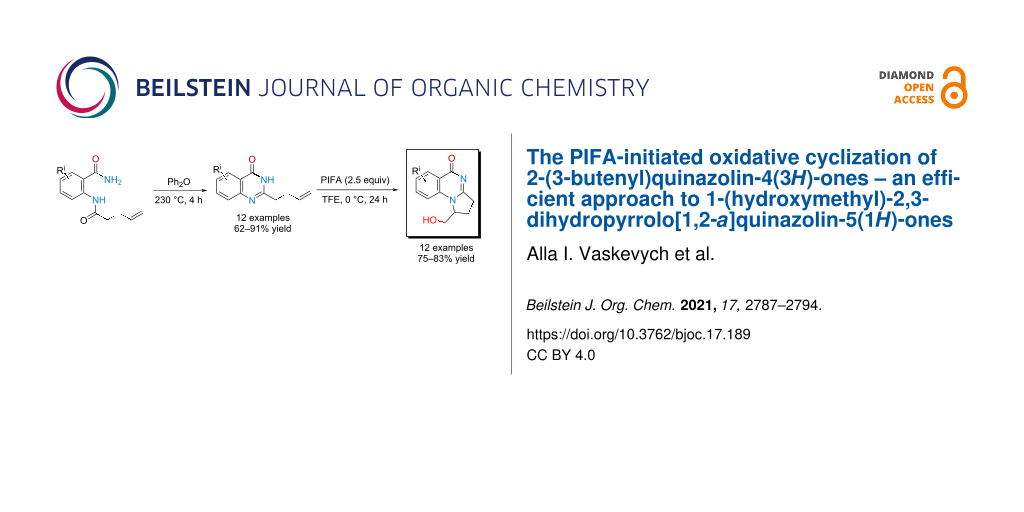
Abstract
A regioselective method for the synthesis of 1-(hydroxymethyl)-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-ones – close structural analogs of naturally occurring vasicinone alkaloids – is described. The procedure is based on PIFA-initiated oxidative 5-exo-trig cyclization of 2-(3-butenyl)quinazolin-4(3Н)-ones, in turn prepared by thermal cyclocondensation of the corresponding 2-(pent-4-enamido)benzamides. The products obtained have a good natural product likeness (NPL) score and therefore can be useful for the design of natural product-like compound libraries.

Graphical Abstract
Introduction
An important design concept in current drug discovery includes structural modifications of naturally occurring compounds to provide novel, sp3-enriched scaffolds with increased propensity to generate potent lead structures with favorable physicochemical properties [1-4]. In light of this, synthesis of compounds that are close analogs of natural compounds is an important task for the synthetic organic chemistry, and this approach has already provided successful results in the view of reaching biological activity. Thus, angular pyrrolo[1,2-a]quinazolines of type 1 – analogs of naturally occurring vasicinone alkaloids bearing an isomeric linear pyrrolo[2,1-b]quinazoline core [5-8] – demonstrated anti-inflammatory [9], antibacterial [10], antiarrythmic [11] activity; some representatives are CNS suppressors and poly-ADP ribose polymerase (PARP) inhibitors [12] (see Figure 1).
![[1860-5397-17-189-1]](/bjoc/content/figures/1860-5397-17-189-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Pyrrolo[1,2-a]quinazoline derivatives – analogs of vasicinone alkaloids and their biological activity.
Figure 1: Pyrrolo[1,2-a]quinazoline derivatives – analogs of vasicinone alkaloids and their biological activi...
Several approaches to obtain 2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one derivatives of type 1 are known in the literature to date. One of them is based on the construction of the central pyrimidine ring starting from ortho-disubstituted aromatic compounds 2 bearing a pyrrolidine moiety (see Scheme 1A). Examples include cyclocondensation of (2-pyrrolidin-1-yl)benzaldehyde and aniline occurring in the presence of TsOH [13], as well as Ir-catalyzed intramolecular dehydrative cross-coupling of 2-(pyrrolidine-1-yl)benzamide [14].
![[1860-5397-17-189-i1]](/bjoc/content/inline/1860-5397-17-189-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic approaches to 2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one derivatives.
Scheme 1: Synthetic approaches to 2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one derivatives.
Another approach to compounds of type 1 that relies on a cascade formation of the pyrimidine and pyrrole rings have found much wider application (see Scheme 1B). One of its variations includes the reaction of readily available anthranilic amides or hydrazides with pent-4-yn-1-ols or -carboxylic acids promoted by PdCl4 [15], Au(І) [16-18] or Cu(ІІ) [19] salts. In another version, the target compounds were obtained by iodine-catalyzed reaction of the aforementioned anthranilic acid derivatives with 4-chloroketones or 4-oxocarboxylic acids in ionic liquids [20-23] or refluxing acetic acid [24,25]. An alternative approach involves cascade cyclization of anthranilamides with 4-chlorobutanoyl chloride [26,27]. Also, reductive cyclocondensation of 2-nitrobenzamides and halogenoketones or ketocarboxylic acids upon action of SnCl2·H2O, TiCl4/Zn, or Fe/CH3COOH can be mentioned [28-30].
1-Aryl-substituted tetrahydropyrrolo[1,2-a]quinazolin-5-ones can be also obtained by a Brønsted acid-catalyzed annulation of arylcyclopropane aldehydes and N′-anthranilic hydrazides [31], as well as by Sm(OTf)3-catalyzed stereoselective [3 + 2] cycloaddition of bis-silyldienediolate and imines, in turn synthesized from anthranylamides and benzaldehydes [32].
A promising approach to the synthesis of 2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one derivatives substituted at the pyrrolidine ring (especially with functional groups) is annulation of the latter moiety to the quinazoline ring. Thus, a series of 1,5-disubstituted pyrroloquinazolines 3 were obtained by a three-component Sonogashira-type coupling of 2-chloro-4-substituted quinazolines 4, propargylic alcohol, and secondary amines (see Scheme 1C) [10]. Appropriately 2-functionalized quinazolinones of type 5 might be good starting materials for the intramolecular cyclization providing the target compounds (see Scheme 1D); however, this approach was rarely used [33-36], possibly due to low regioselectivity of the ring formation at the two competing nucleophilic centers.
Results and Discussion
In this work, we propose a regioselective approach to the synthesis of 2,3-dihydropyrroloquinazolin-5(1H)-ones 6 functionalized with a hydroxymethyl group by oxidative cyclization of hereto unknown 2-(buten-3-yl)quinazolin-4(3H)-ones 7 upon action of bis(trifluoroacetoxy)iodobenzene (PIFA) (see Scheme 1D). A part from the well-known applications of hypervalent iodine compounds for oxidative rearrangements, fragmentations, halogenations and hydroxylations [37,38], they were also involved in the synthesis of N-heterocycles [39,40] including from properly functionalized arenes and alkenes [41-53]. In the case of such substrates having oxygen-containing functional groups, PIFA attacked the double bond first [41-43]. With unsaturated amides or hydroxamates, oxidation of the nitrogen atom to nitrenium intermediates occurred initially; further stabilization of these species resulted in the formation of hydroxylated lactams [44-48], azaspirocycles [49-53], or tricyclic nitrogen-containing heterocycles [44,53].
Our study commenced with the synthesis of key intermediates 7 bearing a homoallyl substituent at the C-2 position. Most of the methods for the preparation of 2-akyl-substitued quinazolin-4(3H)-ones [54] require the use of hardly available starting materials, expensive catalysts, and/or harsh reaction conditions [55], often intolerant to the unsaturated moieties. The known literature exceptions included synthesis of 2-alkynylquinazolines via acylation of anthranilamides with alkynylcarboxylic acids and further cyclocondensation under alkaline conditions [36], as well as formation of 2-alkenyl counterparts by a one-pot Yb(OTf)3-catalyzed microwave- or ultrasound-assisted reaction of 2-aminobenzonitrile and alkenoyl chlorides [56].
We have found that substrates 7 can be obtained efficiently by a two-step reaction sequence commencing from acylation of anthranilamides 8 with α-allylacetyl chloride 9 leading to benzamides 10. These intermediates appeared to be stable towards heating and underwent intramolecular cyclocondensation performed in diphenyl ether at 230 °C giving target products 7 in good to high yields (see Table 1).
Table 1: Synthesis of 2-(buten-3-yl)quinazolin-4(3H)-ones 7.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-189-i3.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Amide | Ri a | Product 10 | Product 10 yield [%]b | Product 7 | Product 7 yield [%]b |
| 1 | 8a | H | 10a | 74 | 7a | 71 |
| 2 | 8b | 5-NO2 | 10b | 91 | 7b | 62 |
| 3 | 8c | 5-F | 10c | 87 | 7c | 63 |
| 4 | 8d | 6-Me | 10d | 75 | 7d | 69 |
| 5 | 8e | 6-OMe | 10e | 71 | 7e | 76 |
| 6 | 8f | 6-Cl | 10f | 89 | 7f | 87 |
| 7 | 8g | 6-NO2 | 10g | 76 | 7g | 91 |
| 8 | 8h | 6,7-(OMe)2 | 10h | 67 | 7h | 71 |
| 9 | 8i | 7-Cl | 10i | 83 | 7i | 79 |
| 10 | 8j | 8-Me | 10j | 74 | 7j | 72 |
| 11 | 8k | 8-Br | 10k | 68 | 7k | 66 |
| 12 | 8l | 8-F | 10l | 76 | 7l | 63 |
aAtom numbering as in product 7. bOver two steps.
Since it was not possible to predict a priori which of the nitrogen atoms of 7 would participate in PIFA-promoted heterocyclization, optimization of the reaction conditions was performed. The solvent, reaction temperature and time, as well as the reagent ratio were varied. Since CH2Cl2, CH2Cl2–TFA, and 2,2,2-trifluoroethanol (CF3CH2OH, TFE) are used most often for the reactions with PIFA, these solvents were evaluated in the study (see Table 2). It was found the reaction did not proceed with 1.5 equiv of PIFA in CH2Cl2 or CH2Cl2–TFA at 0 °C over 2 h (Table 2, entries 1 and 2), while in TFE, the conversion was 37% (Table 2, entry 3). To achieve full conversion of the starting material 7a in TFE at 0 °C, 2.5 equiv of the oxidant and 24 h reaction time were necessary (Table 2, entry 10).
Table 2: Optimization of the reaction conditionsa.
| Entry | Solvent | Amount of PIFA [equiv] | Temperature [°C] | Time [h] | Conversion [%]b |
| 1 | CH2Cl2 | 1.5 | 0 | 2 | 0 |
| 2 | CH2Cl2–TFA | 1.5 | 0 | 2 | 0 |
| 3 | TFE | 1.5 | 0 | 2 | 37 |
| 4 | TFE | 1.5 | 0 | 6 | 46 |
| 5 | TFE | 1.5 | 25 | 6 | 100c |
| 6 | TFE | 2.5 | 0 | 6 | 57 |
| 7 | TFE | 3.5 | 0 | 6 | 56 |
| 8 | TFE | 2.5 | 0 | 12 | 85 |
| 9 | TFE | 2.5 | 0 | 18 | 96 |
| 10 | TFE | 2.5 | 0 | 24 | 100 |
| 11 | TFE | 2.5 | −15 | 240 | 88 |
| 12 | TFE | 3.5 | −15 | 240 | 100 |
aReaction conditions: 4a (0.25 mmol), solvent (10 mL). bThe conversion was determined by LC–MS. cThe target product content was 70%.
The optimized conditions were applied to all quinazolones 7a–l, and target 1-(hydroxymethyl)-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-ones 6a–l were obtained in 75–83% yield (see Table 3). Their structural analysis showed that the reaction proceeded with high regioselectivity as 5-exo-trig cyclization. The substituent in the quinazoline ring had virtually no effect at the yield or selectivity of the heterocyclization.
Table 3: Synthesis of 1-(hydroxymethyl)-2,3-dihydropyrroloquinazolin-5(1H)-ones 6.
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-189-i4.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Starting compound | Ri a | Product | Yield [%] | NPL scoreb |
| 1 | 7a | H | 6a | 80 | 0.42 |
| 2 | 7b | 5-NO2 | 6b | 82 | −0.10 |
| 3 | 7c | 5-F | 6c | 83 | 0.33 |
| 4 | 7d | 6-Me | 6d | 79 | 0.54 |
| 5 | 7e | 6-OMe | 6e | 75 | 0.57 |
| 6 | 7f | 6-Cl | 6f | 81 | 0.24 |
| 7 | 7g | 6-NO2 | 6g | 75 | 0.01 |
| 8 | 7h | 6,7-(OMe)2 | 6h | 76 | 0.69 |
| 9 | 7i | 7-Cl | 6i | 80 | 0.23 |
| 10 | 7j | 8-Me | 6j | 81 | 0.81 |
| 11 | 7k | 8-Br | 6k | 76 | 0.55 |
| 12 | 7l | 8-F | 6l | 82 | 0.64 |
aAtom numbering as in starting compound 7. bErtl’s natural product likeness (NPL) score calculated using an online NaPLeS tool [4].
Two pathways seem to be possible for the oxidative heterocyclization of quinazolinones 7 (see Scheme 2). The first of them (pathway a) includes the formation of the nitrene cation 11 [41,42,44,45,53] under action of PIFA as an oxidant. A subsequent electrophilic attack at the double bond provides aziridinium cation 12 that undergoes selective ring opening with the trifluoroacetate anion to give intermediate 13. The formation of the last-mentioned can be achieved by an initial PIFA attack on the homoallyl C=C bond [41,42,57,58] through the possible intermediates 14 and 15 alternatively (pathway b). Finally, alkaline hydrolysis of 13 upon work-up of the reaction mixture leads to the formation of target product 6.
![[1860-5397-17-189-i2]](/bjoc/content/inline/1860-5397-17-189-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Plausible mechanism for the formation of 6.
Scheme 2: Plausible mechanism for the formation of 6.
The structure of products 6 was confirmed by NMR spectroscopy; in addition to that, X-ray diffraction studies were performed with single crystals of compound 6f (see Figure 2).
![[1860-5397-17-189-2]](/bjoc/content/figures/1860-5397-17-189-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray crystal structure of compound 6f.
Figure 2: X-ray crystal structure of compound 6f.
Since the application of the obtained compounds in early drug discovery is anticipated, it is important to assess their suitability for the synthesis of compound libraries relevant to medicinal chemistry. While many chemoinformatic tools are available for that purpose, we have turned our attention to Ertl’s natural product likeness (NPL) score since the target compounds were designed as natural product analogs [1-4]. In its essence, the NPL score for any molecule estimates its similarity to natural products vs synthetic molecules; it is based on the occurrence frequencies of the corresponding molecular fragments in the two series mentioned above. Zero value of the score is characteristic for the compounds equally similar to natural products and synthetic compounds, while positive values – for those more similar to natural products [1,4].
It was found that most of the compounds 6a–l had the score values (–0.10 to 0.81; 0.46 on average, see Table 3) – even somewhat higher than natural vasicinone (0.30) and deoxyvasicinone (−0.01). Therefore, the products obtained in this work indeed fit into the natural product-like chemical space.
Conclusion
A novel PIFA-initiated oxidative cyclization of 2-(but-3-en-1-yl)quinazolin-4(3H)-ones into 1-(hydroxymethyl)-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-ones was found. The reaction proceeds with high regioselectivity when 2.5 equivalents of PIFA are used in 2,2,2-trifluoroethanol solution at 0 °С over 24 h. The synthesized compounds represent a new class of functionalized pyrroloquinazolinones – close analogs of naturally occurring vasicinone alkaloids – that can be used as building blocks to obtain natural product-like compound libraries of potential biologically active compounds.
Experimental
Commercially available reagents and solvents were used without further purification. The IR spectra of the compounds obtained were recorded on a Bruker Vertex 70 spectrometer in KBr pellets. The NMR spectra were recorded with Varian VXR-300 (400, 500, 600) instruments (300, 400, 600 MHz for 1H, 188 MHz for 19F and 100, 125, 150 MHz for 13C) in CDCl3 and DMSO-d6 solutions, with TMS as an internal standard. Multiplets were assigned as s (singlet), d (doublet), t (triplet), dd (doublet of doublet), q (quartet), m (multiplet) and br s (broad singlet). LC–MS spectra were recorded on an Agilent 1100 Series high-performance liquid chromatograph equipped with a diode matrix with an Agilent LC\MSD SL mass selective detector. Mass spectrometric detection of samples were performed with an Infinity 1260 UHPLC system (Agilent Technologies, Waldbronn, Germany) coupled to a 6224 Accurate Mass TOF LC–MS system (Agilent Technologies, Singapore).
General procedure for the synthesis of 2-(but-3-en-1-yl)quinazolin-4(3H)-ones 7: A solution of amide 10 (1 mmol) in diphenyl ether (10 mL) was heated at 230 °C for 4 h, then cooled to rt, and diluted with hexanes (20 mL). The precipitate was filtered, washed with t-BuOMe/hexanes (1:3), and dried to give product 7. An analytical sample was obtained by recrystallization from t-BuOMe.
2-(But-3-en-1-yl)quinazolin-4(3H)-one (7a) [59]: Prepared using amide 10a (218 mg, 1 mmol, 1 equiv). Light brown powder (142 mg, 0.71 mmol, 71%). Mp: 177–178 °C; 1H NMR (400 MHz, DMSO-d6) δ 12.18 (s, 1H, NH), 8.08 (d, J = 8.0 Hz, 1H, ArH), 7.77 (t, J = 8.0 Hz, 1H, ArH), 7.60 (d, J = 8.0 Hz, 1H, ArH), 7.46 (t, J = 7.6 Hz, 1H, ArH),5.92–5.82 (m, 1H, CH), 5.07 (d, J = 17.2 Hz, 1H, =CH2), 4.98 (d, J = 10.4 Hz, 1H, =CH2), 2.70 (t, J = 7.2 Hz, 2H, CH2), 2.49–2.47 (m, 2H, CH2); 13C NMR (125 MHz, DMSO-d6) δ 161.8, 156.7, 148.9, 137.2, 134.2, 126.8, 125.9, 125.7, 120.9, 115.5, 33.7, 30.6; HRMS–ESI (m/z): [M + H]+ calcd for C12H12N2O+, 201.1023; found, 201.1024.
General procedure for the PIFA-mediated cyclization of compounds 7a–l: To a solution of quinazoline 7 (0.65 mmol) in TFE (5 mL), a solution of PIFA (0.70 g, 1.63 mmol) in TFE (20 mL) was added at 0 °С, and the mixture was stirred for 24 h at 0 °С. Saturated aq NaHCO3 (20 mL) was added, the precipitate was filtered off, the filtrates were extracted with CH2Cl2 (3 × 25 mL), dried over Na2SO4, and the solvent was removed in vacuo. The residue was recrystallized from MeOH to give product 6.
1-(Hydroxymethyl)-2,3-dihydropyrrolo[1,2-a]quinazolin-5(1H)-one (6a): Prepared using quinazoline 7a (130 mg, 0.65 mmol, 1 equiv). White solid (112 mg, 0.52 mmol, 80%). Mp 253–255 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.07 (d, J = 7.6 Hz, 1H, ArH), 7.77 (t, J = 7.6 Hz, 1H, ArH), 7.59 (d, J = 7.6 Hz, 1H, ArH), 7.46 (t, J = 7.6 Hz, 1H, ArH), 5.02 (t, J = 5.6 Hz, 1H, OH), 4.92–4.90 (m, 1H, CH), 3.88–3.82 (m, 1H, CH2), 3.67–3.61 (m, 1H, CH2), 3.22–3.15 (m, 1H, CH2), 2.83–2.81 (m, 1H, CH2), 2.44–2.33 (m, 1H, CH2), 2.22–2.16 (m, 1H, CH2); 13C NMR (125 MHz, DMSO-d6) δ 169.2, 167.4, 138.3, 133.4, 127.6, 125.3, 118.7, 116.1, 62.2, 61.3, 32.2, 22.7; HRMS–ESI (m/z): [M + H]+ calcd for C12H12N2O2+, 217.0972; found, 217.0973.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures for all compounds and precursors, X-ray structure determination, 1H/13C/19F NMR spectra for all compounds. | ||
| Format: PDF | Size: 5.7 MB | Download |
References
-
Ertl, P.; Roggo, S.; Schuffenhauer, A. J. Chem. Inf. Model. 2008, 48, 68–74. doi:10.1021/ci700286x
Return to citation in text: [1] [2] [3] -
Kumar, K.; Waldmann, H. Isr. J. Chem. 2019, 59, 41–51. doi:10.1002/ijch.201800105
Return to citation in text: [1] [2] -
Grygorenko, O. O.; Volochnyuk, D. M.; Ryabukhin, S. V.; Judd, D. B. Chem. – Eur. J. 2020, 26, 1196–1237. doi:10.1002/chem.201903232
Return to citation in text: [1] [2] -
Sorokina, M.; Steinbeck, C. J. Cheminf. 2019, 11, 55. doi:10.1186/s13321-019-0378-z
Return to citation in text: [1] [2] [3] [4] -
Dumitrascu, F.; Georgescu, F.; Georgescu, E.; Caira, M. R. Adv. Heterocycl. Chem. 2019, 129, 155–244. doi:10.1016/bs.aihch.2019.01.004
Return to citation in text: [1] -
Mhaske, S. B.; Argade, N. P. Tetrahedron 2006, 62, 9787–9826. doi:10.1016/j.tet.2006.07.098
Return to citation in text: [1] -
Shang, X.-F.; Morris-Natschke, S. L.; Liu, Y.-Q.; Guo, X.; Xu, X.-S.; Goto, M.; Li, J.-C.; Yang, G.-Z.; Lee, K.-H. Med. Res. Rev. 2018, 38, 775–828. doi:10.1002/med.21466
Return to citation in text: [1] -
Shang, X.-F.; Morris-Natschke, S. L.; Yang, G.-Z.; Liu, Y.-Q.; Guo, X.; Xu, X.-S.; Goto, M.; Li, J.-C.; Zhang, J.-Y.; Lee, K.-H. Med. Res. Rev. 2018, 38, 1614–1660. doi:10.1002/med.21492
Return to citation in text: [1] -
Stavytskyi, V.; Antypenko, O.; Nosulenko, I.; Berest, G.; Voskoboinik, O.; Kovalenko, S. Anti-Inflammatory Anti-Allergy Agents Med. Chem. 2021, 20, 75–88. doi:10.2174/1871523019666200505073232
Return to citation in text: [1] -
Kazemi, S. S.; Keivanloo, A.; Nasr-Isfahani, H.; Bamoniri, A. RSC Adv. 2016, 6, 92663–92669. doi:10.1039/c6ra21219k
Return to citation in text: [1] [2] -
Ostersehlt, B.; Schlecker, R.; Rendenbach, B.; von Philipsborn, G.; Franke, A. Tetracyclic quinazoline derivatives, effective as antiarrythmic agents. U.S. Patent US5,214,047A, May 25, 1993.
Return to citation in text: [1] -
Dumitrascu, F.; Popa, M. M. ARKIVOC 2014, No. i, 428–452. doi:10.3998/ark.5550190.p008.699
Return to citation in text: [1] -
Mori, K.; Ohshima, Y.; Ehara, K.; Akiyama, T. Chem. Lett. 2009, 38, 524–525. doi:10.1246/cl.2009.524
Return to citation in text: [1] -
Sun, X.; Hu, Y.; Nie, S.-z.; Yan, Y.-y.; Zhang, X.-j.; Yan, M. Adv. Synth. Catal. 2013, 355, 2179–2184. doi:10.1002/adsc.201300455
Return to citation in text: [1] -
Patil, N. T.; Kavthe, R. D.; Raut, V. S.; Shinde, V. S.; Sridhar, B. J. Org. Chem. 2010, 75, 1277–1280. doi:10.1021/jo902293f
Return to citation in text: [1] -
Feng, E.; Zhou, Y.; Zhang, D.; Zhang, L.; Sun, H.; Jiang, H.; Liu, H. J. Org. Chem. 2010, 75, 3274–3282. doi:10.1021/jo100228u
Return to citation in text: [1] -
Patil, N. T.; Lakshmi, P. G. V. V.; Sridhar, B.; Patra, S.; Bhadra, M. P.; Patra, C. R. Eur. J. Org. Chem. 2012, 1790–1799. doi:10.1002/ejoc.201101822
Return to citation in text: [1] -
Jia, X.; Li, P.; Liu, X.; Lin, J.; Chu, Y.; Yu, J.; Wang, J.; Liu, H.; Zhao, F. Molecules 2019, 24, 988. doi:10.3390/molecules24050988
Return to citation in text: [1] -
Naidu, S.; Reddy, S. R. ChemistrySelect 2017, 2, 1196–1201. doi:10.1002/slct.201601872
Return to citation in text: [1] -
Zhou, J.-X.; Lu, L.; Li, T.-J.; Yao, C.-S.; Wang, X.-S. J. Heterocycl. Chem. 2014, 51, 1472–1475. doi:10.1002/jhet.1768
Return to citation in text: [1] -
Lu, L.; Yang, K.; Zhang, M.-M.; Wang, X.-S. J. Heterocycl. Chem. 2014, 51, 841–845. doi:10.1002/jhet.1116
Return to citation in text: [1] -
Zhang, W.-T.; Qiang, W.-W.; Yao, C.-S.; Wang, X.-S. Tetrahedron 2016, 72, 2178–2185. doi:10.1016/j.tet.2016.03.018
Return to citation in text: [1] -
Liu, J.-Q.; Zhang, W.-T.; Wang, X.-S. J. Heterocycl. Chem. 2018, 55, 1906–1916. doi:10.1002/jhet.3228
Return to citation in text: [1] -
Iminov, R. T.; Tverdokhlebov, A. V.; Tolmachev, A. A.; Volovenko, Y. M.; Shishkina, S. V.; Shishkin, O. V. Tetrahedron 2009, 65, 8582–8586. doi:10.1016/j.tet.2009.07.059
Return to citation in text: [1] -
Zicāne, D.; Tetere, Z.; Rāviņa, I.; Turks, M. Chem. Heterocycl. Compd. 2013, 49, 310–316. doi:10.1007/s10593-013-1248-7
Return to citation in text: [1] -
Sutherell, C. L.; Tallant, C.; Monteiro, O. P.; Yapp, C.; Fuchs, J. E.; Fedorov, O.; Siejka, P.; Müller, S.; Knapp, S.; Brenton, J. D.; Brennan, P. E.; Ley, S. V. J. Med. Chem. 2016, 59, 5095–5101. doi:10.1021/acs.jmedchem.5b01997
Return to citation in text: [1] -
Sutherell, C. L.; Ley, S. V. Synthesis 2017, 49, 135–144. doi:10.1055/s-0035-1562792
Return to citation in text: [1] -
Wang, M.; Dou, G.; Shi, D. J. Comb. Chem. 2010, 12, 582–586. doi:10.1021/cc100062e
Return to citation in text: [1] -
Zhao, X.; Shi, D.-Q. J. Heterocycl. Chem. 2011, 48, 634–638. doi:10.1002/jhet.637
Return to citation in text: [1] -
Bunce, R. A.; Nammalwar, B. J. Heterocycl. Chem. 2011, 48, 991–997. doi:10.1002/jhet.672
Return to citation in text: [1] -
Singh, P.; Kaur, N.; Banerjee, P. J. Org. Chem. 2020, 85, 3393–3406. doi:10.1021/acs.joc.9b03170
Return to citation in text: [1] -
Boomhoff, M.; Ukis, R.; Schneider, C. J. Org. Chem. 2015, 80, 8236–8244. doi:10.1021/acs.joc.5b01293
Return to citation in text: [1] -
Volovenko, Y. M.; Resnyanskaya, E. V. Mendeleev Commun. 2002, 12, 119–120. doi:10.1070/mc2002v012n03abeh001580
Return to citation in text: [1] -
Mhaske, S. B.; Argade, N. P. J. Org. Chem. 2001, 66, 9038–9040. doi:10.1021/jo010727l
Return to citation in text: [1] -
Fang, J.; Zhou, J. Org. Biomol. Chem. 2012, 10, 2389–2391. doi:10.1039/c2ob07178a
Return to citation in text: [1] -
Wang, H.; Jiao, S.; Chen, K.; Zhang, X.; Zhao, L.; Liu, D.; Zhou, Y.; Liu, H. Beilstein J. Org. Chem. 2015, 11, 416–424. doi:10.3762/bjoc.11.47
Return to citation in text: [1] [2] -
Stang, P. J.; Zhdankin, V. V. Chem. Rev. 1996, 96, 1123–1178. doi:10.1021/cr940424+
Return to citation in text: [1] -
Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2002, 102, 2523–2584. doi:10.1021/cr010003+
Return to citation in text: [1] -
Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547
Return to citation in text: [1] -
Yang, Y. O.; Wang, X.; Xiao, J.; Li, Y.; Sun, F.; Du, Y. Curr. Org. Chem. 2021, 25, 68–132. doi:10.2174/1385272822999201117154919
Return to citation in text: [1] -
Tellitu, I.; Domínguez, E. Trends Heterocycl. Chem. 2011, 15, 23–32.
Return to citation in text: [1] [2] [3] [4] -
Tellitu, I.; Urrejola, A.; Serna, S.; Moreno, I.; Herrero, M. T.; Domínguez, E.; SanMartin, R.; Correa, A. Eur. J. Org. Chem. 2007, 437–444. doi:10.1002/ejoc.200600782
Return to citation in text: [1] [2] [3] [4] -
Saito, A.; Anzai, T.; Matsumoto, A.; Hanzawa, Y. Tetrahedron Lett. 2011, 52, 4658–4661. doi:10.1016/j.tetlet.2011.06.117
Return to citation in text: [1] [2] -
Wardrop, D. J.; Bowen, E. G.; Forslund, R. E.; Sussman, A. D.; Weerasekera, S. L. J. Am. Chem. Soc. 2010, 132, 1188–1189. doi:10.1021/ja9069997
Return to citation in text: [1] [2] [3] [4] -
Bowen, E. G.; Wardrop, D. J. Org. Lett. 2010, 12, 5330–5333. doi:10.1021/ol102371x
Return to citation in text: [1] [2] [3] -
Wardrop, D. J.; Bowen, E. G. Org. Lett. 2011, 13, 2376–2379. doi:10.1021/ol2006117
Return to citation in text: [1] [2] -
Serna, S.; Tellitu, I.; Domı́nguez, E.; Moreno, I.; SanMartı́n, R. Tetrahedron Lett. 2003, 44, 3483–3486. doi:10.1016/s0040-4039(03)00670-1
Return to citation in text: [1] [2] -
Correa, A.; Tellitu, I.; Domínguez, E.; Moreno, I.; SanMartin, R. J. Org. Chem. 2005, 70, 2256–2264. doi:10.1021/jo047872u
Return to citation in text: [1] [2] -
Wardrop, D. J.; Basak, A. Org. Lett. 2001, 3, 1053–1056. doi:10.1021/ol015626o
Return to citation in text: [1] [2] -
Wardrop, D. J.; Zhang, W. Org. Lett. 2001, 3, 2353–2356. doi:10.1021/ol0161514
Return to citation in text: [1] [2] -
Wardrop, D. J.; Burge, M. S.; Zhang, W.; Ortı́z, J. A. Tetrahedron Lett. 2003, 44, 2587–2591. doi:10.1016/s0040-4039(03)00227-2
Return to citation in text: [1] [2] -
Wardrop, D. J.; Zhang, W.; Landrie, C. L. Tetrahedron Lett. 2004, 45, 4229–4231. doi:10.1016/j.tetlet.2004.04.028
Return to citation in text: [1] [2] -
Bhattacharjee, A.; Gerasimov, M. V.; DeJong, S.; Wardrop, D. J. Org. Lett. 2017, 19, 6570–6573. doi:10.1021/acs.orglett.7b03283
Return to citation in text: [1] [2] [3] [4] -
He, L.; Li, H.; Chen, J.; Wu, X.-F. RSC Adv. 2014, 4, 12065–12077. doi:10.1039/c4ra00351a
Return to citation in text: [1] -
Ramana, D. V.; Sundaram, N.; Yuvaraj, T. E.; Babu, B. G. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 1999, 38, 905–908.
Return to citation in text: [1] -
Fiorito, S.; Taddeo, V. A.; Epifano, F.; Genovese, S. ARKIVOC 2016, No. ii, 68–75. doi:10.3998/ark.5550190.p009.710
Return to citation in text: [1] -
Das, M.; Rodríguez, A.; Lo, P. K. T.; Moran, W. J. Adv. Synth. Catal. 2021, 363, 1646–1650. doi:10.1002/adsc.202001451
Return to citation in text: [1] -
Butt, S. E.; Das, M.; Sotiropoulos, J.-M.; Moran, W. J. J. Org. Chem. 2019, 84, 15605–15613. doi:10.1021/acs.joc.9b02623
Return to citation in text: [1] -
Hamasharif, M. S.; Smith, O. E. P.; Curran, C. J.; Hemming, K. ACS Omega 2017, 2, 1222–1231. doi:10.1021/acsomega.7b00211
Return to citation in text: [1]
| 55. | Ramana, D. V.; Sundaram, N.; Yuvaraj, T. E.; Babu, B. G. Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem. 1999, 38, 905–908. |
| 36. | Wang, H.; Jiao, S.; Chen, K.; Zhang, X.; Zhao, L.; Liu, D.; Zhou, Y.; Liu, H. Beilstein J. Org. Chem. 2015, 11, 416–424. doi:10.3762/bjoc.11.47 |
| 56. | Fiorito, S.; Taddeo, V. A.; Epifano, F.; Genovese, S. ARKIVOC 2016, No. ii, 68–75. doi:10.3998/ark.5550190.p009.710 |
| 1. | Ertl, P.; Roggo, S.; Schuffenhauer, A. J. Chem. Inf. Model. 2008, 48, 68–74. doi:10.1021/ci700286x |
| 2. | Kumar, K.; Waldmann, H. Isr. J. Chem. 2019, 59, 41–51. doi:10.1002/ijch.201800105 |
| 3. | Grygorenko, O. O.; Volochnyuk, D. M.; Ryabukhin, S. V.; Judd, D. B. Chem. – Eur. J. 2020, 26, 1196–1237. doi:10.1002/chem.201903232 |
| 4. | Sorokina, M.; Steinbeck, C. J. Cheminf. 2019, 11, 55. doi:10.1186/s13321-019-0378-z |
| 11. | Ostersehlt, B.; Schlecker, R.; Rendenbach, B.; von Philipsborn, G.; Franke, A. Tetracyclic quinazoline derivatives, effective as antiarrythmic agents. U.S. Patent US5,214,047A, May 25, 1993. |
| 28. | Wang, M.; Dou, G.; Shi, D. J. Comb. Chem. 2010, 12, 582–586. doi:10.1021/cc100062e |
| 29. | Zhao, X.; Shi, D.-Q. J. Heterocycl. Chem. 2011, 48, 634–638. doi:10.1002/jhet.637 |
| 30. | Bunce, R. A.; Nammalwar, B. J. Heterocycl. Chem. 2011, 48, 991–997. doi:10.1002/jhet.672 |
| 10. | Kazemi, S. S.; Keivanloo, A.; Nasr-Isfahani, H.; Bamoniri, A. RSC Adv. 2016, 6, 92663–92669. doi:10.1039/c6ra21219k |
| 31. | Singh, P.; Kaur, N.; Banerjee, P. J. Org. Chem. 2020, 85, 3393–3406. doi:10.1021/acs.joc.9b03170 |
| 9. | Stavytskyi, V.; Antypenko, O.; Nosulenko, I.; Berest, G.; Voskoboinik, O.; Kovalenko, S. Anti-Inflammatory Anti-Allergy Agents Med. Chem. 2021, 20, 75–88. doi:10.2174/1871523019666200505073232 |
| 24. | Iminov, R. T.; Tverdokhlebov, A. V.; Tolmachev, A. A.; Volovenko, Y. M.; Shishkina, S. V.; Shishkin, O. V. Tetrahedron 2009, 65, 8582–8586. doi:10.1016/j.tet.2009.07.059 |
| 25. | Zicāne, D.; Tetere, Z.; Rāviņa, I.; Turks, M. Chem. Heterocycl. Compd. 2013, 49, 310–316. doi:10.1007/s10593-013-1248-7 |
| 1. | Ertl, P.; Roggo, S.; Schuffenhauer, A. J. Chem. Inf. Model. 2008, 48, 68–74. doi:10.1021/ci700286x |
| 4. | Sorokina, M.; Steinbeck, C. J. Cheminf. 2019, 11, 55. doi:10.1186/s13321-019-0378-z |
| 5. | Dumitrascu, F.; Georgescu, F.; Georgescu, E.; Caira, M. R. Adv. Heterocycl. Chem. 2019, 129, 155–244. doi:10.1016/bs.aihch.2019.01.004 |
| 6. | Mhaske, S. B.; Argade, N. P. Tetrahedron 2006, 62, 9787–9826. doi:10.1016/j.tet.2006.07.098 |
| 7. | Shang, X.-F.; Morris-Natschke, S. L.; Liu, Y.-Q.; Guo, X.; Xu, X.-S.; Goto, M.; Li, J.-C.; Yang, G.-Z.; Lee, K.-H. Med. Res. Rev. 2018, 38, 775–828. doi:10.1002/med.21466 |
| 8. | Shang, X.-F.; Morris-Natschke, S. L.; Yang, G.-Z.; Liu, Y.-Q.; Guo, X.; Xu, X.-S.; Goto, M.; Li, J.-C.; Zhang, J.-Y.; Lee, K.-H. Med. Res. Rev. 2018, 38, 1614–1660. doi:10.1002/med.21492 |
| 26. | Sutherell, C. L.; Tallant, C.; Monteiro, O. P.; Yapp, C.; Fuchs, J. E.; Fedorov, O.; Siejka, P.; Müller, S.; Knapp, S.; Brenton, J. D.; Brennan, P. E.; Ley, S. V. J. Med. Chem. 2016, 59, 5095–5101. doi:10.1021/acs.jmedchem.5b01997 |
| 27. | Sutherell, C. L.; Ley, S. V. Synthesis 2017, 49, 135–144. doi:10.1055/s-0035-1562792 |
| 59. | Hamasharif, M. S.; Smith, O. E. P.; Curran, C. J.; Hemming, K. ACS Omega 2017, 2, 1222–1231. doi:10.1021/acsomega.7b00211 |
| 15. | Patil, N. T.; Kavthe, R. D.; Raut, V. S.; Shinde, V. S.; Sridhar, B. J. Org. Chem. 2010, 75, 1277–1280. doi:10.1021/jo902293f |
| 19. | Naidu, S.; Reddy, S. R. ChemistrySelect 2017, 2, 1196–1201. doi:10.1002/slct.201601872 |
| 41. | Tellitu, I.; Domínguez, E. Trends Heterocycl. Chem. 2011, 15, 23–32. |
| 42. | Tellitu, I.; Urrejola, A.; Serna, S.; Moreno, I.; Herrero, M. T.; Domínguez, E.; SanMartin, R.; Correa, A. Eur. J. Org. Chem. 2007, 437–444. doi:10.1002/ejoc.200600782 |
| 57. | Das, M.; Rodríguez, A.; Lo, P. K. T.; Moran, W. J. Adv. Synth. Catal. 2021, 363, 1646–1650. doi:10.1002/adsc.202001451 |
| 58. | Butt, S. E.; Das, M.; Sotiropoulos, J.-M.; Moran, W. J. J. Org. Chem. 2019, 84, 15605–15613. doi:10.1021/acs.joc.9b02623 |
| 14. | Sun, X.; Hu, Y.; Nie, S.-z.; Yan, Y.-y.; Zhang, X.-j.; Yan, M. Adv. Synth. Catal. 2013, 355, 2179–2184. doi:10.1002/adsc.201300455 |
| 20. | Zhou, J.-X.; Lu, L.; Li, T.-J.; Yao, C.-S.; Wang, X.-S. J. Heterocycl. Chem. 2014, 51, 1472–1475. doi:10.1002/jhet.1768 |
| 21. | Lu, L.; Yang, K.; Zhang, M.-M.; Wang, X.-S. J. Heterocycl. Chem. 2014, 51, 841–845. doi:10.1002/jhet.1116 |
| 22. | Zhang, W.-T.; Qiang, W.-W.; Yao, C.-S.; Wang, X.-S. Tetrahedron 2016, 72, 2178–2185. doi:10.1016/j.tet.2016.03.018 |
| 23. | Liu, J.-Q.; Zhang, W.-T.; Wang, X.-S. J. Heterocycl. Chem. 2018, 55, 1906–1916. doi:10.1002/jhet.3228 |
| 1. | Ertl, P.; Roggo, S.; Schuffenhauer, A. J. Chem. Inf. Model. 2008, 48, 68–74. doi:10.1021/ci700286x |
| 2. | Kumar, K.; Waldmann, H. Isr. J. Chem. 2019, 59, 41–51. doi:10.1002/ijch.201800105 |
| 3. | Grygorenko, O. O.; Volochnyuk, D. M.; Ryabukhin, S. V.; Judd, D. B. Chem. – Eur. J. 2020, 26, 1196–1237. doi:10.1002/chem.201903232 |
| 4. | Sorokina, M.; Steinbeck, C. J. Cheminf. 2019, 11, 55. doi:10.1186/s13321-019-0378-z |
| 13. | Mori, K.; Ohshima, Y.; Ehara, K.; Akiyama, T. Chem. Lett. 2009, 38, 524–525. doi:10.1246/cl.2009.524 |
| 4. | Sorokina, M.; Steinbeck, C. J. Cheminf. 2019, 11, 55. doi:10.1186/s13321-019-0378-z |
| 12. | Dumitrascu, F.; Popa, M. M. ARKIVOC 2014, No. i, 428–452. doi:10.3998/ark.5550190.p008.699 |
| 16. | Feng, E.; Zhou, Y.; Zhang, D.; Zhang, L.; Sun, H.; Jiang, H.; Liu, H. J. Org. Chem. 2010, 75, 3274–3282. doi:10.1021/jo100228u |
| 17. | Patil, N. T.; Lakshmi, P. G. V. V.; Sridhar, B.; Patra, S.; Bhadra, M. P.; Patra, C. R. Eur. J. Org. Chem. 2012, 1790–1799. doi:10.1002/ejoc.201101822 |
| 18. | Jia, X.; Li, P.; Liu, X.; Lin, J.; Chu, Y.; Yu, J.; Wang, J.; Liu, H.; Zhao, F. Molecules 2019, 24, 988. doi:10.3390/molecules24050988 |
| 41. | Tellitu, I.; Domínguez, E. Trends Heterocycl. Chem. 2011, 15, 23–32. |
| 42. | Tellitu, I.; Urrejola, A.; Serna, S.; Moreno, I.; Herrero, M. T.; Domínguez, E.; SanMartin, R.; Correa, A. Eur. J. Org. Chem. 2007, 437–444. doi:10.1002/ejoc.200600782 |
| 44. | Wardrop, D. J.; Bowen, E. G.; Forslund, R. E.; Sussman, A. D.; Weerasekera, S. L. J. Am. Chem. Soc. 2010, 132, 1188–1189. doi:10.1021/ja9069997 |
| 45. | Bowen, E. G.; Wardrop, D. J. Org. Lett. 2010, 12, 5330–5333. doi:10.1021/ol102371x |
| 53. | Bhattacharjee, A.; Gerasimov, M. V.; DeJong, S.; Wardrop, D. J. Org. Lett. 2017, 19, 6570–6573. doi:10.1021/acs.orglett.7b03283 |
| 33. | Volovenko, Y. M.; Resnyanskaya, E. V. Mendeleev Commun. 2002, 12, 119–120. doi:10.1070/mc2002v012n03abeh001580 |
| 34. | Mhaske, S. B.; Argade, N. P. J. Org. Chem. 2001, 66, 9038–9040. doi:10.1021/jo010727l |
| 35. | Fang, J.; Zhou, J. Org. Biomol. Chem. 2012, 10, 2389–2391. doi:10.1039/c2ob07178a |
| 36. | Wang, H.; Jiao, S.; Chen, K.; Zhang, X.; Zhao, L.; Liu, D.; Zhou, Y.; Liu, H. Beilstein J. Org. Chem. 2015, 11, 416–424. doi:10.3762/bjoc.11.47 |
| 32. | Boomhoff, M.; Ukis, R.; Schneider, C. J. Org. Chem. 2015, 80, 8236–8244. doi:10.1021/acs.joc.5b01293 |
| 10. | Kazemi, S. S.; Keivanloo, A.; Nasr-Isfahani, H.; Bamoniri, A. RSC Adv. 2016, 6, 92663–92669. doi:10.1039/c6ra21219k |
| 44. | Wardrop, D. J.; Bowen, E. G.; Forslund, R. E.; Sussman, A. D.; Weerasekera, S. L. J. Am. Chem. Soc. 2010, 132, 1188–1189. doi:10.1021/ja9069997 |
| 53. | Bhattacharjee, A.; Gerasimov, M. V.; DeJong, S.; Wardrop, D. J. Org. Lett. 2017, 19, 6570–6573. doi:10.1021/acs.orglett.7b03283 |
| 54. | He, L.; Li, H.; Chen, J.; Wu, X.-F. RSC Adv. 2014, 4, 12065–12077. doi:10.1039/c4ra00351a |
| 44. | Wardrop, D. J.; Bowen, E. G.; Forslund, R. E.; Sussman, A. D.; Weerasekera, S. L. J. Am. Chem. Soc. 2010, 132, 1188–1189. doi:10.1021/ja9069997 |
| 45. | Bowen, E. G.; Wardrop, D. J. Org. Lett. 2010, 12, 5330–5333. doi:10.1021/ol102371x |
| 46. | Wardrop, D. J.; Bowen, E. G. Org. Lett. 2011, 13, 2376–2379. doi:10.1021/ol2006117 |
| 47. | Serna, S.; Tellitu, I.; Domı́nguez, E.; Moreno, I.; SanMartı́n, R. Tetrahedron Lett. 2003, 44, 3483–3486. doi:10.1016/s0040-4039(03)00670-1 |
| 48. | Correa, A.; Tellitu, I.; Domínguez, E.; Moreno, I.; SanMartin, R. J. Org. Chem. 2005, 70, 2256–2264. doi:10.1021/jo047872u |
| 49. | Wardrop, D. J.; Basak, A. Org. Lett. 2001, 3, 1053–1056. doi:10.1021/ol015626o |
| 50. | Wardrop, D. J.; Zhang, W. Org. Lett. 2001, 3, 2353–2356. doi:10.1021/ol0161514 |
| 51. | Wardrop, D. J.; Burge, M. S.; Zhang, W.; Ortı́z, J. A. Tetrahedron Lett. 2003, 44, 2587–2591. doi:10.1016/s0040-4039(03)00227-2 |
| 52. | Wardrop, D. J.; Zhang, W.; Landrie, C. L. Tetrahedron Lett. 2004, 45, 4229–4231. doi:10.1016/j.tetlet.2004.04.028 |
| 53. | Bhattacharjee, A.; Gerasimov, M. V.; DeJong, S.; Wardrop, D. J. Org. Lett. 2017, 19, 6570–6573. doi:10.1021/acs.orglett.7b03283 |
| 41. | Tellitu, I.; Domínguez, E. Trends Heterocycl. Chem. 2011, 15, 23–32. |
| 42. | Tellitu, I.; Urrejola, A.; Serna, S.; Moreno, I.; Herrero, M. T.; Domínguez, E.; SanMartin, R.; Correa, A. Eur. J. Org. Chem. 2007, 437–444. doi:10.1002/ejoc.200600782 |
| 43. | Saito, A.; Anzai, T.; Matsumoto, A.; Hanzawa, Y. Tetrahedron Lett. 2011, 52, 4658–4661. doi:10.1016/j.tetlet.2011.06.117 |
| 44. | Wardrop, D. J.; Bowen, E. G.; Forslund, R. E.; Sussman, A. D.; Weerasekera, S. L. J. Am. Chem. Soc. 2010, 132, 1188–1189. doi:10.1021/ja9069997 |
| 45. | Bowen, E. G.; Wardrop, D. J. Org. Lett. 2010, 12, 5330–5333. doi:10.1021/ol102371x |
| 46. | Wardrop, D. J.; Bowen, E. G. Org. Lett. 2011, 13, 2376–2379. doi:10.1021/ol2006117 |
| 47. | Serna, S.; Tellitu, I.; Domı́nguez, E.; Moreno, I.; SanMartı́n, R. Tetrahedron Lett. 2003, 44, 3483–3486. doi:10.1016/s0040-4039(03)00670-1 |
| 48. | Correa, A.; Tellitu, I.; Domínguez, E.; Moreno, I.; SanMartin, R. J. Org. Chem. 2005, 70, 2256–2264. doi:10.1021/jo047872u |
| 49. | Wardrop, D. J.; Basak, A. Org. Lett. 2001, 3, 1053–1056. doi:10.1021/ol015626o |
| 50. | Wardrop, D. J.; Zhang, W. Org. Lett. 2001, 3, 2353–2356. doi:10.1021/ol0161514 |
| 51. | Wardrop, D. J.; Burge, M. S.; Zhang, W.; Ortı́z, J. A. Tetrahedron Lett. 2003, 44, 2587–2591. doi:10.1016/s0040-4039(03)00227-2 |
| 52. | Wardrop, D. J.; Zhang, W.; Landrie, C. L. Tetrahedron Lett. 2004, 45, 4229–4231. doi:10.1016/j.tetlet.2004.04.028 |
| 53. | Bhattacharjee, A.; Gerasimov, M. V.; DeJong, S.; Wardrop, D. J. Org. Lett. 2017, 19, 6570–6573. doi:10.1021/acs.orglett.7b03283 |
| 41. | Tellitu, I.; Domínguez, E. Trends Heterocycl. Chem. 2011, 15, 23–32. |
| 42. | Tellitu, I.; Urrejola, A.; Serna, S.; Moreno, I.; Herrero, M. T.; Domínguez, E.; SanMartin, R.; Correa, A. Eur. J. Org. Chem. 2007, 437–444. doi:10.1002/ejoc.200600782 |
| 43. | Saito, A.; Anzai, T.; Matsumoto, A.; Hanzawa, Y. Tetrahedron Lett. 2011, 52, 4658–4661. doi:10.1016/j.tetlet.2011.06.117 |
| 37. | Stang, P. J.; Zhdankin, V. V. Chem. Rev. 1996, 96, 1123–1178. doi:10.1021/cr940424+ |
| 38. | Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2002, 102, 2523–2584. doi:10.1021/cr010003+ |
| 39. | Yoshimura, A.; Zhdankin, V. V. Chem. Rev. 2016, 116, 3328–3435. doi:10.1021/acs.chemrev.5b00547 |
| 40. | Yang, Y. O.; Wang, X.; Xiao, J.; Li, Y.; Sun, F.; Du, Y. Curr. Org. Chem. 2021, 25, 68–132. doi:10.2174/1385272822999201117154919 |
© 2021 Vaskevych et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.