
Abstract
The first immobilization of a MacMillan’s first generation organocatalyst onto dendritic support is described. A modified tyrosine-based imidazolidin-4-one was grafted to a soluble high-loading hyperbranched polyglycerol via a copper-catalyzed alkyne–azide cycloaddition (CuAAC) reaction and readily purified by dialysis. The efficiency of differently functionalized multivalent organocatalysts 4a–c was tested in the asymmetric Friedel–Crafts alkylation of N-methylpyrrole with α,β-unsaturated aldehydes. A variety of substituted enals was investigated to explore the activity of the catalytic system which was also compared with monovalent analogues. The catalyst 4b showed excellent turnover rates and no loss of activity due to immobilization, albeit moderate enantioselectivities were observed. Moreover, easy recovery by selective precipitation allowed the reuse of the catalyst for three cycles.
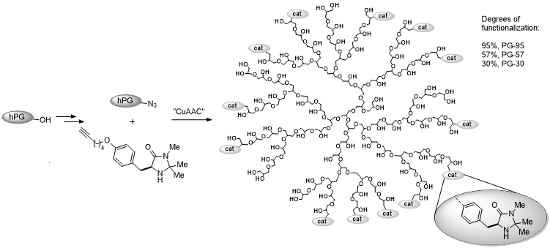
Graphical Abstract
Introduction
In nature, multivalent architectures, e.g., enzymes, bacteria or viruses, are responsible for cooperative interactions between different interfaces or molecules [1]. The realization of the concept of multivalency has attracted attention from different fields ranging from medicine and biochemistry [2] to supramolecular chemistry [3,4] and materials sciences [5]. However, applications in catalysis are still limited [6-8]. Recently, the use of polymeric support has stimulated the development of multivalent architectures for catalytic applications [9]. In general, both linear and various families of branched polymers such as dendrimers, dendritic-hybrid and hyperbranched polymers are used as macromolecular support for catalysis [10-12]. Linear polymers such as poly(ethylene glycol) (PEG) [13] or non-cross-linked polystyrene (NCPS) [14] are readily available but suffer from poor loading capacity, while in the case of dendrimers, the highest loading can be achieved due to their extraordinary branching [15]. These well-defined molecules are soluble in many organic solvents and can combine the advantages of hetero- and homogeneous catalysis [16,17]. However, their tedious and multistep syntheses using either divergent or convergent approaches are arguably the reason for their limited use as support in organic synthesis [18]. To overcome these obstacles, a hybrid dendron-polymer might constitute a valuable alternative for high-loading platforms [19], despite the use of solid support such as polystyrene may lead to the disadvantage of operating in heterogeneous media. In contrast to the stepwise syntheses of dendrimers and dendron hybrids, the hyperbranched polymers can be easily obtained in kilogram scale through one-pot reactions [10], maintaining properties such as high loading capacity combined with the solubility characteristics of the respective dendrimers [20,21]. As a macromolecule, the supported catalyst can be recovered from the reaction media by selective precipitation, dialysis or filtration techniques, depending on its particular physical properties. Hyperbranched polymers like polytriallylsilane or polyglycerol have been used in a wide range of transformations including aldol condensations [22], Suzuki cross-couplings [23] and Diels–Alder reactions [24], to name a few, with metal complexes as catalytically active principle.
The advent of organocatalysis has allowed for selective C–C bond formation by using small organic molecules [25-31]. In contrast to metal complexes, chiral or achiral organocatalysts are easily attached on supports. They do not suffer from metal leaching and they can be reused more readily [32-36]. Moreover, their stability allows to perform reactions under mild and aerobic conditions and in the presence of water, both as co-solvent or the only solvent [37]. In the last years, several reports on water effects in organocatalytic reactions were published [38-42]. The use of supported catalyst has proven beneficial with regard to rate acceleration and increased selectivity due to formation of an aqueous microenvironment favored by the swelling properties of polymeric materials [43]. Particularly, in the case of dendritic proline derivatives [44-46] and N-alkylimidazole decorated dendron-hybrids [47], the presence of water was crucial for aldol and Baylis–Hillman reactions, as recently reported by Miller and Portnoy [48].
To the best of our knowledge, the immobilization of chiral organocatalysts on hyperbranched polymeric support has remained unexplored. Therefore, we decided to use hyperbranched polyglycerol (hPG) [49] as a polymeric support. The high local concentration of hydrophilic functionality present on its periphery is especially attractive since it might promote water coordination. These properties prompted us to investigate the effects of high-loading support in asymmetric organocatalysis.
The use of chiral imidazolidinones in organocatalysis has been extensively reported for a wide range of enantioselective reactions involving α,β-unsaturated aldehydes, such as the Diels–Alder reactions [50], 1,3-dipolar cycloadditions [51] and Friedel–Crafts alkylations [52,53]. To date, heterogenizations have been applied mainly in Diels–Alder reactions [54-61]. Nevertheless, Friedel–Crafts alkylations are recently emerging as a compelling field of study as reported by Pericàs [62] and others [58,60]. The simple approach providing an enantioselective entry to new C–C bonds allows for the use of readily available starting materials and can typically be carried out in THF–water mixtures. Our aim was to employ this transformation as a benchmark in order to explore the efficiency of novel multivalent architectures.
Herein, we describe the first immobilization of imidazolidin-4-one onto hyperbranched polyglycerol (hPG) and its application as multivalent organocatalyst.
Results and Discussion
To explore the synthetic utility of hPG in organocatalysis, we here report the synthesis and application of a series of three multivalent dendronized imidazolidin-4-ones PG-95 (4a), PG-57 (4b) and PG-30 (4c) representing different degrees of functionalization: 95% (4a), 57% (4b) and 30% (4c), respectively. An (S)-tyrosine-derived imidazolidin-4-one 5 was anchored to the polymeric support through a CuAAC reaction. Following the same strategy, a monovalent analog 8 bearing a G1 glycerol dendron tail was also prepared for comparison with the multivalent systems 4a–c and evaluation of the possible presence of cooperative effects (Scheme 1).
![[1860-5397-11-83-i1]](/bjoc/content/inline/1860-5397-11-83-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of hyperbranched polyglycerol-supported and G1 dendronized imidazolidin-4-ones 4a–c and 8 using a CuAAC reaction. Reaction conditions: (a) 1 (1.0 equiv), MsCl (1.2 equiv, with respect to degrees of functionalization), pyridine, 25 °C, 16 h, 76% 2a, 82% 2b and 87% 2c. (b) 2a–c (1.0 equiv), NaN3 (3.0 equiv), DMF, 65 °C, 72 h, 72% 3a, 81% 3b and 86% 3c. (c) 3a–c (1.0 equiv), 5 (2.0 equiv), CuSO4·5H2O (0.2 equiv), sodium ascorbate (2.0 equiv), THF/H2O 3:1 (v/v), 25 °C, 48 h, 71% 4a, 40% 4b and 35% 4c. (d) 6 (1.1 equiv), 5 (1.0 equiv), CuSO4·5H2O (0.1 equiv), sodium ascorbate (0.2 equiv), DIPEA (0.1 equiv), THF/H2O 3:1 (v/v), 25 °C, 12 h, 70%. (e) 7, Dowex 50, MeOH, reflux, 12 h, 95%.
Scheme 1: Synthesis of hyperbranched polyglycerol-supported and G1 dendronized imidazolidin-4-ones 4a–c and 8...
Polyglycerol 1 (Mn = 9000 g/mol, loading OH = 13.5 mmol/g, PDI = 1.87) was obtained following a previously reported procedure by a one-step ring opening anionic polymerization (ROAP) [49]. The controlled mesylation on hPG 1 yielded 2a–c (95%, 57% and 30% of functionalization, respectively (for details see Supporting Information File 1)), which were converted to the corresponding azides 3a–c [63,64]. Azide 6 was prepared according to well-established protocols [65]. Consequently, we adopted the Sharpless–Fokin modification for the Huisgen azide–alkyne cycloaddition [66] to achieve the final immobilization of the modified imidazolidin-4-one onto the hyperbranched polymer and on the G1 dendron [65]. The progress of the reaction was monitored by IR spectroscopy and TLC. Purification of the products 4a–c was carried out by washing with aqueous saturated EDTA solution followed by dialysis in methanol/chloroform mixture for 24 h, and then in methanol and chloroform, respectively, for additional 12 h each. The catalyst structures were confirmed by 1H and 13C NMR spectroscopy and the functionalization degrees of 4a–c were determined by correlating the aromatic with the polyglycerol backbone protons (for details see Supporting Information File 1).
The synthesis of modified imidazolidin-4-one 5 started with (S)-tyrosine methyl ester hydrochloride (9). Following a protocol by Zhang and co-workers [58], 10 was obtained in good yield and subsequent anchoring of the linker was realized through O-alkylation on phenol 10, leading to linkable catalyst 5 in excellent yield (Scheme 2).
![[1860-5397-11-83-i2]](/bjoc/content/inline/1860-5397-11-83-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of tyrosine-based imidazolidin-4-one 5. Reaction conditions: (a) 9 (1.0 equiv), MeNH2 (5.0 equiv), EtOH, 25 °C, 20 h. (b) PTSA (0.01 equiv), acetone, MeOH, reflux, 18 h, 79% (2 steps). (c) 10 (1.0 equiv), NaH (1.1 equiv), 6-chloro-1-hexyne (1.3 equiv), TBAI (0.01 equiv), DMF, 25 °C, 16 h, 88%.
Scheme 2: Synthesis of tyrosine-based imidazolidin-4-one 5. Reaction conditions: (a) 9 (1.0 equiv), MeNH2 (5....
The reactivity of the multivalent catalysts 4a–c was investigated in the Friedel–Crafts alkylation of N-methylpyrrole (11) with α,β-unsaturated aldehydes reported by MacMillan [53]. To make the results comparable, we normalized the loading of the multivalent catalysts 4a–c with respect to the number of single anchored imidazolidin-4-ones. Therefore, a constant number of catalytic units for each degree of functionalization was maintained. Initially, we decided to perform the reaction using trans-cinnamaldehyde (12) as a model substrate and trifluoroacetic acid (TFA) as an additive. In a preliminary survey on the water influence, a catalyst loading of 3.5 mol % in THF was selected to allow 4a and 4b to operate under homogeneous conditions, while in the same solvent 4c proved to be less soluble (Table 1).
Table 1: Initial screening on the Friedel–Crafts alkylation of N-methylpyrrole (11) with trans-cinnamaldehyde (12).a
![[Graphic 1]](/bjoc/content/inline/1860-5397-11-83-i3.svg?max-width=637&scale=1.0)
|
||||
| Entry | Catalyst | THF/H2O (v/v) | Yield (%)b | ee (%)c |
|---|---|---|---|---|
| 1 | PG-95 (4a) | 100:0 | 38 | 66 |
| 2 | PG-57 (4b) | 100:0 | 56 | 69 |
| 3 | PG-30 (4c) | 100:0 | 26 | 56 |
| 4 | PG-95 (4a) | 95:5 | 62 | 68 |
| 5 | PG-57 (4b) | 95:5 | 68 | 66 |
| 6 | PG-30 (4c) | 95:5 | 32 | 59 |
| 7 | PG-95 (4a) | 90:10 | 42 | 59 |
| 8 | PG-57 (4b) | 90:10 | 38 | 60 |
| 9 | PG-30 (4c) | 90:10 | 45 | 54 |
| 10 | PG-95 (4a) | 0:100 | –d | – |
| 11 | PG-57 (4b) | 0:100 | –d | – |
| 12 | PG-30 (4c) | 0:100 | –d | – |
aReaction conditions: trans-cinnamaldehyde (12, 0.25 mmol, 1.0 equiv), N-methylpyrrole (11, 1.25 mmol, 5.0 equiv), catalyst 4a–c (3.5 mol %), aq TFA (5 M; 3.5 mol %), 0.63 M with respect to trans-cinnamaldehyde (12), 25 °C, 20 h. bIsolated yield. cDetermined by chiral GC. dComplex mixture of products.
Moderate conversion of 13 were achieved using only THF as a solvent and in presence of substoichiometric amounts of water (0.4 equiv) [41]. Addition of water as co-solvent proved beneficial for the formation of product 13. Notably, PG-95 (4a) and PG-57 (4b) exhibited comparable trends and the best yield and ee were observed when 5 vol % of water was added to the reaction mixture (Table 1, entries 4 and 5). Increasing the water content to 10 vol % resulted in incomplete conversion to 13 and lower ee values of the product (Table 1, entries 7 and 8). In case of the more hydrophilic PG-30 (4c) the activity increased with the amount of water in the reaction medium; yields and selectivities remained moderate. Attempts to carry out the reaction in water as the only solvent were unsuccessful (Table 1, entries 10–12). As expected, the outcomes of this reaction were strongly dependent on the solvent/water ratio and the catalysts 4a–c exhibited different activity with changes on the degrees of functionalization. In general, catalysts 4a and 4b were found to be more efficient in comparison to the less functionalized 4c. Probably, the poor ability of 4c to catalyze the model transformation might be explained by its low solubility in the reaction medium, most likely due to the large number of free hydroxy groups on the periphery of the multivalent catalyst. Instead, catalysts 4a–c demonstrated to be completely soluble in chloroform and methanol. Unfortunately, the use of these solvents led to decreased yields and selectivities of 13. Therefore, we decided to further investigate the superior catalysts PG-95 (4a) and PG-57 (4b) in THF/H2O mixture.
As reported in the literature, immobilization of chiral imidazolidin-4-ones on polymeric support might affect the formation of the desired products and lead to decreased enantioselectivities [58]. Indeed, in all the experiments reported in Table 1 the enantiomeric excess of 13 was lower compared to MacMillan’s original experiments [53]. In an attempt to improve the enantiomeric ratios, we studied the influence of temperature using the optimized conditions obtained for 4a and 4b in Table 1 (for results, see Table 2).
Table 2: Influence of temperature in the Friedel–Crafts alkylation.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-11-83-i4.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Catalyst | T (°C) | t (h) | Yield (%)b | ee (%)c |
|---|---|---|---|---|---|
| 1 | PG-95 (4a) | 25 | 20 | 62 | 68 |
| 2 | PG-57 (4b) | 25 | 20 | 68 | 66 |
| 3 | PG-95 (4a) | 4 | 35 | 60 | 68 |
| 4 | PG-57 (4b) | 4 | 35 | 64 | 68 |
| 5 | PG-95 (4a) | –24 | 48 | 46 | 76 |
| 6 | PG-57 (4b) | –24 | 48 | 25 | 78 |
aReaction conditions: trans-cinnamaldehyde (12, 0.25 mmol, 1.0 equiv), N-methylpyrrole (11, 1.25 mmol, 5.0 equiv), catalyst 4a,b (3.5 mol %), aq TFA (5 M; 3.5 mol %), THF/H2O 95:5 (v/v), 0.63 M with respect to trans-cinnamaldehyde (12). bIsolated yield. cDetermined by chiral GC.
To our dismay, running the transformation at lower temperatures did not lead to any significant improvements, although slight changes were observed. Carrying out the reactions at 4 °C gave similar ee values (Table 2, entries 3 and 4), whereas at −24 °C the alkylation led to marginally increased selectivities, at the cost of a drop in the yield (Table 2, entries 5 and 6). Nevertheless, the observed enantiomeric excess of the product 13 is still low when compared with those (93% ee, at −30 °C) originally reported in the case of the traditional (S)-phenylalanine-based imidazolidin-4-one [53].
Using the optimized solvent system (Table 1), we then turned our attention to study the catalyst loading and further prove the efficiency of multivalent 4a and 4b (Table 3).
Table 3: Catalyst loading study.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-11-83-i5.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Catalyst | Load. (mol %) | THF/H2O (v/v) | Yield (%)b | ee (%)c |
|---|---|---|---|---|---|
| 1 | PG-95 (4a) | 2 | 95:5 | 43 | 59 |
| 2 | PG-57 (4b) | 2 | 95:5 | 62 | 64 |
| 3 | PG-95 (4a) | 2 | 97:3 | 66 | 68 |
| 4 | PG-57 (4b) | 2 | 97:3 | 65 | 67 |
| 5 | PG-95 (4a) | 1 | 98.5:1.5 | 46 | 67 |
| 6 | PG-57 (4b) | 1 | 98.5:1.5 | 50 | 74 |
aReaction conditions: trans-cinnamaldehyde (12, 0.50 mmol, 1.0 equiv), N-methylpyrrole (11, 2.50 mmol, 5.0 equiv), catalyst 4a,b (2 or 1 mol %), aq TFA (5 M; 2 mol %, entries 1–4 or 1 mol %, entries 5 and 6), 0.63 M with respect to trans-cinnamaldehyde (12), 25 °C, 48 h. bIsolated yield. cDetermined by chiral GC.
Initial attempts with 2 mol % of the multivalent 4a and 4b, using 5 vol % of water in THF led to the isolation of 13 in moderate yield and slightly lower enantioselectivies, a result even more pronounced in the case of PG-95 (4a) (Table 3, entries 1 and 2). Next, we questioned if in addition to a catalyst loading reduction also a concomitant reduction of the water amount was necessary to maintain yield and enantiomeric ratio. Consistently, we reduced the water amount from 5 to 3 vol % and observed higher conversion to 13 and improved ee values (Table 3, entries 3 and 4). Therefore, in the following experiments the catalyst/water ratio was kept constant. The excellent efficiency of the catalyst was confirmed with moderate to good yields of 13 even though using 1 mol % of 4a and 4b, respectively (Table 3, entries 5 and 6). Considering, for the supported case, a typical catalyst loading for this transformation to be 10 mol % in order to achieve good conversion [62], the loadings reported in Table 3 could be decreased by one order of magnitude.
After solvent and temperature screening, our studies were focused on dilution experiments (Table 4).
Table 4: Dilution experiments.a
![[Graphic 4]](/bjoc/content/inline/1860-5397-11-83-i6.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Catalyst | Conc. (M)b | THF/H2O (v/v) | Yield (%)c | ee (%)d |
|---|---|---|---|---|---|
| 1e | PG-95 (4a) | 0.63 | 97:3 | 66 | 68 |
| 2e | PG-57 (4b) | 0.63 | 97/3 | 65 | 67 |
| 3 | PG-95 (4a) | 0.30 | 98.5:1.5 | 70 | 68 |
| 4 | PG-57 (4b) | 0.30 | 98.5:1.5 | 87 | 70 |
| 5 | PG-95 (4a) | 0.10 | 99.5:0.5 | <1f | n.d.g |
| 6 | PG-57 (4b) | 0.10 | 99.5:0.5 | 29f | n.d.g |
aReaction conditions: trans-cinnamaldehyde (12, 0.25 mmol, 1.0 equiv), N-methylpyrrole (11, 1.25 mmol, 5.0 equiv), catalyst 4a,b (2 mol %), aq TFA (5 M; 2 mol %), 25 °C, 48 h. bWith respect to trans-cinnamaldehyde (12). cIsolated yield. dDetermined by chiral GC. etrans-Cinnamaldehyde (12, 0.50 mmol, 1.0 equiv), N-methylpyrrole (11, 2.50 mmol, 5.0 equiv). fDetermined by 1H NMR. gn.d. = not determined.
The best yield and enantioselectivity of 13 was obtained using PG-57 (4b) and lowering the concentration from 0.63 to 0.30 M (Table 4, entry 4). Contrarily, PG-95 (4a) did not lead to any appreciable improvement (Table 4, entry 3). By reducing the concentration to 0.10 M, 4b gave only poor to moderate yields while the efficiency of 4a decreased even more sharply and only traces of product 13 were observed (Table 4, entries 5 and 6). On the other hand, the enantioselectivities remained unchanged passing from concentration of 0.63 M to more diluted conditions (0.30 M). This outcome might be attributed to the constant local neighborhood in the polymer periphery where the catalytic centers are located, therefore the concentration may not affect the chiral induction [24].
After the completion of our systematic optimization of the reaction parameters using the model transformation, the most active catalyst 4b was selected for a screening of different enals in the alkylation reaction of N-methylpyrrole (11). A study on the substrate scope was further carried out under the established conditions (see Table 4, entry 4). A variety of substituted α,β-unsaturated aldehydes 14a–e was employed using 2 mol % PG-57 (4b) in THF/H2O 98.5:1.5 (v/v) (Table 5).
Table 5: Substrate scope.a
![[Graphic 5]](/bjoc/content/inline/1860-5397-11-83-i7.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | Substrate | Product | t (h) | Yield (%)b | ee (%)c | ||
|---|---|---|---|---|---|---|---|
| 1 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-11-83-i8.svg?max-width=637&scale=1.0)
|
14a |
![[Graphic 7]](/bjoc/content/inline/1860-5397-11-83-i9.svg?max-width=637&scale=1.0)
|
15a | 24 | 86 | 69 |
| 2 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-11-83-i10.svg?max-width=637&scale=1.0)
|
14b |
![[Graphic 9]](/bjoc/content/inline/1860-5397-11-83-i11.svg?max-width=637&scale=1.0)
|
15b | 24 | 83 | 68 |
| 3 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-11-83-i12.svg?max-width=637&scale=1.0)
|
14c |
![[Graphic 11]](/bjoc/content/inline/1860-5397-11-83-i13.svg?max-width=637&scale=1.0)
|
15c | 48 | 80 | 56 |
| 4 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-11-83-i14.svg?max-width=637&scale=1.0)
|
14d |
![[Graphic 13]](/bjoc/content/inline/1860-5397-11-83-i15.svg?max-width=637&scale=1.0)
|
15d | 48 | 86 | 71 |
| 5 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-11-83-i16.svg?max-width=637&scale=1.0)
|
14e |
![[Graphic 15]](/bjoc/content/inline/1860-5397-11-83-i17.svg?max-width=637&scale=1.0)
|
15e | 48 | 99 | 78 |
aReaction conditions: aldehyde 14a–e (0.25 mmol, 1.0 equiv), N-methylpyrrole (11, 1.25 mmol, 5.0 equiv), catalyst 4b (2 mol %), aq TFA (5 M; 2 mol %), THF/H2O 98.5:1.5 (v/v), 0.30 M with respect to aldehyde 14a–e, 25 °C. bIsolated yield. cDetermined by chiral GC.
Multivalent catalyst 4b showed good to excellent activities in a range of substrates and moderate to good enantiomeric ratios for the formation of products 15a–e, as shown in Table 5. Electron-deficient aromatic enals 14d,e afforded higher yields and selectivities, confirming the strong influence of the substituent (Table 5, entries 4 and 5). Contrarily, aliphatic enals 14a,b were well-tolerated and the outcomes were not affected significantly (Table 5, entries 1 and 2).
In our studies on the utilization of hPG as a soluble support in organocatalysis, hyperbranched PG-95 (4a) and PG-57 (4b) were finally compared with the monovalent G1-dendron imidazolidin-4-one 8 previously prepared and the original MacMillan’s first generation catalyst 16 using the optimum conditions (Table 6).
Table 6: Comparison of hPG catalysts 4a,b with monovalent analogue 8 and MacMillan’s first generation 16.a
![[Graphic 16]](/bjoc/content/inline/1860-5397-11-83-i18.svg?max-width=637&scale=1.0)
|
|||
| Entry | Catalyst | Yield (%)b | ee (%)c |
|---|---|---|---|
| 1 | PG-95 (4a) | 70 | 68 |
| 2 | PG-57 (4b) | 87 | 70 |
| 3 | 8 | 83 | 67 |
| 4 | 16 | 64 | 77 |
aReaction conditions: trans-cinnamaldehyde (12, 0.25 mmol, 1.0 equiv), N-methylpyrrole (11, 1.25 mmol, 5.0 equiv), cat. (2 mol %), aq TFA (5 M; 2 mol %), THF/H2O 98.5:1.5 (v/v), 0.30 M with respect to trans-cinnamaldehyde (12), 25 °C, 48 h. bIsolated yield. cDetermined by chiral GC.
As shown in Table 6, multivalent 4b and monovalent 8 afforded similar results (Table 6, entries 2 and 3), probably due to their comparable high hydrophilicity. This outcome did not indicate additional cooperative effects between the active catalytic sites. Increased activities were observed compared to MacMillan’s catalyst 16, albeit with lower enantioselectivity (Table 6, entries 2, 3 and 4). Catalyst 4a showed turnover rates comparable with the traditional imidizolidin-4-one 16 (Table 6, entries 1 and 4). In conclusion, high- (PG-95, 4a) or low- (PG-30, 4c) loaded support were less active when compared to an intermediate degree of functionalization (PG-57, 4b). In the case of PG-57 (4b) a good compromise between hydrophilicity and solubility was achieved. The results reported in Table 6 point out that catalyst 4b was not suffering from diminished reactivity as often observed with immobilizations. Additionally the polymeric support was found to be responsible for enhanced reactivity with respect to the original imidazolidin-4-one 16. The presence of anchimeric assistance by hydroxy groups, in the hydrolysis step of the iminium intermediate, might account for the observed improved turnover rates.
To complete our studies on the generality of hPG catalysts, finally, recycling of the polymer was studied. Heterogeneous catalysis allowed for simple separations of the immobilized species from the reaction media. Indeed, working under homogeneous conditions did not enable separation by simple filtration. On the other hand, the multivalent catalysts 4a and 4b showed poor solubility in solvents with low polarity, thus allowing for an easy recovery in 60–70% yield after selective precipitation using Et2O. The utility of our soluble support was examined in the catalytic efficiency of recovered PG-57 (4b) (Table 7).
![[Graphic 17]](/bjoc/content/inline/1860-5397-11-83-i19.svg?max-width=637&scale=1.0)
aReaction conditions: trans-cinnamaldehyde (12, 1.0 equiv), N-methylpyrrole (11, 5.0 equiv), catalyst 4b (2 mol %), THF/H2O 97:3 (v/v), 0.63 M with respect to trans-cinnamaldehyde (12), 25 °C, 48 h. bIsolated yield. cDetermined by chiral GLC. dAq TFA (5 M; 2 mol %) was added to the reaction mixture.
Catalyst 4b was used three times in the asymmetric alkylation reaction. The experiment showed constant enantiomeric ratios although decreased activity and yields were observed. The lower yields exhibited after each cycle might be attributed to the decreased solubility of the recovered polymer. For the same reason, early attempts using the optimized parameters (conc. 0.30 M) were not successful; therefore the same PG-57 (4b) was subjected to more concentrated conditions (conc. 0.63 M). Moreover, addition of the acidic co-catalyst was crucial to establish the reactivity of the imidazolidin-4-one in the third cycle. Attempts to elucidate the reason of the decreased reactivity and analysis of the recovered polymer by 1H NMR indicated the leakage of the imidazolidin-4-one moiety. Nevertheless, studies focussing on improved catalyst stability and recycling are in progress.
Conclusion
In summary, we have successfully employed a CuAAC strategy in the first immobilization of a chiral imidazolidin-4-one onto hyperbranched polyglycerol support and examined its efficiency in organocatalysis. Catalyst 4c proved to be less soluble in the reaction media compared to 4a and 4b, and showed poor activity and selectivity. The soluble polymers 4a and 4b enabled homogeneous reactions without loss of efficiency due to immobilization. The activity of multivalent catalyst 4a was comparable with that exhibited by the traditional MacMillan’s catalyst, while 4b was shown to be superior. Nevertheless, erosion in enantioselectivity was observed, probably as a consequence of high local concentration effects on the periphery of the dendritic architecture, where the catalytic sites are located. The novel multivalent system 4b achieved good conversion to afford product 13, even with low polymer loading (1 mol %) compared to common loadings of 10 mol % required for the supported imidazolidin-4-ones. Moreover, 4b was shown to be well-tolerated in a range of α,β-unsaturated aldehydes. The improved efficiency shown by 4b might derive from an anchimeric assistance in the hydrolysis step of the iminium ion. Interestingly, the presence of such an effect might offer opportunities for further studies. One of the advantages of the multivalent catalyst 4b was demonstrated to be its easy separability from the reaction media and its reuse for three consecutive times, whereas further investigations will be necessary on recycling of the polymeric support.
Supporting Information
| Supporting Information File 1: Experimental procedures, analytical data, copies of NMR spectra and GC reports. | ||
| Format: PDF | Size: 2.2 MB | Download |
References
-
Fasting, C.; Schalley, C. A.; Weber, M.; Seitz, O.; Hecht, S.; Koksch, B.; Dernedde, J.; Graf, C.; Knapp, E.-W.; Haag, R. Angew. Chem., Int. Ed. 2012, 51, 10472–10498. doi:10.1002/anie.201201114
Return to citation in text: [1] -
Mammen, M.; Choi, S.-K.; Whitesides, G. M. Angew. Chem., Int. Ed. 1998, 37, 2754–2794. doi:10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3
Return to citation in text: [1] -
Mulder, A.; Huskens, J.; Reinhoudt, D. N. Org. Biomol. Chem. 2004, 2, 3409–3424. doi:10.1039/B413971B
Return to citation in text: [1] -
Badjić, J. D.; Nelson, A.; Cantrill, S. J.; Turnbull, W. B.; Stoddart, J. F. Acc. Chem. Res. 2005, 38, 723–732. doi:10.1021/ar040223k
Return to citation in text: [1] -
Dallas, P.; Sharma, V. K.; Zboril, R. Adv. Colloid Interface Sci. 2011, 166, 119–135. doi:10.1016/j.cis.2011.05.008
Return to citation in text: [1] -
Park, J.; Hong, S. Chem. Soc. Rev. 2012, 41, 6931–6943. doi:10.1039/c2cs35129c
Return to citation in text: [1] -
Shibasaki, M.; Yoshikawa, N. Chem. Rev. 2002, 102, 2187–2210. doi:10.1021/cr010297z
Return to citation in text: [1] -
van den Beuken, E. K.; Feringa, B. L. Tetrahedron 1998, 54, 12985–13011. doi:10.1016/S0040-4020(98)00319-6
Return to citation in text: [1] -
Haag, R.; Roller, S. Top. Curr. Chem. 2004, 242, 1–42.
Return to citation in text: [1] -
Hajji, C.; Haag, R. Top. Organomet. Chem. 2006, 20, 149–176. doi:10.1007/3418_035
Return to citation in text: [1] [2] -
Wang, D.; Astruc, D. Coord. Chem. Rev. 2013, 257, 2317–2334. doi:10.1016/j.ccr.2013.03.032
Return to citation in text: [1] -
Bergbreiter, D. E.; Tian, J.; Hongfa, C. Chem. Rev. 2009, 109, 530–582. doi:10.1021/cr8004235
Return to citation in text: [1] -
Chinnusamy, T.; Hilgers, P.; Reiser, O. Catalysts Bound to Soluble Polymers. In Recoverable and Recyclable Catalysts; Benaglia, M., Ed.; John Wiley & Sons: Chichester, UK, 2009; pp 77–100.
Return to citation in text: [1] -
Chen, J.; Yang, G.; Zhang, H.; Chen, Z. React. Funct. Polym. 2006, 66, 1434–1451. doi:10.1016/j.reactfunctpolym.2006.04.008
Return to citation in text: [1] -
Caminade, A.-M.; Turrin, C.-O.; Laurent, R.; Ouali, A.; Delavaux-Nicot, B., Eds. Dendrimers: Towards Catalytic, Material and Biomedical Uses; John Wiley & Sons: Chichester, UK, 2011.
Return to citation in text: [1] -
Helms, B.; Fréchet, J. M. J. Adv. Synth. Catal. 2006, 348, 1125–1148. doi:10.1002/adsc.200606095
Return to citation in text: [1] -
Gade, L. H., Ed. Dendrimer Catalysis; Top. Organomet. Chem., Vol. 20; Springer: Berlin, 2006. doi:10.1007/11603788
Return to citation in text: [1] -
Haag, R.; Roller, S. Dendritic Polymers as High-Loading Supports for Organic Synthesis and Catalysis. In Polymeric Materials in Organic Synthesis and Catalysis; Buchmeiser, M. R., Ed.; Wiley-VCH: Weinheim, Germany, 2003; pp 305–344.
Return to citation in text: [1] -
Carlmark, A.; Hawker, C.; Hult, A.; Malkoch, M. Chem. Soc. Rev. 2009, 38, 352–362. doi:10.1039/B711745K
Return to citation in text: [1] -
Gao, C.; Yan, D. Prog. Polym. Sci. 2004, 29, 183–275. doi:10.1016/j.progpolymsci.2003.12.002
Return to citation in text: [1] -
Jikei, M.; Kakimoto, M. Prog. Polym. Sci. 2001, 26, 1233–1285. doi:10.1016/S0079-6700(01)00018-1
Return to citation in text: [1] -
Schlenk, C.; Kleij, A. W.; Frey, H.; van Koten, G. Angew. Chem., Int. Ed. 2000, 39, 3445–3447. doi:10.1002/1521-3773(20001002)39:19<3445::AID-ANIE3445>3.0.CO;2-8
Return to citation in text: [1] -
Hebel, A.; Haag, R. J. Org. Chem. 2002, 67, 9452–9455. doi:10.1021/jo026076q
Return to citation in text: [1] -
Hajji, C.; Roller, S.; Beigi, M.; Liese, A.; Haag, R. Adv. Synth. Catal. 2006, 348, 1760–1771. doi:10.1002/adsc.200606168
Return to citation in text: [1] [2] -
Seayad, J.; List, B. Org. Biomol. Chem. 2005, 3, 719–724. doi:10.1039/B415217B
Return to citation in text: [1] -
Dalko, P. I.; Moisan, L. Angew. Chem., Int. Ed. 2001, 40, 3726–3748. doi:10.1002/1521-3773(20011015)40:20<3726::AID-ANIE3726>3.0.CO;2-D
Return to citation in text: [1] -
Dalko, P. I.; Moisan, L. Angew. Chem., Int. Ed. 2004, 43, 5138–5175. doi:10.1002/anie.200400650
Return to citation in text: [1] -
Bertelsen, S.; Jørgensen, K. A. Chem. Soc. Rev. 2009, 38, 2178–2189. doi:10.1039/B903816G
Return to citation in text: [1] -
Enders, D.; Grondal, C.; Hüttl, M. R. S. Angew. Chem., Int. Ed. 2007, 46, 1570–1581. doi:10.1002/anie.200603129
Return to citation in text: [1] -
Scheffler, U.; Mahrwald, R. Chem. – Eur. J. 2013, 19, 14346–14396. doi:10.1002/chem.201301996
Return to citation in text: [1] -
Dondoni, A.; Massi, A. Angew. Chem., Int. Ed. 2008, 47, 4638–4660. doi:10.1002/anie.200704684
Return to citation in text: [1] -
Kristensen, T. E.; Hansen, T. Eur. J. Org. Chem. 2010, 3179–3204. doi:10.1002/ejoc.201000319
Return to citation in text: [1] -
Cozzi, F. Adv. Synth. Catal. 2006, 348, 1367–1390. doi:10.1002/adsc.200606096
Return to citation in text: [1] -
Benaglia, M.; Puglisi, A.; Cozzi, F. Chem. Rev. 2003, 103, 3401–3430. doi:10.1021/cr010440o
Return to citation in text: [1] -
Trindade, A. F.; Gois, P. M. P.; Afonso, C. A. M. Chem. Rev. 2009, 109, 418–514. doi:10.1021/cr800200t
Return to citation in text: [1] -
Gruttadauria, M.; Giacalone, F.; Noto, R. Chem. Soc. Rev. 2008, 37, 1666–1688. doi:10.1039/B800704G
Return to citation in text: [1] -
Wu, Y.; Zhang, Y.; Yu, M.; Zhao, G.; Wang, S. Org. Lett. 2006, 8, 4417–4420. doi:10.1021/ol061418q
Return to citation in text: [1] -
Gruttadauria, M.; Giacalone, F.; Noto, R. Adv. Synth. Catal. 2009, 351, 33–57. doi:10.1002/adsc.200800731
Return to citation in text: [1] -
Hernàndez, J. G.; Juaristi, E. Chem. Commun. 2012, 48, 5396–5409. doi:10.1039/C2CC30951C
Return to citation in text: [1] -
Mase, N.; Barbas, C. F., III. Org. Biomol. Chem. 2010, 8, 4043–4050. doi:10.1039/C004970K
Return to citation in text: [1] -
Hayashi, Y. Angew. Chem., Int. Ed. 2006, 45, 8103–8104. doi:10.1002/anie.200603378
Return to citation in text: [1] [2] -
Mlynarski, J.; Paradowska, J. Chem. Soc. Rev. 2008, 37, 1502–1511. doi:10.1039/B710577K
Return to citation in text: [1] -
Font, D.; Sayalero, S.; Bastero, A.; Jimeno, C.; Pericàs, M. A. Org. Lett. 2008, 10, 337–340. doi:10.1021/ol702901z
Return to citation in text: [1] -
Bellis, E.; Kokotos, G. J. Mol. Catal. A: Chem. 2005, 241, 166–174. doi:10.1016/j.molcata.2005.05.047
Return to citation in text: [1] -
Kehat, T.; Portnoy, M. Chem. Commun. 2007, 2823–2825. doi:10.1039/B703016A
Return to citation in text: [1] -
Mitsui, K.; Hyatt, S. A.; Turner, D. A.; Hadad, C. M.; Parquette, J. R. Chem. Commun. 2009, 3261–3263. doi:10.1039/B902960E
Return to citation in text: [1] -
Goren, K.; Portnoy, M. Chem. Commun. 2010, 46, 1965–1967. doi:10.1039/B915577E
Return to citation in text: [1] -
Goren, K.; Karabline-Kuks, J.; Shiloni, Y.; Barak-Kulbak, E.; Miller, S. J.; Portnoy, M. Chem. – Eur. J. 2015, 21, 1191–1197. doi:10.1002/chem.201404560
Return to citation in text: [1] -
Sunder, A.; Mülhaupt, R.; Haag, R.; Frey, H. Adv. Mater. 2000, 12, 235–239. doi:10.1002/(SICI)1521-4095(200002)12:3<235::AID-ADMA235>3.0.CO;2-Y
Return to citation in text: [1] [2] -
Ahrendt, K. A.; Borths, C. J.; MacMillan, D. W. C. J. Am. Chem. Soc. 2000, 122, 4243–4244. doi:10.1021/ja000092s
Return to citation in text: [1] -
Puglisi, A.; Benaglia, M.; Cinquini, M.; Cozzi, F.; Celentano, G. Eur. J. Org. Chem. 2004, 567–573. doi:10.1002/ejoc.200300571
Return to citation in text: [1] -
Terrasson, V.; Marcia de Figueiredo, R.; Campagne, J. M. Eur. J. Org. Chem. 2010, 2635–2655. doi:10.1002/ejoc.200901492
Return to citation in text: [1] -
Paras, N. A.; MacMillan, D. W. C. J. Am. Chem. Soc. 2001, 123, 4370–4371. doi:10.1021/ja015717g
Return to citation in text: [1] [2] [3] [4] -
Selkälä, S. A.; Tois, J.; Pihko, P. M.; Koskinen, A. M. P. Adv. Synth. Catal. 2002, 344, 941–945. doi:10.1002/1615-4169(200210)344:9<941::AID-ADSC941>3.0.CO;2-M
Return to citation in text: [1] -
Mitsudome, T.; Nose, K.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Tetrahedron Lett. 2008, 49, 5464–5466. doi:10.1016/j.tetlet.2008.07.011
Return to citation in text: [1] -
Chu, Q.; Zhang, W.; Curran, D. P. Tetrahedron Lett. 2006, 47, 9287–9290. doi:10.1016/j.tetlet.2006.10.101
Return to citation in text: [1] -
Park, J. K.; Sreekanth, P.; Kim, B. M. Adv. Synth. Catal. 2004, 346, 49–52. doi:10.1002/adsc.200303167
Return to citation in text: [1] -
Zhang, Y.; Zhao, L.; Lee, S. S.; Ying, J. Y. Adv. Synth. Catal. 2006, 348, 2027–2032. doi:10.1002/adsc.200600240
Return to citation in text: [1] [2] [3] [4] -
Benaglia, M.; Celentano, G.; Cinquini, M.; Puglisi, A.; Cozzi, F. Adv. Synth. Catal. 2002, 344, 149–152. doi:10.1002/1615-4169(200202)344:2<149::AID-ADSC149>3.0.CO;2-U
Return to citation in text: [1] -
Chiroli, V.; Benaglia, M.; Puglisi, A.; Porta, R.; Jumde, R. P.; Mandoli, A. Green Chem. 2014, 16, 2798–2806. doi:10.1039/C4GC00031E
Return to citation in text: [1] [2] -
Pecinovsky, C. S.; Nicodemus, G. D.; Gin, D. L. Chem. Mater. 2005, 17, 4889–4891. doi:10.1021/cm0514995
Return to citation in text: [1] -
Riente, P.; Yadav, J.; Pericàs, M. A. Org. Lett. 2012, 14, 3668–3671. doi:10.1021/ol301515d
Return to citation in text: [1] [2] -
Paez, J. I.; Brunetti, V.; Strumia, M. C.; Becherer, T.; Solomun, T.; Miguel, J.; Hermanns, C. F.; Calderón, M.; Haag, R. J. Mater. Chem. 2012, 22, 19488–19497. doi:10.1039/c2jm32486e
Return to citation in text: [1] -
Roller, S.; Zhou, H.; Haag, R. Mol. Diversity 2005, 9, 305–316. doi:10.1007/s11030-005-8117-y
Return to citation in text: [1] -
Wyszogrodzka, M.; Haag, R. Chem. – Eur. J. 2008, 14, 9202–9214. doi:10.1002/chem.200800892
Return to citation in text: [1] [2] -
Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4
Return to citation in text: [1]
| 50. | Ahrendt, K. A.; Borths, C. J.; MacMillan, D. W. C. J. Am. Chem. Soc. 2000, 122, 4243–4244. doi:10.1021/ja000092s |
| 51. | Puglisi, A.; Benaglia, M.; Cinquini, M.; Cozzi, F.; Celentano, G. Eur. J. Org. Chem. 2004, 567–573. doi:10.1002/ejoc.200300571 |
| 52. | Terrasson, V.; Marcia de Figueiredo, R.; Campagne, J. M. Eur. J. Org. Chem. 2010, 2635–2655. doi:10.1002/ejoc.200901492 |
| 53. | Paras, N. A.; MacMillan, D. W. C. J. Am. Chem. Soc. 2001, 123, 4370–4371. doi:10.1021/ja015717g |
| 1. | Fasting, C.; Schalley, C. A.; Weber, M.; Seitz, O.; Hecht, S.; Koksch, B.; Dernedde, J.; Graf, C.; Knapp, E.-W.; Haag, R. Angew. Chem., Int. Ed. 2012, 51, 10472–10498. doi:10.1002/anie.201201114 |
| 6. | Park, J.; Hong, S. Chem. Soc. Rev. 2012, 41, 6931–6943. doi:10.1039/c2cs35129c |
| 7. | Shibasaki, M.; Yoshikawa, N. Chem. Rev. 2002, 102, 2187–2210. doi:10.1021/cr010297z |
| 8. | van den Beuken, E. K.; Feringa, B. L. Tetrahedron 1998, 54, 12985–13011. doi:10.1016/S0040-4020(98)00319-6 |
| 20. | Gao, C.; Yan, D. Prog. Polym. Sci. 2004, 29, 183–275. doi:10.1016/j.progpolymsci.2003.12.002 |
| 21. | Jikei, M.; Kakimoto, M. Prog. Polym. Sci. 2001, 26, 1233–1285. doi:10.1016/S0079-6700(01)00018-1 |
| 66. | Rostovtsev, V. V.; Green, L. G.; Fokin, V. V.; Sharpless, K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. doi:10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4 |
| 5. | Dallas, P.; Sharma, V. K.; Zboril, R. Adv. Colloid Interface Sci. 2011, 166, 119–135. doi:10.1016/j.cis.2011.05.008 |
| 22. | Schlenk, C.; Kleij, A. W.; Frey, H.; van Koten, G. Angew. Chem., Int. Ed. 2000, 39, 3445–3447. doi:10.1002/1521-3773(20001002)39:19<3445::AID-ANIE3445>3.0.CO;2-8 |
| 65. | Wyszogrodzka, M.; Haag, R. Chem. – Eur. J. 2008, 14, 9202–9214. doi:10.1002/chem.200800892 |
| 3. | Mulder, A.; Huskens, J.; Reinhoudt, D. N. Org. Biomol. Chem. 2004, 2, 3409–3424. doi:10.1039/B413971B |
| 4. | Badjić, J. D.; Nelson, A.; Cantrill, S. J.; Turnbull, W. B.; Stoddart, J. F. Acc. Chem. Res. 2005, 38, 723–732. doi:10.1021/ar040223k |
| 19. | Carlmark, A.; Hawker, C.; Hult, A.; Malkoch, M. Chem. Soc. Rev. 2009, 38, 352–362. doi:10.1039/B711745K |
| 63. | Paez, J. I.; Brunetti, V.; Strumia, M. C.; Becherer, T.; Solomun, T.; Miguel, J.; Hermanns, C. F.; Calderón, M.; Haag, R. J. Mater. Chem. 2012, 22, 19488–19497. doi:10.1039/c2jm32486e |
| 64. | Roller, S.; Zhou, H.; Haag, R. Mol. Diversity 2005, 9, 305–316. doi:10.1007/s11030-005-8117-y |
| 2. | Mammen, M.; Choi, S.-K.; Whitesides, G. M. Angew. Chem., Int. Ed. 1998, 37, 2754–2794. doi:10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3 |
| 10. | Hajji, C.; Haag, R. Top. Organomet. Chem. 2006, 20, 149–176. doi:10.1007/3418_035 |
| 65. | Wyszogrodzka, M.; Haag, R. Chem. – Eur. J. 2008, 14, 9202–9214. doi:10.1002/chem.200800892 |
| 14. | Chen, J.; Yang, G.; Zhang, H.; Chen, Z. React. Funct. Polym. 2006, 66, 1434–1451. doi:10.1016/j.reactfunctpolym.2006.04.008 |
| 16. | Helms, B.; Fréchet, J. M. J. Adv. Synth. Catal. 2006, 348, 1125–1148. doi:10.1002/adsc.200606095 |
| 17. | Gade, L. H., Ed. Dendrimer Catalysis; Top. Organomet. Chem., Vol. 20; Springer: Berlin, 2006. doi:10.1007/11603788 |
| 58. | Zhang, Y.; Zhao, L.; Lee, S. S.; Ying, J. Y. Adv. Synth. Catal. 2006, 348, 2027–2032. doi:10.1002/adsc.200600240 |
| 60. | Chiroli, V.; Benaglia, M.; Puglisi, A.; Porta, R.; Jumde, R. P.; Mandoli, A. Green Chem. 2014, 16, 2798–2806. doi:10.1039/C4GC00031E |
| 13. | Chinnusamy, T.; Hilgers, P.; Reiser, O. Catalysts Bound to Soluble Polymers. In Recoverable and Recyclable Catalysts; Benaglia, M., Ed.; John Wiley & Sons: Chichester, UK, 2009; pp 77–100. |
| 18. | Haag, R.; Roller, S. Dendritic Polymers as High-Loading Supports for Organic Synthesis and Catalysis. In Polymeric Materials in Organic Synthesis and Catalysis; Buchmeiser, M. R., Ed.; Wiley-VCH: Weinheim, Germany, 2003; pp 305–344. |
| 49. | Sunder, A.; Mülhaupt, R.; Haag, R.; Frey, H. Adv. Mater. 2000, 12, 235–239. doi:10.1002/(SICI)1521-4095(200002)12:3<235::AID-ADMA235>3.0.CO;2-Y |
| 10. | Hajji, C.; Haag, R. Top. Organomet. Chem. 2006, 20, 149–176. doi:10.1007/3418_035 |
| 11. | Wang, D.; Astruc, D. Coord. Chem. Rev. 2013, 257, 2317–2334. doi:10.1016/j.ccr.2013.03.032 |
| 12. | Bergbreiter, D. E.; Tian, J.; Hongfa, C. Chem. Rev. 2009, 109, 530–582. doi:10.1021/cr8004235 |
| 54. | Selkälä, S. A.; Tois, J.; Pihko, P. M.; Koskinen, A. M. P. Adv. Synth. Catal. 2002, 344, 941–945. doi:10.1002/1615-4169(200210)344:9<941::AID-ADSC941>3.0.CO;2-M |
| 55. | Mitsudome, T.; Nose, K.; Mizugaki, T.; Jitsukawa, K.; Kaneda, K. Tetrahedron Lett. 2008, 49, 5464–5466. doi:10.1016/j.tetlet.2008.07.011 |
| 56. | Chu, Q.; Zhang, W.; Curran, D. P. Tetrahedron Lett. 2006, 47, 9287–9290. doi:10.1016/j.tetlet.2006.10.101 |
| 57. | Park, J. K.; Sreekanth, P.; Kim, B. M. Adv. Synth. Catal. 2004, 346, 49–52. doi:10.1002/adsc.200303167 |
| 58. | Zhang, Y.; Zhao, L.; Lee, S. S.; Ying, J. Y. Adv. Synth. Catal. 2006, 348, 2027–2032. doi:10.1002/adsc.200600240 |
| 59. | Benaglia, M.; Celentano, G.; Cinquini, M.; Puglisi, A.; Cozzi, F. Adv. Synth. Catal. 2002, 344, 149–152. doi:10.1002/1615-4169(200202)344:2<149::AID-ADSC149>3.0.CO;2-U |
| 60. | Chiroli, V.; Benaglia, M.; Puglisi, A.; Porta, R.; Jumde, R. P.; Mandoli, A. Green Chem. 2014, 16, 2798–2806. doi:10.1039/C4GC00031E |
| 61. | Pecinovsky, C. S.; Nicodemus, G. D.; Gin, D. L. Chem. Mater. 2005, 17, 4889–4891. doi:10.1021/cm0514995 |
| 15. | Caminade, A.-M.; Turrin, C.-O.; Laurent, R.; Ouali, A.; Delavaux-Nicot, B., Eds. Dendrimers: Towards Catalytic, Material and Biomedical Uses; John Wiley & Sons: Chichester, UK, 2011. |
| 62. | Riente, P.; Yadav, J.; Pericàs, M. A. Org. Lett. 2012, 14, 3668–3671. doi:10.1021/ol301515d |
| 25. | Seayad, J.; List, B. Org. Biomol. Chem. 2005, 3, 719–724. doi:10.1039/B415217B |
| 26. | Dalko, P. I.; Moisan, L. Angew. Chem., Int. Ed. 2001, 40, 3726–3748. doi:10.1002/1521-3773(20011015)40:20<3726::AID-ANIE3726>3.0.CO;2-D |
| 27. | Dalko, P. I.; Moisan, L. Angew. Chem., Int. Ed. 2004, 43, 5138–5175. doi:10.1002/anie.200400650 |
| 28. | Bertelsen, S.; Jørgensen, K. A. Chem. Soc. Rev. 2009, 38, 2178–2189. doi:10.1039/B903816G |
| 29. | Enders, D.; Grondal, C.; Hüttl, M. R. S. Angew. Chem., Int. Ed. 2007, 46, 1570–1581. doi:10.1002/anie.200603129 |
| 30. | Scheffler, U.; Mahrwald, R. Chem. – Eur. J. 2013, 19, 14346–14396. doi:10.1002/chem.201301996 |
| 31. | Dondoni, A.; Massi, A. Angew. Chem., Int. Ed. 2008, 47, 4638–4660. doi:10.1002/anie.200704684 |
| 58. | Zhang, Y.; Zhao, L.; Lee, S. S.; Ying, J. Y. Adv. Synth. Catal. 2006, 348, 2027–2032. doi:10.1002/adsc.200600240 |
| 24. | Hajji, C.; Roller, S.; Beigi, M.; Liese, A.; Haag, R. Adv. Synth. Catal. 2006, 348, 1760–1771. doi:10.1002/adsc.200606168 |
| 53. | Paras, N. A.; MacMillan, D. W. C. J. Am. Chem. Soc. 2001, 123, 4370–4371. doi:10.1021/ja015717g |
| 41. | Hayashi, Y. Angew. Chem., Int. Ed. 2006, 45, 8103–8104. doi:10.1002/anie.200603378 |
| 48. | Goren, K.; Karabline-Kuks, J.; Shiloni, Y.; Barak-Kulbak, E.; Miller, S. J.; Portnoy, M. Chem. – Eur. J. 2015, 21, 1191–1197. doi:10.1002/chem.201404560 |
| 49. | Sunder, A.; Mülhaupt, R.; Haag, R.; Frey, H. Adv. Mater. 2000, 12, 235–239. doi:10.1002/(SICI)1521-4095(200002)12:3<235::AID-ADMA235>3.0.CO;2-Y |
| 44. | Bellis, E.; Kokotos, G. J. Mol. Catal. A: Chem. 2005, 241, 166–174. doi:10.1016/j.molcata.2005.05.047 |
| 45. | Kehat, T.; Portnoy, M. Chem. Commun. 2007, 2823–2825. doi:10.1039/B703016A |
| 46. | Mitsui, K.; Hyatt, S. A.; Turner, D. A.; Hadad, C. M.; Parquette, J. R. Chem. Commun. 2009, 3261–3263. doi:10.1039/B902960E |
| 24. | Hajji, C.; Roller, S.; Beigi, M.; Liese, A.; Haag, R. Adv. Synth. Catal. 2006, 348, 1760–1771. doi:10.1002/adsc.200606168 |
| 47. | Goren, K.; Portnoy, M. Chem. Commun. 2010, 46, 1965–1967. doi:10.1039/B915577E |
| 38. | Gruttadauria, M.; Giacalone, F.; Noto, R. Adv. Synth. Catal. 2009, 351, 33–57. doi:10.1002/adsc.200800731 |
| 39. | Hernàndez, J. G.; Juaristi, E. Chem. Commun. 2012, 48, 5396–5409. doi:10.1039/C2CC30951C |
| 40. | Mase, N.; Barbas, C. F., III. Org. Biomol. Chem. 2010, 8, 4043–4050. doi:10.1039/C004970K |
| 41. | Hayashi, Y. Angew. Chem., Int. Ed. 2006, 45, 8103–8104. doi:10.1002/anie.200603378 |
| 42. | Mlynarski, J.; Paradowska, J. Chem. Soc. Rev. 2008, 37, 1502–1511. doi:10.1039/B710577K |
| 53. | Paras, N. A.; MacMillan, D. W. C. J. Am. Chem. Soc. 2001, 123, 4370–4371. doi:10.1021/ja015717g |
| 43. | Font, D.; Sayalero, S.; Bastero, A.; Jimeno, C.; Pericàs, M. A. Org. Lett. 2008, 10, 337–340. doi:10.1021/ol702901z |
| 62. | Riente, P.; Yadav, J.; Pericàs, M. A. Org. Lett. 2012, 14, 3668–3671. doi:10.1021/ol301515d |
| 32. | Kristensen, T. E.; Hansen, T. Eur. J. Org. Chem. 2010, 3179–3204. doi:10.1002/ejoc.201000319 |
| 33. | Cozzi, F. Adv. Synth. Catal. 2006, 348, 1367–1390. doi:10.1002/adsc.200606096 |
| 34. | Benaglia, M.; Puglisi, A.; Cozzi, F. Chem. Rev. 2003, 103, 3401–3430. doi:10.1021/cr010440o |
| 35. | Trindade, A. F.; Gois, P. M. P.; Afonso, C. A. M. Chem. Rev. 2009, 109, 418–514. doi:10.1021/cr800200t |
| 36. | Gruttadauria, M.; Giacalone, F.; Noto, R. Chem. Soc. Rev. 2008, 37, 1666–1688. doi:10.1039/B800704G |
| 58. | Zhang, Y.; Zhao, L.; Lee, S. S.; Ying, J. Y. Adv. Synth. Catal. 2006, 348, 2027–2032. doi:10.1002/adsc.200600240 |
| 37. | Wu, Y.; Zhang, Y.; Yu, M.; Zhao, G.; Wang, S. Org. Lett. 2006, 8, 4417–4420. doi:10.1021/ol061418q |
| 53. | Paras, N. A.; MacMillan, D. W. C. J. Am. Chem. Soc. 2001, 123, 4370–4371. doi:10.1021/ja015717g |
© 2015 Pecchioli et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)
