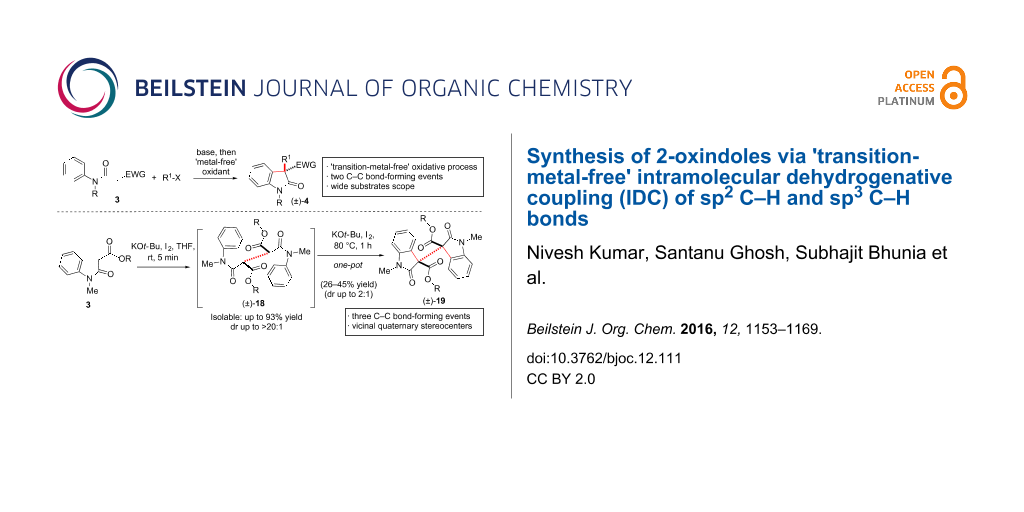
Abstract
The synthesis of a variety of 2-oxindoles bearing an all-carbon quaternary center at the pseudo benzylic position has been achieved via a ‘transition-metal-free’ intramolecular dehydrogenative coupling (IDC). The construction of 2-oxindole moieties was carried out through formation of carbon–carbon bonds using KOt-Bu-catalyzed one pot C-alkylation of β-N-arylamido esters with alkyl halides followed by a dehydrogenative coupling. Experimental evidences indicated toward a radical-mediated path for this reaction.

Graphical Abstract
Introduction
The C−H functionalization is an attractive synthetic strategy used in organic synthesis for the development of atom- and step-economical routes [1-10]. In recent years it was witnessed a mushrooming growth in the number of reports in the literature owing to the efficiency of the oxidative coupling of two C–H bonds [also termed as cross-dehydrogenative-coupling (CDC)] in the formation of C–C bonds [11-16]. This was facilitated by the introduction of transition metals in organic synthesis providing an amazing tool to explore these oxidative coupling reactions in an efficient manner. However, despite the associated advantages, these methodologies require one or two metal catalysts for efficient reactions, which are sometimes undesirable [17-21]. Therefore, an alternate strategy to carry out these transformations under 'transition-metal-free' conditions has recently gained immense importance.
2-Oxindoles having all carbon quaternary centres at the pseudobenzylic position are common structural scaffolds in many naturally occurring alkaloids of biological relevance [22-25]. These heterocyclic motifs especially exist in indole alkaloids with a wide spectrum of biological and pharmacological properties and hence are very attractive as well as challenging synthetic targets [26]. Selected examples for the synthesis of 2-oxindole include an intramolecular homolytic aromatic substitution on the aryl ring by an amidyl radical formed by homolysis of a C–X bond [27-30], single electron transfer (SET) to a α-halo anilides followed by halide elimination [31,32], and the formation of an aryl radical followed by a 1,5-hydrogen atom translocation [33,34]. Out of these strategies, the initial two require specifically functionalized precursors such as the presence of an o-halogen, an o-selenium, or an o-xanthate, respectively. One of the direct approaches to 2-oxindoles could be a one-electron oxidation of an amide enolate as shown in Scheme 1. Toward this end, in 2009, Kündig and co-workers have developed a novel route to 3,3-disubstituted-2-oxindoles while working on asymmetric synthesis of 3,3-disubstituted-2-oxindoles via a Pd-catalyzed (chiral N-heterocyclic carbene as ligands) intramolecular α-arylation of an amide [35-37]. For this 'intramolecular dehydrogenative coupling' (IDC) of Csp2-H and Csp3-H they used 2.2 equiv of CuCl2 and 5 equiv of NaOt-Bu [38,39].
![[1860-5397-12-111-i1]](/bjoc/content/inline/1860-5397-12-111-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 2-oxindoles via oxidative processes.
Scheme 1: Synthesis of 2-oxindoles via oxidative processes.
In the same year, Taylor and co-workers independently reported synthesis of 2-oxindoles in the presence of Cu(OAc)2·H2O as oxidant (Scheme 1) [40-44]. Experimental evidence suggests involvement of a free-radical process in the addition of α-carbonylalkyl radicals to the phenyl ring. The α-carbonylalkyl radicals were formed by Cu(II)-mediated oxidation of the respective enolate precursors. In 2010, Yu and co-workers have reported the synthesis of 3-acetyloxindoles via Ag2O-mediated intramolecular oxidative coupling [45]. For the past few years, our group is engaged in the development of efficient methodologies for the synthesis of 2-oxindoles with intriguing ring systems. To this end, recently, we have reported a transition-metal-free ‘intramolecular-dehydrogenative-coupling' (IDC) strategy to access such 2-oxindole moieties through a C-alkylation followed by an oxidative construction of the C–C bond (Scheme 1) [46]. Applying the aforementioned strategy, we were able to synthesize several 3-alkyl-2-oxindoles bearing ester functionalities at the pseudobenzylic position from β-N-arylamido allyl, methallyl, dimethylallyl, and geranyl esters. Here, in this article, we disclose the scope and limitations of 'transition-metal-free' IDC of Csp2-H and Csp3-H using iodine and N-iodosuccinimide (NIS) as oxidants. In addition, we have also demonstrated the synthetic utility of oxidative coupling products in the syntheses of 3-substituted-2-oxindoles, via a decarboxylative protonation on 2-oxindoles bearing an benzylester or para-methoxybenzyl ester at the 3-position in presence of a catalytic amount of Pd on activated charcoal. We have also shown the direct installation of allyl, prenyl, reverse-prenyl, or geranyl groups at the 3-position of 2-oxindole using Pd-catalyzed decarboxylative strategies [47].
Results and Discussion
We decided to use iodine as an oxidant for the synthesis of 2-oxindoles [48-53], starting from β-N-arylamido ester 3a and methyl iodide as the substrates (Table 1). An elaborate optimization study suggested that the methylation can be done in the presence of 1.2 equivalents of KOt-Bu and 1.1 equivalents of methyl iodide. This was accompanied with an oxidative coupling using 1.2 equivalents of KOt-Bu and iodine to afford the desired product in 65% yield (Table 1, entries 1 and 2). Optimization studies in search of suitable solvent, potential bases, oxidants etc. yielded the desired product in good yields i.e. 85%, 88%, and 90% in THF, dioxane, and DMSO, respectively (Table 1, entries 3, 5, and 8). However, in non-polar aromatic solvents like xylene, benzene, and toluene, poor yields (43–49%, Table 1, entries 4, 6, and 7) of products were observed with reactions being unclean (mixture of products) [54]. KOt-Bu was superior over other bases used in this reaction like NaH, NaOMe, K2CO3, Cs2CO3, and NaOt-Bu (Table 1, entries 9–13). Among other metal-free oxidants, iodosobenzenediacetate (PIDA), DBDMH (1,3-dibromo-5,5-dimethylhydantoin), and ICl afforded 2-oxindole 4a in 82%, 16%, and 69%, respectively (Table 1, entries 17–19). Later, we turned our attention to N-halo succinimides as potential oxidants in our methodology [53]. Interestingly, N-iodosuccinimide (NIS), N-bromosuccinimide (NBS), and N-chlorosuccinimide (NCS) afforded 4a in 84%, 75%, and 58% yields, respectively (Table 1, entries 20–22). However, trichloroisocyanuric acid (TCICA) is found to be inefficient in oxidative coupling and afforded 56–62% yields of 4a (Table 1, entries 23 and 24). In the absence of iodine or NIS, no product was formed. Eventually, the combination of 1.2 equivalents of iodine (conditions A) or NIS (conditions B) were found to be the best and chosen for further studies (Table 1, entries 14 and 20).
Table 1: Optimization of intramolecular-dehydrogenative-coupling (IDC)a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-111-i12.svg?max-width=637&scale=1.0)
|
||||||
| entry | solvent | base | Alkylations at 25 °C | oxidants | time | % 4ab,c |
|---|---|---|---|---|---|---|
| 1. | DMF | t-BuOK | 20 min | 1.5 equiv I2 | 6 h | 65% |
| 2. | DMF | t-BuOK | 20 min | 1.2 equiv I2 | 3 h | 62% |
| 3. | THF | t-BuOK | 30 min | 1.2 equiv I2 | 3 h | 85% |
| 4. | xylene | t-BuOK | 45 min | 1.2 equiv I2 | 1 h | 49%d |
| 5. | dioxane | t-BuOK | 20 min | 1.2 equiv I2 | 2 h | 88% |
| 6. | benzene | t-BuOK | 50 min | 1.2 equiv I2 | 2 h | 43%d |
| 7. | toluene | t-BuOK | 45 min | 1.2 equiv I2 | 1 h | 45%d |
| 8. | DMSO | t-BuOK | 20 min | 1.2 equiv I2 | 30 min | 90% |
| 9. | DMSO | NaH | 20 min | 1.2 equiv I2 | 30 min | 33% |
| 10. | DMSO | NaOMe | 2 h | 1.2 equiv I2 | 30 min | –e |
| 11. | DMSO | K2CO3 | 1 hf | – | – | – |
| 12. | DMSO | CS2CO3 | 2 h | 1.5 equiv I2 | 30 min | 26%g |
| 13. | DMSO | t-BuONa | 30 min | 1.5 equiv I2 | 30 min | –g |
| 14. | DMSO | t-BuOK | 15 min | 1.2 equiv I2 | 30 min | 90% |
| 15. | DMSO | t-BuOK | 15 min | 0.6 equiv I2 | 1 h | 54% |
| 16. | DMSO | t-BuOK | 15 min | 0.3 equiv I2 | 1 h | 29% |
| 17. | DMSO | t-BuOK | 15 min | 1.2 equiv PIDA | 30 min | 82% |
| 18. | DMSO | t-BuOK | 15 min | 1.2 equiv DBDMHh | 30 min | 16%g |
| 19. | DMSO | t-BuOK | 15 min | 1.2 equivICl | 30 min | 69% |
| 20. | DMSO | t-BuOK | 15 min | 1.2 equiv NIS | 30 min | 84% |
| 21. | DMSO | t-BuOK | 15 min | 1.2 equiv NBS | 30 min | 75% |
| 22. | DMSO | t-BuOK | 15 min | 1.2 equiv NCS | 30 min | 58% |
| 23. | DMSO | t-BuOK | 15 min | 1.0 equivTCICAi | 30 min | 62% |
| 24. | DMSO | t-BuOK | 15 min | 0.5 equivTCICAi | 30 min | 56% |
a Entries 1–18 have been reproduced from our preliminary communication (reference [46]. bReactions were carried out on a 0.25 mmol of 3a with 0.275 mmol of methyl iodide in presence of 0.30 mmol of base in 1 mL of solvent at 25 °C for specified time for alkylations and 0.275 mmol of oxidant in presence of 0.30 mmol of base under heating at 110 °C for oxidative coupling steps, unless noted otherwise. cIsolated yields of 4a after column chromatography. dMixture of products were observed for rest of the mass balance. eC-methylation as major product. fStarting material was recovered (92%). gDecomposition of starting materials. hDBDMH (1,3-dibromo-5,5-dimethylhydantoin) as oxidant. iTCICA (trichloroisocyanuric acid).
Next, the substrate scope of the reaction was explored as shown in Figure 1. A variety of substrates were prepared by a coupling reaction of N-methyl arylamines and monoalkyl malonates/cyanoacetic acids. Under optimized conditions A and B, various β-N-arylamido esters and nitriles (3) were subjected to a one-pot alkylations using 1.2 equivalents of KOt-Bu to produce C-alkylated intermediate 5 followed by oxidative coupling using 1.2 equivalents iodine or NIS. Gratifyingly, it was found that a range of β-N-arylamido esters (3a–s) and β-N-arylamido ketones (3t–x) underwent intramolecular dehydrogenative coupling (IDC) under both conditions A and B to afford a wide range of 2-oxindoles (4a–x) having an all-carbon quaternary center in high yields. However, we observed that in case of 2-oxindoles 4v and 4k, a two-step protocol is necessary, where in first step C-alkylation of β-N-arylamido ketone was carried out using 1.2 equivalents of KOt-Bu to afford products 5v and 5k, respectively (Figure 1), followed by a second oxidative coupling reaction in the presence of iodine or NIS.
![[1860-5397-12-111-1]](/bjoc/content/figures/1860-5397-12-111-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Substrates scope of one-pot ‘transition-metal-free’ IDC. The syntheses of compounds 4a–s according to method A have been reproduced from reference [46]. Conditions A: KOt-Bu, iodine; conditions B: KOt-Bu, NIS.
Figure 1: Substrates scope of one-pot ‘transition-metal-free’ IDC. The syntheses of compounds 4a–s according ...
We envisioned that the oxidative coupling products containing benzyl or p-methoxybenzyl ester could be effective intermediates for the synthesis of 3-monosubstituted 2-oxindoles via deprotection of the benzyl group followed by decarboxylative protonation in presence of a catalytic amount of Pd on activated charcoal under hydrogenolysis. Thus, we explored the substrate scope using β-N-arylamido benzyl ester or β-N-arylamido p-methoxybenzyl ester as starting materials for the oxidative coupling reaction shown in Figure 2. Towards this end, β-N-aryl amido benzylester or β-N-arylamido p-methoxybenzyl ester 3 were subjected to an one pot alkylation to generate the intermediate 7 followed by oxidative coupling reaction using our optimized conditions A and B to furnish products of type (±)-6 in good yields (Figure 2). For the synthesis of compound (±)-6g, we followed a two-step protocol: In first step a C-alkylation of β-N-arylamido benzylester in presence of 1.2 equivalents of NaH and alkylating agent afford compound (±)-7g in good yields (74%), followed by an oxidative coupling in presence of 1.2 equivalents of KOt-Bu and iodine or NIS as oxidant.
![[1860-5397-12-111-2]](/bjoc/content/figures/1860-5397-12-111-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Further substrates scope of one-pot ‘transition metal-free’ IDC. Conditions A: KOt-Bu, iodine; conditions B: KOt-Bu, NIS.
Figure 2: Further substrates scope of one-pot ‘transition metal-free’ IDC. Conditions A: KOt-Bu, iodine; cond...
Next, we focussed our attention to prenylated, reverse-prenylated, and geranylated hexahydropyrrolo[2,3-b]indole alkaloids showing broad biological activities [55-61]. For the synthesis of these compounds, we thought of utilizing the Pd-catalyzed decarboxylative strategy to install the prenyl, reverse-prenyl, or geranyl group at the 3-position of 2-oxindole starting from the corresponding β-amido esters such as 8 [47]. This further extended the methodology to a variety of β-N-arylamido esters containing allyl, methallyl, dimethylallyl, and geranyl ester groups (9). It is noteworthy that, the substrate of type 9 could undergo smooth IDC in the presence of iodine (conditions A) to provide an access to compounds 8 in synthetically useful yields (Figure 3).
![[1860-5397-12-111-3]](/bjoc/content/figures/1860-5397-12-111-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Substrates scope of ‘transition-metal-free’ IDC using KOt-Bu/I2. Reproduced from [46].
Figure 3: Substrates scope of ‘transition-metal-free’ IDC using KOt-Bu/I2. Reproduced from [46].
Noticeably, we could directly construct the 2-oxindoles with a geranyl group at the 3-position using geranyl bromide as an alkylating agent. Upon a subsequent oxidative coupling step, products in good yields (8q–s, Figure 3) were formed using conditions A. Later, the IDC was extended to substrates having β-N-arylamido geranyl esters to afford compounds 8t–v (Figure 3). These compounds could be excellent substrates for carrying out Tsuji–Trost decarboxylative geranylations/reverse-geranylations [62,63]. However, conditions B (NIS) were found unsuccessful in case of β-N-arylamidoallyl, methallyl, dimethylallyl, and geranyl esters 9. We speculate that the olefin functionality of substrates might be reacting with NIS (conditions B) faster than iodine (conditions A). Although our iodine-mediated IDC is successful in most of the cases, however, in few cases we have seen moderate yields of products. Thus, we decided to carry out IDC in the presence of organic bases as well.
Thus, for an alternative approach to 2-oxindoles bearing allyl, methallyl, dimethylallyl, and geranyl esters, we were interested for IDC using simple organic bases such as triethylamine, pyridine, and DABCO (Table 2) [64]. It was found that IDC can operate in the presence of organic bases to afford products only in 25–34% yields of 2-oxindoles (Table 2, entries 1, 2 and 4). These reactions were always associated with unreacted starting material (28–51%) and decomposition of the rest of the mass balance. Interestingly, when the base was changed to DBU (using 1.5 equiv DBU and 1.2 equiv of iodine) the desired 2-oxindole was isolated in 82% (conditions C).
Table 2: IDC in the presence of organic bases. Reproduced from [46].
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-111-i13.svg?max-width=637&scale=1.0)
|
||||
| entry | base | time | % of 8j | % of 10j |
|---|---|---|---|---|
| 1. | pyridine | 12 h | 29 | 30 |
| 2. | Et3N | 12 h | 25 | 28 |
| 3. | DBU | 40 min | 82 | – |
| 4. | DABCO | 12 h | 34 | 51 |
aReactions were carried out on a 0.25 mmol of 10j using 0.50 mmol of base and 0.275 mmol of iodine in 1 mL of solvent for specified time.
With this result in hand, we thought of exploring IDC using C-alkylated substrates 10. For this purpose, a variety of C-alkylated β-N-arylamidoallyl, methallyl, dimethylallyl, and geranyl esters 10 were synthesized in good yields as per Figure 4. These substrates were then utilized in IDC-promoted by DBU/I2 and the results are summarized in Figure 5. Interestingly, under this conditions, we can synthesize a variety of 2-oxindoles 8 in moderate to good yields.
![[1860-5397-12-111-4]](/bjoc/content/figures/1860-5397-12-111-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: C-Alkylation of anilides using KOt-Bu.
Figure 4: C-Alkylation of anilides using KOt-Bu.
![[1860-5397-12-111-5]](/bjoc/content/figures/1860-5397-12-111-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Substrates scope of ‘transition-metal-free’ IDC of C-alkylated anilides using DBU/I2.
Figure 5: Substrates scope of ‘transition-metal-free’ IDC of C-alkylated anilides using DBU/I2.
There are a large number of indole alkaloids bearing a 3-arylated-2-oxindole moiety that are known for their various biological activities [65-67]. In a quest for such structural scaffolds, C-arylated substrates (±)-11a–d were subjected to standard reaction conditions to afford compound 12a–d (Scheme 2). To our pleasure, C-arylated β-N-arylamidoesters (±)-11a–d afforded products (±)-12a–d in 59–89% yield after 1 h under conditions A and B.
![[1860-5397-12-111-i2]](/bjoc/content/inline/1860-5397-12-111-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Oxidative coupling of C-arylated anilides (±)-11a–d. The synthesis of 12b as per method A has been reproduced from reference [46].
Scheme 2: Oxidative coupling of C-arylated anilides (±)-11a–d. The synthesis of 12b as per method A has been ...
Our synthetic methodology was further explored in the construction of spiro-fused oxindole ring systems (Scheme 3). The spiro-fused oxindoles such as coerulescine (15a) [68-72], horsfiline (15b) [73], and elacomine (16), are prevalently found in a huge number of indole-based alkaloids having analgesic properties. Our oxidative methodology offered us a direct access to the core structures of these alkaloids under the optimized IDC conditions in high yields (Scheme 3).
![[1860-5397-12-111-i3]](/bjoc/content/inline/1860-5397-12-111-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of spirocyclic product through IDC The synthesis of 14 as per method A has been reproduced from reference [46]. Conditions A: KOt-Bu, iodine; conditions B: KOt-Bu, NIS.
Scheme 3: Synthesis of spirocyclic product through IDC The synthesis of 14 as per method A has been reproduce...
Next, we thought of carrying out the IDC without alkylations of compounds 3a and b and 17a and b (Scheme 4). Unfortunately, we could not isolate products due to decomposition under optimized IDC conditions. It was noticed that changing the solvent to THF effected very fast (within 5 minutes) dimerization of 3a and b at room temperature to afford 18a and b as sole products in 91–93% yield and in up to >20:1 dr (Scheme 4). This shows that formation of a stabilized tertiary radical probably facilitates the IDC process for the syntheses of 2-oxindoles.
![[1860-5397-12-111-i4]](/bjoc/content/inline/1860-5397-12-111-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Dimerization of β-N-aryl-amidoesters 3a and b. Reproduced from [46].
Scheme 4: Dimerization of β-N-aryl-amidoesters 3a and b. Reproduced from [46].
However, if a tertiary radical is responsible for the oxidative process, then one would realize the formation of dimeric 2-oxindoles sharing vicinal all-carbon quaternary centers from dimeric β-N-arylamidoesters 18a and b (Scheme 4). The reason behind our interest towards this direction was due to the prevalence of various dimeric cyclotryptamine alkaloids containing 3a,3a'-bis-pyrrolo[2,3-b]indole subunits (core structure of alkaloids 22a and b, see, Scheme 5) [74-77], sharing a vicinal all-carbon quaternary stereogenic centers with extreme steric congestion at the C3a–C3a' σ-bond as well as the attendant lability of this linkage. Under the optimized conditions, one-pot dimerization of β-N-arylamido ester 3a and b and 9a took place in the presence of 1.2 equivalents of KOt-Bu and I2 followed by a double IDC on treatment with 1.2 equivalents of KOt-Bu and I2 affording the dimeric 2-oxindoles (±)-19a–c in poor to moderate yields (26–45% yield and 2:1 dr) along with 15–18% isolation of dimeric β-N-arylamidoesters (±)-18a–c (Scheme 5). This transformation is an efficient one-pot formation of three consecutive carbon–carbon bonds. X-ray crystal structure determination of (±)-19b proved the outcome of the reaction unambiguously. It was noteworthy to observe Pd-catalyzed highly enantio-, chemo-, and diastereoselective double decarboxylative allylations on dimeric β-N-arylamido allyl ester 19c to yield the enantiopure compounds of type 20a and b in good yields [78-81]. Especially, enantioenriched 20b is the advanced intermediate for the total syntheses of 3a,3a'-bispyrrolo[2,3-b]indole alkaloids, chimonanthine (22a), folicanthine (22b), and their rearranged skeleton such as calycanthine (22c) (Scheme 5) [78].
![[1860-5397-12-111-i5]](/bjoc/content/inline/1860-5397-12-111-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of dimeric 2-oxindoles utilizing IDC. The syntheses of 19a and b have been reproduced from [46].
Scheme 5: Synthesis of dimeric 2-oxindoles utilizing IDC. The syntheses of 19a and b have been reproduced fro...
In all the cases, IDC was feasible with substrates having substituents at the carbon atom α- to the amides. This gave a clue for a radical-mediated process where a single electron transfer (SET) mechanism might be operating. A tentative mechanism has been proposed in Scheme 6, the reaction can adopt a SET mechanism leading to the intermediate 23a, after C-alkylation. Compound 23a in turn gets converted into intermediate aryl radical 23b. From this intermediate another intermediate aryl carbocation 23c is formed by transferring a single electron to the oxidant. Carbocation 23c is stabilized by the amide nitrogen as shown in 23d. Eventually, in the presence of base, rearomatization of 23d takes place to afford the final product of the oxidative coupling reaction.
![[1860-5397-12-111-i6]](/bjoc/content/inline/1860-5397-12-111-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Plausible mechanism of ‘transition-metal-free’ IDC The mechanistic consideration in Scheme 6 has been reproduced from [46].
Scheme 6: Plausible mechanism of ‘transition-metal-free’ IDC The mechanistic consideration in Scheme 6 has been repro...
Kündig et al. in their oxidative coupling process using 2.2 equivalent of CuCl2 showed that it is important to have a tertiary carbon α- to the amide for the process to be radical mediated [38,39]. Also, it is well evident from literature that the oxidation processes using Mn(OAc)3 as oxidant follow a radical pathway [82-86]. In fact, the reaction of 3a also afforded 2-oxindole 4a in 69% yield when the oxidative coupling was carried out in presence of 1.2 equiv of Mn(OAc)3 (Scheme 7). A similar result was also observed when reaction was carried out using C-methyl β-N-arylamido ester 5a (Scheme 7) [82-86].
![[1860-5397-12-111-i7]](/bjoc/content/inline/1860-5397-12-111-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Intramolecular-dehydrogenative-coupling (IDC) of 3a and 5a. Reproduced from [46].
Scheme 7: Intramolecular-dehydrogenative-coupling (IDC) of 3a and 5a. Reproduced from [46].
However, one can’t rule out the possibility of a substitution reaction on C-iodo product 24 from the adjacent aryl group (Scheme 8). Thus, the possibilty of the addition at the 2-position of electron-rich N-acylated aniline 3a to the tertiary iodide intermediate was also investigated. Towards this, we thought of synthesizing the C-iodo intermediate using N-iodosuccinimide (NIS) or ICl in the presence of a base. Surprisingly, our all effort to prepare C-iodo compound 24 in the presence of KOt-Bu as a base only led to formation of 2-oxindole 4a in 72% and 69% yields, respectively (Scheme 8). Along the same line, C-methyl β-N-arylamido ester 5a also afforded product 4a in 80–85% yield when the reaction was carried out at elevated temperature (Scheme 8). We thought there could be the possibility of a substitution reaction of iodide compound 24 prepared in-situ to form directly 2-oxindole 4a under elevated temperature. Thus, it was decided to carry out the C-iodination at room temperature, where substitution reactions would be unlikely, considering the fact that the substitution has to occur at the sterically congested tertiary iodide 24. However, to our surprise, when C-iodination of 5a was carried out at rt, we found that it also afforded 2-oxindole 4a in 30–39% yield along with 43–52% of recovered starting material (Scheme 8) and no trace of C-iodide 24 was observed. These results suggest that, NIS and ICl also acts as oxidants and helping in a single electron transfer (SET) in the oxidative coupling reaction [87,88]. It is also well evident in the literature that, these can also be used as oxidants in variety of oxidative coupling reactions [87,88]. Further, oxidative coupling of 5a was carried out in presence of well-known t-BuOI, which generally goes through a radical-mediated pathway [89,90]. Towards this, when the oxidative coupling was carried out in presence of in situ generated t-BuOI [91], the reaction afforded oxidative coupling product (±)-4a in 68% yield, which is also probably indicating a radical pathway of the reaction (Scheme 8).
![[1860-5397-12-111-i8]](/bjoc/content/inline/1860-5397-12-111-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: IDC of 3a and 5a using different oxidants. Reproduced from [46].
Scheme 8: IDC of 3a and 5a using different oxidants. Reproduced from [46].
Shifting our attention towards the synthetic application of our IDC methodology, we put forward our effort towards the synthesis of 3-alkylated or arylated 2-oxindoles. Towards this, we subjected to react, the oxidative coupling products (±)-6, (±)-12c and d having benzyl (Bn) or p-methoxybenzyl (PMB) esters with a catalytic amount of Pd on activated charcoal (10% Pd on charcoal) under atmospheric pressure of hydrogen gas in MeOH/EtOH (Scheme 9).
![[1860-5397-12-111-i9]](/bjoc/content/inline/1860-5397-12-111-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Synthesis of 3-substituted-2-oxindoles from benzyl esters.
Scheme 9: Synthesis of 3-substituted-2-oxindoles from benzyl esters.
Interestingly, we observed that the oxidative coupling products undergo deprotection of benzyl or p-methoxybenzyl group and provided the intermediate carboxylic acid, followed by decarboxylative protonation in the same pot gave us the desired products (±)-25a–h in excellent yields (Scheme 9 and Scheme 10).
![[1860-5397-12-111-i10]](/bjoc/content/inline/1860-5397-12-111-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: 3-Substituted-2-oxindoles from p-methoxybenzyl esters.
Scheme 10: 3-Substituted-2-oxindoles from p-methoxybenzyl esters.
Later, we envisioned that the oxidative coupling products having allyl, methallyl, dimethylallyl esters after Trost–Tsuji decarboxylative allylations could serve as an interesting platform for complex natural product synthesis after further synthetic elaboration and functionalization. A few substrates were treated under decarboxylative allylation (DcA) conditions in the presence of 10 mol % of Pd(PPh3)4 in refluxing tetrahydrofuran (7–8 h), which afforded products 26a–d in up to 99% yield (Scheme 11). Interestingly, oxidative coupling products with dimethylallyl esters 8j underwent smooth decarboxylative prenylation and reverse-prenylation in the presence of 5 mol % of Pd2(dba)3 and 15 mol % dppp in refluxing toluene (7–8 h, 96% yields) to afford prenylated (27a) and reverse-prenylated (27b) structures in 64% and 32% yield, respectively (Scheme 11) [92,93]. These structures commonly occur in many hexahydropyrrolo[2,3-b]indole-based alkaloids.
![[1860-5397-12-111-i11]](/bjoc/content/inline/1860-5397-12-111-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Synthetic elaboration using Tsuji–Trost reactions. Reproduced from [46].
Scheme 11: Synthetic elaboration using Tsuji–Trost reactions. Reproduced from [46].
Conclusion
In summary, we have successfully demonstrated the synthesis of 2-oxindoles bearing an all-carbon quaternary center applying a ‘transition-metal-free’ intramolecular dehydrogenative coupling (IDC) strategy. The methodology has been broadly applied to a wide range of substrates affording 2-oxindoles in good yields in a facile one-pot C-alkylation concomitant with oxidative coupling strategy. These products serve as a great synthetic platform for several indole-based natural products. The methodology demonstrated here has several advantages: (i) C-alkylations can be carried in same pot; (ii) simple oxidants like iodine and N-iodosuccinimide (NIS) could be used in the absence of any transition metal which may be toxic and (iii) substrates with a scope of further functionalization work equally well. The easy handling and the low cost of the reagents involved in this synthetic methodology offers profound opportunities to expand and explore the use of IDC in organic synthesis. Further applications of this strategy are under active investigation in our laboratory.
Supporting Information
| Supporting Information File 1: Copies of 1H, and13C NMR spectra for all new compounds. | ||
| Format: PDF | Size: 9.2 MB | Download |
Acknowledgements
Financial support from the Department of Science and Technology (DST) through FAST-TRACK scheme (SB/FT/CS-54/2011) and the Council of Scientific and Industrial Research (CSIR) [02(0013)/11/EMR-II], Govt. of India is gratefully acknowledged. N.K., S.G., and S.B. thank the CSIR, New Delhi, for predoctoral fellowships. We sincerely thank Dr. Subahdip De, Dr. Badrinath N. Kakde, and Dr. Amit Adhikary for preliminary studies. Facilities from the Department of Chemistry, IISER Bhopal is gratefully acknowledged.
References
-
Dyker, G., Ed. Handbook of C-H Transformations: Applications in Organic Synthesis; 2005; Vol. 2.
Return to citation in text: [1] -
Stuart, D. R.; Fagnou, K. Science 2007, 316, 1172. doi:10.1126/science.1141956
Return to citation in text: [1] -
Ashenhurst, J. A. Chem. Soc. Rev. 2010, 39, 540. doi:10.1039/B907809F
Return to citation in text: [1] -
Le Bras, J.; Muzart, J. Chem. Rev. 2011, 111, 1170. doi:10.1021/cr100209d
Return to citation in text: [1] -
Rossi, R.; Bellina, F.; Lessi, M. Synthesis 2010, 4131. doi:10.1055/s-0030-1258262
Return to citation in text: [1] -
Scheuermann, C. J. Chem. – Asian J. 2010, 5, 436. doi:10.1002/asia.200900487
Return to citation in text: [1] -
You, S.-l.; Xia, J.-B. Top. Curr. Chem. 2010, 292, 165. doi:10.1007/128_2009_18
Return to citation in text: [1] -
Newhouse, T.; Baran, P. S.; Hoffmann, R. W. Chem. Soc. Rev. 2009, 38, 3010. doi:10.1039/b821200g
Return to citation in text: [1] -
Gaich, T.; Baran, P. S. J. Org. Chem. 2010, 75, 4657. doi:10.1021/jo1006812
Return to citation in text: [1] -
Hendrickson, J. B. J. Am. Chem. Soc. 1975, 97, 5784. doi:10.1021/ja00853a023
Return to citation in text: [1] -
Cho, S. H.; Kim, J. Y.; Kwak, J.; Chang, S. Chem. Soc. Rev. 2011, 40, 5068. doi:10.1039/c1cs15082k
Return to citation in text: [1] -
Yeung, C. S.; Dong, V. M. Chem. Rev. 2011, 111, 1215. doi:10.1021/cr100280d
Return to citation in text: [1] -
McGlacken, G. P.; Bateman, L. M. Chem. Soc. Rev. 2009, 38, 2447. doi:10.1039/b805701j
Return to citation in text: [1] -
Liu, C.; Zhang, H.; Shi, W.; Lei, A. Chem. Rev. 2011, 111, 1780. doi:10.1021/cr100379j
Return to citation in text: [1] -
Siemsen, P.; Livingston, R. C.; Diederich, F. Angew. Chem., Int. Ed. 2000, 39, 2632. doi:10.1002/1521-3773(20000804)39:15<2632::AID-ANIE2632>3.0.CO;2-F
Return to citation in text: [1] -
Li, C.-J. Acc. Chem. Res. 2009, 42, 335. doi:10.1021/ar800164n
Return to citation in text: [1] -
Li, Z.; Bohle, D. S.; Li, C.-J. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 8928. doi:10.1073/pnas.0601687103
Return to citation in text: [1] -
Li, C.-J.; Li, Z. Pure Appl. Chem. 2006, 78, 935. doi:10.1351/pac200678050935
Return to citation in text: [1] -
Yoo, W.-J.; Li, C.-J. Top. Curr. Chem. 2010, 292, 281. doi:10.1007/128_2009_17
Return to citation in text: [1] -
Zhang, Y.; Li, C.-J. J. Am. Chem. Soc. 2006, 128, 4242. doi:10.1021/ja060050p
See for metal-free CDC.
Return to citation in text: [1] -
Uyanik, M.; Ishihara, K. ChemCatChem 2012, 4, 177. doi:10.1002/cctc.201100352
Return to citation in text: [1] -
Marti, C.; Carreira, E. M. Eur. J. Org. Chem. 2003, 2209. doi:10.1002/ejoc.200300050
Return to citation in text: [1] -
Klein, J. E. M. N.; Taylor, R. J. K. Eur. J. Org. Chem. 2011, 6821. doi:10.1002/ejoc.201100836
Return to citation in text: [1] -
Roth, G. J.; Heckel, A.; Colbatzky, F.; Handschuh, S.; Kley, J.; Lehmann-Lintz, T.; Lotz, R.; Tontsch-Grunt, U.; Walter, R.; Hilberg, F. J. Med. Chem. 2009, 52, 4466. doi:10.1021/jm900431g
Return to citation in text: [1] -
Rudrangi, S. R. S.; Bontha, V. K.; Manda, V. R.; Bethi, S. Asian J. Res. Chem. 2011, 4, 335.
Return to citation in text: [1] -
Trost, B. M.; Brennan, M. K. Synthesis 2009, 3003. doi:10.1055/s-0029-1216975
Return to citation in text: [1] -
Nishio, T.; Iseki, K.; Araki, N.; Miyazaki, T. Helv. Chim. Acta 2005, 88, 35. doi:10.1002/hlca.200490294
Return to citation in text: [1] -
Axon, J.; Boiteau, L.; Boivin, J.; Forbes, J. E.; Zard, S. Z. Tetrahedron Lett. 1994, 35, 1719. doi:10.1016/0040-4039(94)88328-9
Return to citation in text: [1] -
Murphy, J. A.; Tripoli, R.; Khan, T. A.; Mail, U. W. Org. Lett. 2005, 7, 3287. doi:10.1021/ol051095i
Return to citation in text: [1] -
Teichert, A.; Jantos, K.; Harms, K.; Studer, A. Org. Lett. 2004, 6, 3477. doi:10.1021/ol048759t
Return to citation in text: [1] -
Zhu, J.; Zhang, W.; Zhang, L.; Liu, J.; Zheng, J.; Hu, J. J. Org. Chem. 2010, 75, 5505. doi:10.1021/jo1005262
Return to citation in text: [1] -
Ju, X.; Liang, Y.; Jia, P.; Li, W.; Yu, W. Org. Biomol. Chem. 2012, 10, 498. doi:10.1039/C1OB06652H
Return to citation in text: [1] -
Khan, T. A.; Tripoli, R.; Crawford, J. J.; Martin, C. G.; Murphy, J. A. Org. Lett. 2003, 5, 2971. doi:10.1021/ol035173i
Return to citation in text: [1] -
Beckwith, A. L. J.; Bowry, V. W.; Bowman, W. R.; Mann, E.; Parr, J.; Storey, J. M. D. Angew. Chem., Int. Ed. 2004, 43, 95. doi:10.1002/anie.200352419
Return to citation in text: [1] -
Kündig, E. P.; Seidel, T. M.; Jia, Y.-X.; Bernardinelli, G. Angew. Chem., Int. Ed. 2007, 46, 8484. doi:10.1002/anie.200703408
Return to citation in text: [1] -
Jia, Y.-X.; Hillgren, J. M.; Watson, E. L.; Marsden, S. P.; Kündig, E. P. Chem. Commun. 2008, 4040. doi:10.1039/b810858g
Return to citation in text: [1] -
Jia, Y.-X.; Katayev, D.; Bernardinelli, G.; Seidel, T. M.; Kündig, E. P. Chem. – Eur. J. 2010, 16, 6300. doi:10.1002/chem.201000031
Return to citation in text: [1] -
Jia, Y. X.; Kündig, E. P. Angew. Chem., Int. Ed. 2009, 48, 1636. doi:10.1002/anie.200805652
See for oxidative coupling using 2.2 equiv of CuCl2.
Return to citation in text: [1] [2] -
Dey, C.; Kündig, E. P. Chem. Commun. 2012, 3064. doi:10.1039/c2cc17871k
Return to citation in text: [1] [2] -
Perry, A.; Taylor, R. J. K. Chem. Commun. 2009, 3249. doi:10.1039/b903516h
See for oxidative coupling using 1.0 equiv of Cu(OAc)2·H2O.
Return to citation in text: [1] -
Pugh, D. S.; Klein, J. E. M. N.; Perry, A.; Taylor, R. J. K. Synlett 2010, 934. doi:10.1055/s-0029-1219392
Return to citation in text: [1] -
Klein, J. E. M. N.; Perry, A.; Pugh, D. S.; Taylor, R. J. K. Org. Lett. 2010, 12, 3446. doi:10.1021/ol1012668
See for coupling using 5–10 mol % of Cu(OAc)2·H2O.
Return to citation in text: [1] -
Moody, C. L.; Franckevičius, V.; Drouhin, P.; Klein, J. E. M. N.; Taylor, R. J. K. Tetrahedron Lett. 2012, 53, 1897. doi:10.1016/j.tetlet.2012.01.120
Return to citation in text: [1] -
Pugh, D. S.; Taylor, R. J. K. Org. Synth. 2012, 89, 438. doi:10.15227/orgsyn.089.0438
Return to citation in text: [1] -
Yu, Z.; Ma, L.; Yu, W. Synlett 2010, 2607. doi:10.1055/s-0030-1258584
Return to citation in text: [1] -
Ghosh, S.; De, S.; Kakde, B. N.; Bhunia, S.; Adhikary, A.; Bisai, A. Org. Lett. 2012, 14, 5864. doi:10.1021/ol302767w
See for a preliminary communication from our group.
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] -
Trost, B. M.; Malhotra, S.; Chan, W. H. J. Am. Chem. Soc. 2011, 133, 7328. doi:10.1021/ja2020873
See for Pd-catalyzed asymmetric prenylations and geranylations.
Return to citation in text: [1] [2] -
Smit, W. A.; Bochkov, A. F.; Caple, R. Organic Synthesis: The Science behind the Art; Royal Society of Chemistry: Cambridge, 1998.
Return to citation in text: [1] -
Corey, E. J.; Cheng, X.-M. The Logic of Chemical Synthesis; John Wiley & Sons: New York, 1995.
Return to citation in text: [1] -
Magdziak, D.; Meek, S. J.; Pettus, T. R. R. Chem. Rev. 2004, 104, 1383. doi:10.1021/cr0306900
Return to citation in text: [1] -
Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2008, 108, 5299. doi:10.1021/cr800332c
Return to citation in text: [1] -
Bisai, A.; West, S. P.; Sarpong, R. J. Am. Chem. Soc. 2008, 130, 7222. doi:10.1021/ja8028069
Return to citation in text: [1] -
West, S. P.; Bisai, A.; Lim, A. D.; Narayan, R. R.; Sarpong, R. J. Am. Chem. Soc. 2009, 131, 11187. doi:10.1021/ja903868n
Return to citation in text: [1] [2] -
Kantak, A. A.; Potavathri, S.; Barham, R. A.; Romano, K. M.; DeBoef, B. J. Am. Chem. Soc. 2011, 133, 19960. doi:10.1021/ja2087085
Due to electron-rich species, aromatic solvents can take part in oxidative coupling reactions. See for an example.
Return to citation in text: [1] -
Jensen, B. S. CNS Drug Rev. 2002, 8, 353. doi:10.1111/j.1527-3458.2002.tb00233.x
Return to citation in text: [1] -
Oleynek, J. J.; Sedlock, D. M.; Barrow, C. J.; Appell, K. C.; Casiano, F.; Haycock, D.; Ward, S. J.; Kaplita, P.; Gillum, A. M. J. Antibiot. 1994, 47, 399. doi:10.7164/antibiotics.47.399
Return to citation in text: [1] -
Yu, Q.-S.; Holloway, H. W.; Flippen-Anderson, J. L.; Hoffman, B.; Brossi, A.; Greig, N. H. J. Med. Chem. 2001, 44, 4062. doi:10.1021/jm010080x
Return to citation in text: [1] -
Yu, Q.-S.; Zhu, X.; Holloway, H. W.; Whittaker, N. F.; Brossi, A.; Greig, N. H. J. Med. Chem. 2002, 45, 3684. doi:10.1021/jm010491d
Return to citation in text: [1] -
Luo, W.; Yu, Q.-S.; Zhan, M.; Parrish, D.; Deschamps, J. R.; Kulkarni, S. S.; Holloway, H. W.; Alley, G. M.; Lahiri, D. K.; Brossi, A.; Greig, N. H. J. Med. Chem. 2005, 48, 986. doi:10.1021/jm049309+
Return to citation in text: [1] -
Kobayashi, J.; Ishibashi, M. Alkaloids 1992, 41, 41.
Return to citation in text: [1] -
Lin, H.; Danishefsky, S. J. Angew. Chem., Int. Ed. 2003, 42, 36. doi:10.1002/anie.200390048
Return to citation in text: [1] -
Okada, M.; Sato, I.; Cho, S. J.; Dubnau, D.; Sakagami, Y. Tetrahedron 2006, 62, 8907. doi:10.1016/j.tet.2006.06.074
See for the isolation of geranylated hexahydropyrrolo[2,3-b]indole alkaloids.
Return to citation in text: [1] -
Rochfort, S. J.; Moore, S.; Craft, C.; Martin, N. H.; Van Wagoner, R. M.; Wright, J. L. C. J. Nat. Prod. 2009, 72, 1773. doi:10.1021/np900282j
Return to citation in text: [1] -
One-pot alkylations followed by dehydrogenative-coupling was unsuccessful, thus, IDC was carried out C-alkylated substrates such as 10.
Return to citation in text: [1] -
Burgett, A. W. G.; Li, Q.; Wei, Q.; Harran, P. G. Angew. Chem., Int. Ed. 2003, 42, 4961. doi:10.1002/anie.200352577
Return to citation in text: [1] -
Nicolaou, K. C.; Hao, J.; Reddy, M. V.; BheemaRao, P.; Rassias, G.; Snyder, S. A.; Huang, X.; Chen, D. Y.-K.; Brenzovich, W. E.; Guiseppone, N.; Giannakakou, P.; O’Brate, A. J. Am. Chem. Soc. 2004, 126, 12897. doi:10.1021/ja040093a
Return to citation in text: [1] -
Wu, Q.-X.; Crew, M. S.; Draskovic, M.; Sohn, J.; Johnson, T. A.; Tenney, K.; Valeriote, F. A.; Yao, X.-J.; Bjeldanes, L. F.; Crews, P. Org. Lett. 2010, 12, 4458. doi:10.1021/ol101396n
Return to citation in text: [1] -
Francke, W.; Kitching, W. Curr. Org. Chem. 2001, 5, 233. doi:10.2174/1385272013375652
Return to citation in text: [1] -
Rosenberg, S.; Leino, R. Synthesis 2009, 2651. doi:10.1055/s-0029-1216892
Return to citation in text: [1] -
Edmondson, S.; Danishefsky, S. J.; Sepp-Lorenzino, L.; Rosen, N. J. Am. Chem. Soc. 1999, 121, 2147. doi:10.1021/ja983788i
Return to citation in text: [1] -
Deppermann, N.; Thomanek, H.; Prenzel, A. H. G. P.; Maison, W. J. Org. Chem. 2010, 75, 5994. doi:10.1021/jo101401z
Return to citation in text: [1] -
Trost, B. M.; Brennan, M. K. Org. Lett. 2006, 8, 2027. doi:10.1021/ol060298j
Return to citation in text: [1] -
De, S.; Das, M. K.; Bhunia, S.; Bisai, A. Org. Lett. 2015, 17, 5922. doi:10.1021/acs.orglett.5b03082
See for our approach using a thiourea catalyzed aldol reaction with paraformaldehyde.
Return to citation in text: [1] -
May, J. A.; Stoltz, B. M. Tetrahedron 2006, 62, 5262. doi:10.1016/j.tet.2006.01.105
Return to citation in text: [1] -
Steven, A.; Overman, L. E. Angew. Chem., Int. Ed. 2007, 46, 5488. doi:10.1002/anie.200700612
Return to citation in text: [1] -
Schmidt, M. A.; Movassaghi, M. Synlett 2008, 313. doi:10.1055/s-2008-1032060
Return to citation in text: [1] -
Ghosh, S.; Chaudhuri, S.; Bisai, A. Org. Lett. 2015, 17, 1373. doi:10.1021/acs.orglett.5b00032
Return to citation in text: [1] -
Ghosh, S.; Chaudhuri, S.; Bisai, A. Chem. – Eur. J. 2015, 21, 17479. doi:10.1002/chem.201502878
See for an enantioselective approach using a key Pd-catalyzed decarboxylative allylation from our group.
Return to citation in text: [1] [2] -
Overman, L. E.; Paone, D. V.; Stearns, B. A. J. Am. Chem. Soc. 1999, 121, 7702. doi:10.1021/ja991714g
See for an asymmetric sequential processes to set a vicinal all-carbon quaternary stereocenter.
Return to citation in text: [1] -
Trost, B. M.; Osipov, M. Angew. Chem., Int. Ed. 2013, 52, 9176. doi:10.1002/anie.201302805
Return to citation in text: [1] -
Ghosh, S.; Bhunia, S.; Kakde, B. N.; De, S.; Bisai, A. Chem. Commun. 2014, 50, 2434. doi:10.1039/c3cc49064e
Return to citation in text: [1] -
Heiba, E. I.; Dessau, R. M.; Koehl, W. J., Jr. J. Am. Chem. Soc. 1968, 90, 2706. doi:10.1021/ja01012a051
See for a Mn(III)-mediated radical process.
Return to citation in text: [1] [2] -
Bush, J. B., Jr.; Finkbeiner, H. J. Am. Chem. Soc. 1968, 90, 5903. doi:10.1021/ja01023a048
Return to citation in text: [1] [2] -
Nikishin, G. I.; Vinogradov, M. G.; Fedorova, T. M. J. Chem. Soc., Chem. Commun. 1973, 693. doi:10.1039/C39730000693
Return to citation in text: [1] [2] -
Corey, E. J.; Kang, M. J. Am. Chem. Soc. 1984, 106, 5384. doi:10.1021/ja00330a076
Return to citation in text: [1] [2] -
Iqbal, J.; Bhatia, B.; Nayyar, N. K. Chem. Rev. 1994, 94, 519. doi:10.1021/cr00026a008
Return to citation in text: [1] [2] -
Newhouse, T.; Lewsi, C. A.; Eastman, K. J.; Baran, P. S. J. Am. Chem. Soc. 2010, 132, 7119. doi:10.1021/ja1009458
Return to citation in text: [1] [2] -
Foo, K.; Newhouse, T.; Mori, I.; Takayama, H.; Baran, P. S. Angew. Chem., Int. Ed. 2011, 50, 2716. doi:10.1002/anie.201008048
Return to citation in text: [1] [2] -
Akhtar, M.; Barton, D. H. R. J. Am. Chem. Soc. 1964, 86, 1528. doi:10.1021/ja01062a016
Return to citation in text: [1] -
Montoro, R.; Wirth, T. Org. Lett. 2003, 5, 4729. doi:10.1021/ol0359012
Return to citation in text: [1] -
Preparation of t-BuOI: A flame-dried round-bottom flask was charged with 0.6 mmol of t-BuONa in 0.5 mL of benzene. To this solution was added 0.6 mmol of I2 at room temperature. This solution was directly used for the oxidative coupling (see, reference [90]).
Return to citation in text: [1] -
Ruchti, J.; Carreira, E. M. J. Am. Chem. Soc. 2014, 136, 16756. doi:10.1021/ja509893s
Return to citation in text: [1] -
Thandavamurthy, K.; Sharma, D.; Porwal, S. K.; Ray, D.; Viswanathan, R. J. Org. Chem. 2014, 79, 10049. doi:10.1021/jo501651z
Return to citation in text: [1]
| 68. | Francke, W.; Kitching, W. Curr. Org. Chem. 2001, 5, 233. doi:10.2174/1385272013375652 |
| 69. | Rosenberg, S.; Leino, R. Synthesis 2009, 2651. doi:10.1055/s-0029-1216892 |
| 70. | Edmondson, S.; Danishefsky, S. J.; Sepp-Lorenzino, L.; Rosen, N. J. Am. Chem. Soc. 1999, 121, 2147. doi:10.1021/ja983788i |
| 71. | Deppermann, N.; Thomanek, H.; Prenzel, A. H. G. P.; Maison, W. J. Org. Chem. 2010, 75, 5994. doi:10.1021/jo101401z |
| 72. | Trost, B. M.; Brennan, M. K. Org. Lett. 2006, 8, 2027. doi:10.1021/ol060298j |
| 73. |
De, S.; Das, M. K.; Bhunia, S.; Bisai, A. Org. Lett. 2015, 17, 5922. doi:10.1021/acs.orglett.5b03082
See for our approach using a thiourea catalyzed aldol reaction with paraformaldehyde. |
| 46. |
Ghosh, S.; De, S.; Kakde, B. N.; Bhunia, S.; Adhikary, A.; Bisai, A. Org. Lett. 2012, 14, 5864. doi:10.1021/ol302767w
See for a preliminary communication from our group. |
| 1. | Dyker, G., Ed. Handbook of C-H Transformations: Applications in Organic Synthesis; 2005; Vol. 2. |
| 2. | Stuart, D. R.; Fagnou, K. Science 2007, 316, 1172. doi:10.1126/science.1141956 |
| 3. | Ashenhurst, J. A. Chem. Soc. Rev. 2010, 39, 540. doi:10.1039/B907809F |
| 4. | Le Bras, J.; Muzart, J. Chem. Rev. 2011, 111, 1170. doi:10.1021/cr100209d |
| 5. | Rossi, R.; Bellina, F.; Lessi, M. Synthesis 2010, 4131. doi:10.1055/s-0030-1258262 |
| 6. | Scheuermann, C. J. Chem. – Asian J. 2010, 5, 436. doi:10.1002/asia.200900487 |
| 7. | You, S.-l.; Xia, J.-B. Top. Curr. Chem. 2010, 292, 165. doi:10.1007/128_2009_18 |
| 8. | Newhouse, T.; Baran, P. S.; Hoffmann, R. W. Chem. Soc. Rev. 2009, 38, 3010. doi:10.1039/b821200g |
| 9. | Gaich, T.; Baran, P. S. J. Org. Chem. 2010, 75, 4657. doi:10.1021/jo1006812 |
| 10. | Hendrickson, J. B. J. Am. Chem. Soc. 1975, 97, 5784. doi:10.1021/ja00853a023 |
| 26. | Trost, B. M.; Brennan, M. K. Synthesis 2009, 3003. doi:10.1055/s-0029-1216975 |
| 48. | Smit, W. A.; Bochkov, A. F.; Caple, R. Organic Synthesis: The Science behind the Art; Royal Society of Chemistry: Cambridge, 1998. |
| 49. | Corey, E. J.; Cheng, X.-M. The Logic of Chemical Synthesis; John Wiley & Sons: New York, 1995. |
| 50. | Magdziak, D.; Meek, S. J.; Pettus, T. R. R. Chem. Rev. 2004, 104, 1383. doi:10.1021/cr0306900 |
| 51. | Zhdankin, V. V.; Stang, P. J. Chem. Rev. 2008, 108, 5299. doi:10.1021/cr800332c |
| 52. | Bisai, A.; West, S. P.; Sarpong, R. J. Am. Chem. Soc. 2008, 130, 7222. doi:10.1021/ja8028069 |
| 53. | West, S. P.; Bisai, A.; Lim, A. D.; Narayan, R. R.; Sarpong, R. J. Am. Chem. Soc. 2009, 131, 11187. doi:10.1021/ja903868n |
| 38. |
Jia, Y. X.; Kündig, E. P. Angew. Chem., Int. Ed. 2009, 48, 1636. doi:10.1002/anie.200805652
See for oxidative coupling using 2.2 equiv of CuCl2. |
| 39. | Dey, C.; Kündig, E. P. Chem. Commun. 2012, 3064. doi:10.1039/c2cc17871k |
| 22. | Marti, C.; Carreira, E. M. Eur. J. Org. Chem. 2003, 2209. doi:10.1002/ejoc.200300050 |
| 23. | Klein, J. E. M. N.; Taylor, R. J. K. Eur. J. Org. Chem. 2011, 6821. doi:10.1002/ejoc.201100836 |
| 24. | Roth, G. J.; Heckel, A.; Colbatzky, F.; Handschuh, S.; Kley, J.; Lehmann-Lintz, T.; Lotz, R.; Tontsch-Grunt, U.; Walter, R.; Hilberg, F. J. Med. Chem. 2009, 52, 4466. doi:10.1021/jm900431g |
| 25. | Rudrangi, S. R. S.; Bontha, V. K.; Manda, V. R.; Bethi, S. Asian J. Res. Chem. 2011, 4, 335. |
| 54. |
Kantak, A. A.; Potavathri, S.; Barham, R. A.; Romano, K. M.; DeBoef, B. J. Am. Chem. Soc. 2011, 133, 19960. doi:10.1021/ja2087085
Due to electron-rich species, aromatic solvents can take part in oxidative coupling reactions. See for an example. |
| 82. |
Heiba, E. I.; Dessau, R. M.; Koehl, W. J., Jr. J. Am. Chem. Soc. 1968, 90, 2706. doi:10.1021/ja01012a051
See for a Mn(III)-mediated radical process. |
| 83. | Bush, J. B., Jr.; Finkbeiner, H. J. Am. Chem. Soc. 1968, 90, 5903. doi:10.1021/ja01023a048 |
| 84. | Nikishin, G. I.; Vinogradov, M. G.; Fedorova, T. M. J. Chem. Soc., Chem. Commun. 1973, 693. doi:10.1039/C39730000693 |
| 85. | Corey, E. J.; Kang, M. J. Am. Chem. Soc. 1984, 106, 5384. doi:10.1021/ja00330a076 |
| 86. | Iqbal, J.; Bhatia, B.; Nayyar, N. K. Chem. Rev. 1994, 94, 519. doi:10.1021/cr00026a008 |
| 17. | Li, Z.; Bohle, D. S.; Li, C.-J. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 8928. doi:10.1073/pnas.0601687103 |
| 18. | Li, C.-J.; Li, Z. Pure Appl. Chem. 2006, 78, 935. doi:10.1351/pac200678050935 |
| 19. | Yoo, W.-J.; Li, C.-J. Top. Curr. Chem. 2010, 292, 281. doi:10.1007/128_2009_17 |
| 20. |
Zhang, Y.; Li, C.-J. J. Am. Chem. Soc. 2006, 128, 4242. doi:10.1021/ja060050p
See for metal-free CDC. |
| 21. | Uyanik, M.; Ishihara, K. ChemCatChem 2012, 4, 177. doi:10.1002/cctc.201100352 |
| 46. |
Ghosh, S.; De, S.; Kakde, B. N.; Bhunia, S.; Adhikary, A.; Bisai, A. Org. Lett. 2012, 14, 5864. doi:10.1021/ol302767w
See for a preliminary communication from our group. |
| 46. |
Ghosh, S.; De, S.; Kakde, B. N.; Bhunia, S.; Adhikary, A.; Bisai, A. Org. Lett. 2012, 14, 5864. doi:10.1021/ol302767w
See for a preliminary communication from our group. |
| 11. | Cho, S. H.; Kim, J. Y.; Kwak, J.; Chang, S. Chem. Soc. Rev. 2011, 40, 5068. doi:10.1039/c1cs15082k |
| 12. | Yeung, C. S.; Dong, V. M. Chem. Rev. 2011, 111, 1215. doi:10.1021/cr100280d |
| 13. | McGlacken, G. P.; Bateman, L. M. Chem. Soc. Rev. 2009, 38, 2447. doi:10.1039/b805701j |
| 14. | Liu, C.; Zhang, H.; Shi, W.; Lei, A. Chem. Rev. 2011, 111, 1780. doi:10.1021/cr100379j |
| 15. | Siemsen, P.; Livingston, R. C.; Diederich, F. Angew. Chem., Int. Ed. 2000, 39, 2632. doi:10.1002/1521-3773(20000804)39:15<2632::AID-ANIE2632>3.0.CO;2-F |
| 16. | Li, C.-J. Acc. Chem. Res. 2009, 42, 335. doi:10.1021/ar800164n |
| 47. |
Trost, B. M.; Malhotra, S.; Chan, W. H. J. Am. Chem. Soc. 2011, 133, 7328. doi:10.1021/ja2020873
See for Pd-catalyzed asymmetric prenylations and geranylations. |
| 46. |
Ghosh, S.; De, S.; Kakde, B. N.; Bhunia, S.; Adhikary, A.; Bisai, A. Org. Lett. 2012, 14, 5864. doi:10.1021/ol302767w
See for a preliminary communication from our group. |
| 35. | Kündig, E. P.; Seidel, T. M.; Jia, Y.-X.; Bernardinelli, G. Angew. Chem., Int. Ed. 2007, 46, 8484. doi:10.1002/anie.200703408 |
| 36. | Jia, Y.-X.; Hillgren, J. M.; Watson, E. L.; Marsden, S. P.; Kündig, E. P. Chem. Commun. 2008, 4040. doi:10.1039/b810858g |
| 37. | Jia, Y.-X.; Katayev, D.; Bernardinelli, G.; Seidel, T. M.; Kündig, E. P. Chem. – Eur. J. 2010, 16, 6300. doi:10.1002/chem.201000031 |
| 40. |
Perry, A.; Taylor, R. J. K. Chem. Commun. 2009, 3249. doi:10.1039/b903516h
See for oxidative coupling using 1.0 equiv of Cu(OAc)2·H2O. |
| 41. | Pugh, D. S.; Klein, J. E. M. N.; Perry, A.; Taylor, R. J. K. Synlett 2010, 934. doi:10.1055/s-0029-1219392 |
| 42. |
Klein, J. E. M. N.; Perry, A.; Pugh, D. S.; Taylor, R. J. K. Org. Lett. 2010, 12, 3446. doi:10.1021/ol1012668
See for coupling using 5–10 mol % of Cu(OAc)2·H2O. |
| 43. | Moody, C. L.; Franckevičius, V.; Drouhin, P.; Klein, J. E. M. N.; Taylor, R. J. K. Tetrahedron Lett. 2012, 53, 1897. doi:10.1016/j.tetlet.2012.01.120 |
| 44. | Pugh, D. S.; Taylor, R. J. K. Org. Synth. 2012, 89, 438. doi:10.15227/orgsyn.089.0438 |
| 78. |
Ghosh, S.; Chaudhuri, S.; Bisai, A. Chem. – Eur. J. 2015, 21, 17479. doi:10.1002/chem.201502878
See for an enantioselective approach using a key Pd-catalyzed decarboxylative allylation from our group. |
| 79. |
Overman, L. E.; Paone, D. V.; Stearns, B. A. J. Am. Chem. Soc. 1999, 121, 7702. doi:10.1021/ja991714g
See for an asymmetric sequential processes to set a vicinal all-carbon quaternary stereocenter. |
| 80. | Trost, B. M.; Osipov, M. Angew. Chem., Int. Ed. 2013, 52, 9176. doi:10.1002/anie.201302805 |
| 81. | Ghosh, S.; Bhunia, S.; Kakde, B. N.; De, S.; Bisai, A. Chem. Commun. 2014, 50, 2434. doi:10.1039/c3cc49064e |
| 33. | Khan, T. A.; Tripoli, R.; Crawford, J. J.; Martin, C. G.; Murphy, J. A. Org. Lett. 2003, 5, 2971. doi:10.1021/ol035173i |
| 34. | Beckwith, A. L. J.; Bowry, V. W.; Bowman, W. R.; Mann, E.; Parr, J.; Storey, J. M. D. Angew. Chem., Int. Ed. 2004, 43, 95. doi:10.1002/anie.200352419 |
| 78. |
Ghosh, S.; Chaudhuri, S.; Bisai, A. Chem. – Eur. J. 2015, 21, 17479. doi:10.1002/chem.201502878
See for an enantioselective approach using a key Pd-catalyzed decarboxylative allylation from our group. |
| 31. | Zhu, J.; Zhang, W.; Zhang, L.; Liu, J.; Zheng, J.; Hu, J. J. Org. Chem. 2010, 75, 5505. doi:10.1021/jo1005262 |
| 32. | Ju, X.; Liang, Y.; Jia, P.; Li, W.; Yu, W. Org. Biomol. Chem. 2012, 10, 498. doi:10.1039/C1OB06652H |
| 46. |
Ghosh, S.; De, S.; Kakde, B. N.; Bhunia, S.; Adhikary, A.; Bisai, A. Org. Lett. 2012, 14, 5864. doi:10.1021/ol302767w
See for a preliminary communication from our group. |
| 27. | Nishio, T.; Iseki, K.; Araki, N.; Miyazaki, T. Helv. Chim. Acta 2005, 88, 35. doi:10.1002/hlca.200490294 |
| 28. | Axon, J.; Boiteau, L.; Boivin, J.; Forbes, J. E.; Zard, S. Z. Tetrahedron Lett. 1994, 35, 1719. doi:10.1016/0040-4039(94)88328-9 |
| 29. | Murphy, J. A.; Tripoli, R.; Khan, T. A.; Mail, U. W. Org. Lett. 2005, 7, 3287. doi:10.1021/ol051095i |
| 30. | Teichert, A.; Jantos, K.; Harms, K.; Studer, A. Org. Lett. 2004, 6, 3477. doi:10.1021/ol048759t |
| 38. |
Jia, Y. X.; Kündig, E. P. Angew. Chem., Int. Ed. 2009, 48, 1636. doi:10.1002/anie.200805652
See for oxidative coupling using 2.2 equiv of CuCl2. |
| 39. | Dey, C.; Kündig, E. P. Chem. Commun. 2012, 3064. doi:10.1039/c2cc17871k |
| 74. | May, J. A.; Stoltz, B. M. Tetrahedron 2006, 62, 5262. doi:10.1016/j.tet.2006.01.105 |
| 75. | Steven, A.; Overman, L. E. Angew. Chem., Int. Ed. 2007, 46, 5488. doi:10.1002/anie.200700612 |
| 76. | Schmidt, M. A.; Movassaghi, M. Synlett 2008, 313. doi:10.1055/s-2008-1032060 |
| 77. | Ghosh, S.; Chaudhuri, S.; Bisai, A. Org. Lett. 2015, 17, 1373. doi:10.1021/acs.orglett.5b00032 |
| 46. |
Ghosh, S.; De, S.; Kakde, B. N.; Bhunia, S.; Adhikary, A.; Bisai, A. Org. Lett. 2012, 14, 5864. doi:10.1021/ol302767w
See for a preliminary communication from our group. |
| 53. | West, S. P.; Bisai, A.; Lim, A. D.; Narayan, R. R.; Sarpong, R. J. Am. Chem. Soc. 2009, 131, 11187. doi:10.1021/ja903868n |
| 82. |
Heiba, E. I.; Dessau, R. M.; Koehl, W. J., Jr. J. Am. Chem. Soc. 1968, 90, 2706. doi:10.1021/ja01012a051
See for a Mn(III)-mediated radical process. |
| 83. | Bush, J. B., Jr.; Finkbeiner, H. J. Am. Chem. Soc. 1968, 90, 5903. doi:10.1021/ja01023a048 |
| 84. | Nikishin, G. I.; Vinogradov, M. G.; Fedorova, T. M. J. Chem. Soc., Chem. Commun. 1973, 693. doi:10.1039/C39730000693 |
| 85. | Corey, E. J.; Kang, M. J. Am. Chem. Soc. 1984, 106, 5384. doi:10.1021/ja00330a076 |
| 86. | Iqbal, J.; Bhatia, B.; Nayyar, N. K. Chem. Rev. 1994, 94, 519. doi:10.1021/cr00026a008 |
| 46. |
Ghosh, S.; De, S.; Kakde, B. N.; Bhunia, S.; Adhikary, A.; Bisai, A. Org. Lett. 2012, 14, 5864. doi:10.1021/ol302767w
See for a preliminary communication from our group. |
| 46. |
Ghosh, S.; De, S.; Kakde, B. N.; Bhunia, S.; Adhikary, A.; Bisai, A. Org. Lett. 2012, 14, 5864. doi:10.1021/ol302767w
See for a preliminary communication from our group. |
| 87. | Newhouse, T.; Lewsi, C. A.; Eastman, K. J.; Baran, P. S. J. Am. Chem. Soc. 2010, 132, 7119. doi:10.1021/ja1009458 |
| 88. | Foo, K.; Newhouse, T.; Mori, I.; Takayama, H.; Baran, P. S. Angew. Chem., Int. Ed. 2011, 50, 2716. doi:10.1002/anie.201008048 |
| 65. | Burgett, A. W. G.; Li, Q.; Wei, Q.; Harran, P. G. Angew. Chem., Int. Ed. 2003, 42, 4961. doi:10.1002/anie.200352577 |
| 66. | Nicolaou, K. C.; Hao, J.; Reddy, M. V.; BheemaRao, P.; Rassias, G.; Snyder, S. A.; Huang, X.; Chen, D. Y.-K.; Brenzovich, W. E.; Guiseppone, N.; Giannakakou, P.; O’Brate, A. J. Am. Chem. Soc. 2004, 126, 12897. doi:10.1021/ja040093a |
| 67. | Wu, Q.-X.; Crew, M. S.; Draskovic, M.; Sohn, J.; Johnson, T. A.; Tenney, K.; Valeriote, F. A.; Yao, X.-J.; Bjeldanes, L. F.; Crews, P. Org. Lett. 2010, 12, 4458. doi:10.1021/ol101396n |
| 46. |
Ghosh, S.; De, S.; Kakde, B. N.; Bhunia, S.; Adhikary, A.; Bisai, A. Org. Lett. 2012, 14, 5864. doi:10.1021/ol302767w
See for a preliminary communication from our group. |
| 64. | One-pot alkylations followed by dehydrogenative-coupling was unsuccessful, thus, IDC was carried out C-alkylated substrates such as 10. |
| 92. | Ruchti, J.; Carreira, E. M. J. Am. Chem. Soc. 2014, 136, 16756. doi:10.1021/ja509893s |
| 93. | Thandavamurthy, K.; Sharma, D.; Porwal, S. K.; Ray, D.; Viswanathan, R. J. Org. Chem. 2014, 79, 10049. doi:10.1021/jo501651z |
| 46. |
Ghosh, S.; De, S.; Kakde, B. N.; Bhunia, S.; Adhikary, A.; Bisai, A. Org. Lett. 2012, 14, 5864. doi:10.1021/ol302767w
See for a preliminary communication from our group. |
| 46. |
Ghosh, S.; De, S.; Kakde, B. N.; Bhunia, S.; Adhikary, A.; Bisai, A. Org. Lett. 2012, 14, 5864. doi:10.1021/ol302767w
See for a preliminary communication from our group. |
| 46. |
Ghosh, S.; De, S.; Kakde, B. N.; Bhunia, S.; Adhikary, A.; Bisai, A. Org. Lett. 2012, 14, 5864. doi:10.1021/ol302767w
See for a preliminary communication from our group. |
| 91. | Preparation of t-BuOI: A flame-dried round-bottom flask was charged with 0.6 mmol of t-BuONa in 0.5 mL of benzene. To this solution was added 0.6 mmol of I2 at room temperature. This solution was directly used for the oxidative coupling (see, reference [90]). |
| 62. |
Okada, M.; Sato, I.; Cho, S. J.; Dubnau, D.; Sakagami, Y. Tetrahedron 2006, 62, 8907. doi:10.1016/j.tet.2006.06.074
See for the isolation of geranylated hexahydropyrrolo[2,3-b]indole alkaloids. |
| 63. | Rochfort, S. J.; Moore, S.; Craft, C.; Martin, N. H.; Van Wagoner, R. M.; Wright, J. L. C. J. Nat. Prod. 2009, 72, 1773. doi:10.1021/np900282j |
| 46. |
Ghosh, S.; De, S.; Kakde, B. N.; Bhunia, S.; Adhikary, A.; Bisai, A. Org. Lett. 2012, 14, 5864. doi:10.1021/ol302767w
See for a preliminary communication from our group. |
| 55. | Jensen, B. S. CNS Drug Rev. 2002, 8, 353. doi:10.1111/j.1527-3458.2002.tb00233.x |
| 56. | Oleynek, J. J.; Sedlock, D. M.; Barrow, C. J.; Appell, K. C.; Casiano, F.; Haycock, D.; Ward, S. J.; Kaplita, P.; Gillum, A. M. J. Antibiot. 1994, 47, 399. doi:10.7164/antibiotics.47.399 |
| 57. | Yu, Q.-S.; Holloway, H. W.; Flippen-Anderson, J. L.; Hoffman, B.; Brossi, A.; Greig, N. H. J. Med. Chem. 2001, 44, 4062. doi:10.1021/jm010080x |
| 58. | Yu, Q.-S.; Zhu, X.; Holloway, H. W.; Whittaker, N. F.; Brossi, A.; Greig, N. H. J. Med. Chem. 2002, 45, 3684. doi:10.1021/jm010491d |
| 59. | Luo, W.; Yu, Q.-S.; Zhan, M.; Parrish, D.; Deschamps, J. R.; Kulkarni, S. S.; Holloway, H. W.; Alley, G. M.; Lahiri, D. K.; Brossi, A.; Greig, N. H. J. Med. Chem. 2005, 48, 986. doi:10.1021/jm049309+ |
| 60. | Kobayashi, J.; Ishibashi, M. Alkaloids 1992, 41, 41. |
| 61. | Lin, H.; Danishefsky, S. J. Angew. Chem., Int. Ed. 2003, 42, 36. doi:10.1002/anie.200390048 |
| 87. | Newhouse, T.; Lewsi, C. A.; Eastman, K. J.; Baran, P. S. J. Am. Chem. Soc. 2010, 132, 7119. doi:10.1021/ja1009458 |
| 88. | Foo, K.; Newhouse, T.; Mori, I.; Takayama, H.; Baran, P. S. Angew. Chem., Int. Ed. 2011, 50, 2716. doi:10.1002/anie.201008048 |
| 47. |
Trost, B. M.; Malhotra, S.; Chan, W. H. J. Am. Chem. Soc. 2011, 133, 7328. doi:10.1021/ja2020873
See for Pd-catalyzed asymmetric prenylations and geranylations. |
| 89. | Akhtar, M.; Barton, D. H. R. J. Am. Chem. Soc. 1964, 86, 1528. doi:10.1021/ja01062a016 |
| 90. | Montoro, R.; Wirth, T. Org. Lett. 2003, 5, 4729. doi:10.1021/ol0359012 |
© 2016 Kumar et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)