:10.1021/ja4031616
Return to citation in text: [1] -
Beauchemin, A. M. Org. Biomol. Chem. 2013, 11, 7039–7050. doi:10.1039/c3ob41172a
Return to citation in text: [1] -
Minatti, A.; Muñiz, K. Chem. Soc. Rev. 2007, 36, 1142–1152. doi:10.1039/b607474j
Return to citation in text: [1] -
Banerjee, D.; Junge, K.; Beller, M. Angew. Chem., Int. Ed. 2014, 53, 1630–1635. doi:10.1002/anie.201308874
Return to citation in text: [1] -
Kanno, O.; Kuriyama, W.; Wang, Z. J.; Toste, F. D. Angew. Chem., Int. Ed. 2011, 50, 9919–9922. doi:10.1002/anie.201104076
Return to citation in text: [1] -
Ye, D.; Zhang, X.; Zhou, Y.; Zhang, D.; Zhang, L.; Wang, H.; Jiang, H.; Liu, H. Adv. Synth. Catal. 2009, 351, 2770–2778. doi:10.1002/adsc.200900505
Return to citation in text: [1] -
Zhang, X.; Zhou, Y.; Wang, H.; Guo, D.; Ye, D.; Xu, Y.; Jiang, H.; Liu, H. Green Chem. 2011, 13, 397–405. doi:10.1039/C0GC00668H
Return to citation in text: [1] -
Zhang, X.; Zhou, Y.; Wang, H.; Guo, D.; Ye, D.; Xu, Y.; Jiang, H.; Liu, H. Adv. Synth. Catal. 2011, 353, 1429–1437. doi:10.1002/adsc.201100038
Return to citation in text: [1]
| 1. | Michael, J. P. Nat. Prod. Rep. 2004, 21, 650–668. doi:10.1039/b310691h |
| 2. | Mhaske, S. B.; Argade, N. P. Tetrahedron 2006, 62, 9787–9826. doi:10.1016/j.tet.2006.07.098 |
| 10. | Eguchi, S.; Suzuki, T.; Okawa, T.; Matsushita, Y.; Yashima, E.; Okamoto, Y. J. Org. Chem. 1996, 61, 7316–7319. doi:10.1021/jo9609283 |
| 9. | Kametani, T.; Loc, C. V.; Higa, T.; Koizumi, M.; Ihara, M.; Fukumoto, K. J. Am. Chem. Soc. 1977, 99, 2306–2309. doi:10.1021/ja00449a047 |
| 7. | Mori, M.; Kobayashi, H.; Kimura, M.; Ban, Y. Heterocycles 1985, 23, 2803–2806. doi:10.3987/R-1985-11-2803 |
| 34. | Ye, D.; Zhang, X.; Zhou, Y.; Zhang, D.; Zhang, L.; Wang, H.; Jiang, H.; Liu, H. Adv. Synth. Catal. 2009, 351, 2770–2778. doi:10.1002/adsc.200900505 |
| 2. | Mhaske, S. B.; Argade, N. P. Tetrahedron 2006, 62, 9787–9826. doi:10.1016/j.tet.2006.07.098 |
| 3. | Amin, A. H.; Mehta, D. R. Nature 1959, 184, 1317. doi:10.1038/1841317a0 |
| 4. | Al-Shamma, A.; Drake, S.; Flynn, D. L.; Mitscher, L. A.; Park, Y. H.; Rao, G. S. R.; Simpson, A.; Swayze, J. K.; Veysoglu, T.; Wu, S. T.-S. J. Nat. Prod. 1981, 44, 745–747. doi:10.1021/np50018a025 |
| 5. | Michael, J. P. Nat. Prod. Rep. 2008, 25, 166–187. doi:10.1039/b612168n |
| 6. | Liu, J.-F.; Ye, P.; Sprague, K.; Sargent, K.; Yohannes, D.; Baldino, C. M.; Wilson, C. J.; Ng, S.-C. Org. Lett. 2005, 7, 3363–3366. doi:10.1021/ol0513084 |
| 7. | Mori, M.; Kobayashi, H.; Kimura, M.; Ban, Y. Heterocycles 1985, 23, 2803–2806. doi:10.3987/R-1985-11-2803 |
| 8. | Johns, S. R.; Lamberton, J. A. Chem. Commun. 1965, 267a. doi:10.1039/c1965000267a |
| 35. | Zhang, X.; Zhou, Y.; Wang, H.; Guo, D.; Ye, D.; Xu, Y.; Jiang, H.; Liu, H. Green Chem. 2011, 13, 397–405. doi:10.1039/C0GC00668H |
| 36. | Zhang, X.; Zhou, Y.; Wang, H.; Guo, D.; Ye, D.; Xu, Y.; Jiang, H.; Liu, H. Adv. Synth. Catal. 2011, 353, 1429–1437. doi:10.1002/adsc.201100038 |
| 2. | Mhaske, S. B.; Argade, N. P. Tetrahedron 2006, 62, 9787–9826. doi:10.1016/j.tet.2006.07.098 |
| 10. | Eguchi, S.; Suzuki, T.; Okawa, T.; Matsushita, Y.; Yashima, E.; Okamoto, Y. J. Org. Chem. 1996, 61, 7316–7319. doi:10.1021/jo9609283 |
| 15. | Müller, T. E.; Hultzsch, K. C.; Yus, M.; Foubelo, F.; Tada, M. Chem. Rev. 2008, 108, 3795–3892. doi:10.1021/cr0306788 |
| 27. | Sevov, C. S.; Zhou, J.; Hartwig, J. F. J. Am. Chem. Soc. 2014, 136, 3200–3207. doi:10.1021/ja412116d |
| 28. | Hesp, K. D. Angew. Chem., Int. Ed. 2014, 53, 2034–2036. doi:10.1002/anie.201309262 |
| 29. | Nguyen, T. M.; Nicewicz, D. A. J. Am. Chem. Soc. 2013, 135, 9588–9591. doi:10.1021/ja4031616 |
| 30. | Beauchemin, A. M. Org. Biomol. Chem. 2013, 11, 7039–7050. doi:10.1039/c3ob41172a |
| 31. | Minatti, A.; Muñiz, K. Chem. Soc. Rev. 2007, 36, 1142–1152. doi:10.1039/b607474j |
| 2. | Mhaske, S. B.; Argade, N. P. Tetrahedron 2006, 62, 9787–9826. doi:10.1016/j.tet.2006.07.098 |
| 13. | Kametani, T.; Higa, T.; Loc, C. V.; Ihara, M.; Koizumi, M.; Fukumoto, K. J. Am. Chem. Soc. 1976, 98, 6186–6188. doi:10.1021/ja00436a019 |
| 32. | Banerjee, D.; Junge, K.; Beller, M. Angew. Chem., Int. Ed. 2014, 53, 1630–1635. doi:10.1002/anie.201308874 |
| 33. | Kanno, O.; Kuriyama, W.; Wang, Z. J.; Toste, F. D. Angew. Chem., Int. Ed. 2011, 50, 9919–9922. doi:10.1002/anie.201104076 |
| 12. | Morris, R. C.; Hanford, W. E.; Adams, R. J. Am. Chem. Soc. 1935, 57, 951–954. doi:10.1021/ja01308a052 |
| 11. | Mhaske, S. B.; Argade, N. P. J. Org. Chem. 2001, 66, 9038–9040. doi:10.1021/jo010727l |
| 14. | Severin, R.; Doye, S. Chem. Soc. Rev. 2007, 36, 1407–1420. doi:10.1039/b600981f |
| 15. | Müller, T. E.; Hultzsch, K. C.; Yus, M.; Foubelo, F.; Tada, M. Chem. Rev. 2008, 108, 3795–3892. doi:10.1021/cr0306788 |
| 16. | Alonso, F.; Beletskaya, I. P.; Yus, M. Chem. Rev. 2004, 104, 3079–3160. doi:10.1021/cr0201068 |
| 17. | Hesp, K. D.; Stradiotto, M. ChemCatChem 2010, 2, 1192–1207. doi:10.1002/cctc.201000102 |
| 18. | Fürstner, A.; Davies, P. W. Angew. Chem., Int. Ed. 2007, 46, 3410–3449. doi:10.1002/anie.200604335 |
| 19. | Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395–3442. doi:10.1021/cr050041j |
| 20. | Arcadi, A. Chem. Rev. 2008, 108, 3266–3325. doi:10.1021/cr068435d |
| 21. | Wang, Y.; Rapakousiou, A.; Latouche, C.; Daran, J.-C.; Singh, A.; Ledoux-Rak, I.; Ruiz, J.; Saillard, J.-Y.; Astruc, D. Chem. Commun. 2013, 49, 5862–5864. doi:10.1039/c3cc42211a |
| 22. | Wong, V. H. L.; Hor, T. S. A.; Hii, K. K. Chem. Commun. 2013, 49, 9272–9274. doi:10.1039/c3cc45500a |
| 23. | Patil, N. T.; Singh, V. J. Organomet. Chem. 2011, 696, 419–432. doi:10.1016/j.jorganchem.2010.10.027 |
| 24. | Kinjo, R.; Donnadieu, B.; Bertrand, G. Angew. Chem., Int. Ed. 2011, 50, 5560–5563. doi:10.1002/anie.201100740 |
| 25. | McNulty, J.; Keskar, K. Eur. J. Org. Chem. 2014, 1622–1629. doi:10.1002/ejoc.201301368 |
| 26. | Müller, T. E.; Grosche, M.; Herdtweck, E.; Pleier, A.-K.; Walter, E.; Yan, Y.-K. Organometallics 1999, 19, 170–183. doi:10.1021/om9906013 |
© 2015 Wang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)