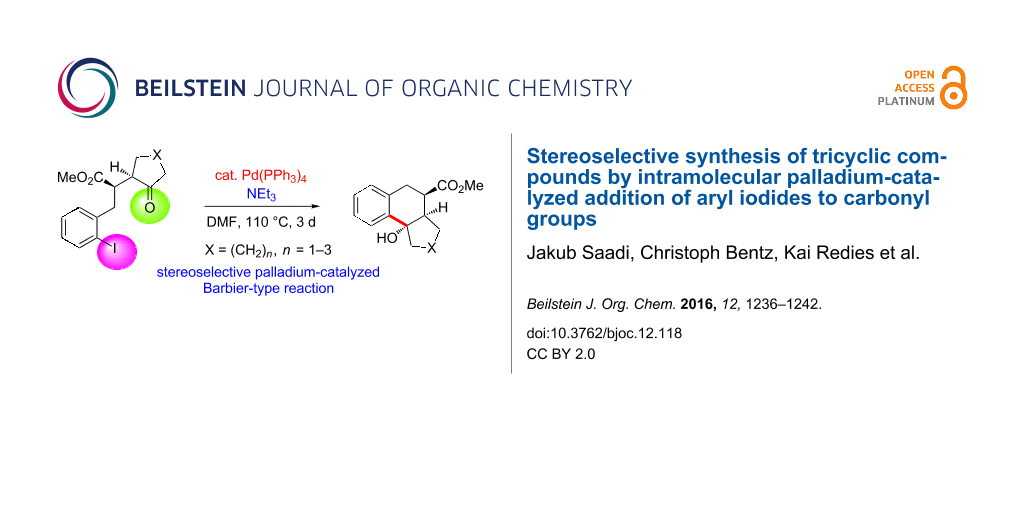
Abstract
Starting from γ-ketoesters with an o-iodobenzyl group we studied a palladium-catalyzed cyclization process that stereoselectively led to bi- and tricyclic compounds in moderate to excellent yields. Four X-ray crystal structure analyses unequivocally defined the structure of crucial cyclization products. The relative configuration of the precursor compounds is essentially transferred to that of the products and the formed hydroxy group in the newly generated cyclohexane ring is consistently in trans-arrangement with respect to the methoxycarbonyl group. A transition-state model is proposed to explain the observed stereochemical outcome. This palladium-catalyzed Barbier-type reaction requires a reduction of palladium(II) back to palladium(0) which is apparently achieved by the present triethylamine.

Graphical Abstract
Introduction
For our systematic studies on samarium diiodide promoted cyclizations leading to benzannulated medium-sized rings [1-4] we required starting materials such as alkenyl-substituted compounds B (Scheme 1). Obvious precursors for B are aryl iodides A that smoothly undergo palladium-catalyzed coupling reactions to provide the desired products. However, in one case [A: R1–R2 = (CH2)4] typical Heck reaction conditions employing styrene as olefin component not only led to the desired styrene derivative B but mainly to the cyclized product C. If the reaction was performed without the olefin it provided only the tertiary alcohol C in reasonable yield [5]. Similar C–C bond forming reactions of aryl halides that involve an insertion of the intermediate aryl palladium species into a carbonyl group are relatively rare (see discussion below). Therefore this serendipitous discovery led us to investigate the reaction in more detail.
![[1860-5397-12-118-i1]](/bjoc/content/inline/1860-5397-12-118-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Planned Heck reaction of A to compound B and serendipitous discovery of the palladium-catalyzed cyclization to products C.
Scheme 1: Planned Heck reaction of A to compound B and serendipitous discovery of the palladium-catalyzed cyc...
Results
The required γ-ketoesters A bearing the aryl iodide substituent were prepared following our well-established route via 2-siloxycyclopropane carboxylates D [6,7] that allows a regioselective introduction of the benzylic substituents at the α-carbon [8,9] to give intermediates E (Scheme 2). After fluoride-promoted ring opening [10] the desired precursor compounds A (1–6) were obtained in reasonable overall efficacies (for details see Supporting Information File 1).
![[1860-5397-12-118-i2]](/bjoc/content/inline/1860-5397-12-118-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of compounds A (1–6) via methyl 2-siloxycyclopropanecarboxylates D, their alkylation to E and fluoride-induced ring opening.
Scheme 2: Synthesis of compounds A (1–6) via methyl 2-siloxycyclopropanecarboxylates D, their alkylation to E...
We start our report with the palladium-catalyzed reactions of simple alkyl ketones 1 and 2 leading to bicyclic products and then continue with the transformations of cyclic ketones 3–6 that led to tricyclic compounds. Methyl ketone 1 provided under the reaction conditions (2 mol % Pd(PPh3)4, 3.5 equivalents NEt3, DMF, 110 °C, 3 d) that had been optimized with compound 4 a moderate yield of the tetralin derivative 7 formed as a single diastereomer (Scheme 3). Although the configurational assignment is ambiguous in this case, the NMR data and the fact that no γ-lactone is formed strongly support the trans-arrangement of the two functional groups as depicted. Under similar conditions (5 mol % Pd(PPh3)4, 90 °C, 3 d) the isopropyl-substituted ketone 2 furnished a mixture of the related trans-compound 8 (11%) together with the de-iodinated product 9 (25%) and the indane derivative 10 as major component (62%). The C–C coupling reaction to 8 seems to be hindered in this case, probably due to the steric bulk of the isopropyl group. The formation of indane derivative 10 occurs by an intramolecular enolate arylation, a reaction that has been discovered by our group some years ago [11,12]. Apparently, under the reaction conditions a ketone enolate of 2 reacts with the iodoarene moiety to form the five-membered ring of 10. The configurational assignments of compounds 7 and 8 are in agreement with those discussed below, where X-ray crystal structure analyses unequivocally confirmed the relative configurations of cyclization products.
![[1860-5397-12-118-i3]](/bjoc/content/inline/1860-5397-12-118-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Palladium-catalyzed reactions of methyl ketone 1 to tetralin derivative 7 and of isopropyl-substituted ketone 2 to compounds 8, 9 and 10.
Scheme 3: Palladium-catalyzed reactions of methyl ketone 1 to tetralin derivative 7 and of isopropyl-substitu...
With the cyclic ketones in part considerably higher yields of tricyclic products could be obtained (Schemes 4–6). The cyclopentanone derivative 3 (Scheme 4) had to be used as a mixture of two diastereomers (ca. 2:1) since these were not separable in our hands. Under the standard reaction conditions this ketone furnished a mixture of two diastereomeric tricyclic products 11a and 11b in 35% combined yield. The configuration of 11a was unequivocally determined by an X-ray crystal structure analysis of the corresponding p-nitrobenzoate 12a obtained by esterification of the tertiary alcohol under standard conditions (Scheme 4, Figure 1) [13]. The configuration of the second product 11b is only tentatively assigned as depicted since the available data do not allow an unambiguous determination. Considering the result obtained with the cycloheptanone derivative where two diastereomers could be assigned by X-ray crystal structure analyses make the proposed trans-annulation of the five- and six-membered rings fairly likely.
![[1860-5397-12-118-i4]](/bjoc/content/inline/1860-5397-12-118-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Palladium-catalyzed cyclization of diastereomeric cyclopentanone derivatives 3a/3b to products 11a and 11b and synthesis of p-nitrobenzoate 12a.
Scheme 4: Palladium-catalyzed cyclization of diastereomeric cyclopentanone derivatives 3a/3b to products 11a ...
![[1860-5397-12-118-1]](/bjoc/content/figures/1860-5397-12-118-1.png?scale=1.6&max-width=1024&background=FFFFFF)
Figure 1: Molecular structure (ORTEP, [14]) of compound 12a (thermal ellipsoids at 50% probability).
Figure 1: Molecular structure (ORTEP, [14]) of compound 12a (thermal ellipsoids at 50% probability).
The palladium-catalyzed cyclization discussed in this report was discovered with cyclohexanone derivative 4 and we therefore tried to optimize the reaction conditions with this substrate. After extensive studies investigating different palladium catalysts, bases, additives, temperatures (with and without microwave) and reaction times we found the recorded conditions to be most reliable. A mixture of the two diastereomers 4a/4b (ca. 1:1) furnished the two isomeric cyclization products 13a and 13b in varying yields and in several experiments only compound 13a was isolated. Fortunately, diastereomers 4a and 4b could be separated by conventional column chromatography and hence a detailed analysis of the stereochemical features of this transformation was possible. Configurationally homogeneous compound 4a furnished diastereomerically pure cyclization product 13a in excellent yield, whereas epimer 4b was converted into product 13b in 80% yield, again formed as a single isomer (Scheme 5). An unequivocal configurational assignment of 13a was possible by the X-ray crystal structure analysis of its corresponding p-nitrobenzoate 14a (Figure 2) [15]. The configuration of compound 13b was tentatively assigned in analogy to its lower and higher homologs.
![[1860-5397-12-118-i5]](/bjoc/content/inline/1860-5397-12-118-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Palladium-catalyzed cyclizations of diastereomeric cyclohexanone derivatives 4a and 4b leading stereoselectively to products 13a and 13b and structure of p-nitrobenzoate 14a.
Scheme 5: Palladium-catalyzed cyclizations of diastereomeric cyclohexanone derivatives 4a and 4b leading ster...
![[1860-5397-12-118-2]](/bjoc/content/figures/1860-5397-12-118-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular structure (ORTEP, [14]) of compound 14a (thermal ellipsoids at 50% probability).
Figure 2: Molecular structure (ORTEP, [14]) of compound 14a (thermal ellipsoids at 50% probability).
With cycloheptanone derivatives 5a/b less efficient cyclizations were observed. The mixture of diastereomers 5a/5b (ca. 1:1) provided a mixture of compounds containing the two isomers 15a and 15b, from which pure 15a could be isolated (Scheme 6). A diastereomerically enriched sample of 5b afforded compound 15b in 45% yield as a single isomer. These results were obtained employing microwave irradiation (400 W) that allowed considerably shorter reaction times, however, the yields were not strongly influenced by this modification. From both product diastereomers crystals suitable for X-ray crystal structure analyses could be obtained (Figure 3 and Figure 4) [16,17]. Again, the configurations of the precursors are reflected in the product structure. Compound 5b provided product 15b with trans-annulation of the two rings and we assume that compound 15a with cis-annulated rings was formed selectively from precursor 5a.
![[1860-5397-12-118-i6]](/bjoc/content/inline/1860-5397-12-118-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Palladium-catalyzed cyclizations of cycloheptanone derivatives 5a and 5b leading to products 15a and 15b.
Scheme 6: Palladium-catalyzed cyclizations of cycloheptanone derivatives 5a and 5b leading to products 15a an...
![[1860-5397-12-118-3]](/bjoc/content/figures/1860-5397-12-118-3.png?scale=1.5&max-width=1024&background=FFFFFF)
Figure 3: Molecular structure (ORTEP, [14]) of compound 15a (thermal ellipsoids at 50% probability).
Figure 3: Molecular structure (ORTEP, [14]) of compound 15a (thermal ellipsoids at 50% probability).
![[1860-5397-12-118-4]](/bjoc/content/figures/1860-5397-12-118-4.png?scale=1.5&max-width=1024&background=FFFFFF)
Figure 4: Molecular structure (ORTEP [14]) of compound 15b (thermal ellipsoids at 50% probability).
Figure 4: Molecular structure (ORTEP [14]) of compound 15b (thermal ellipsoids at 50% probability).
We also briefly studied the palladium-catalyzed cyclization of p-methoxy-substituted aryl iodide 6a/b that led under the standard conditions to a mixture containing compound 16 (Scheme 7). We cannot exclude that other regioisomers or even primarily formed tetralin derivatives are in the crude product mixture, but we isolated only compound 16 in pure form in 24% yield. Not surprisingly, the p-methoxy substituent favored the elimination of water from the primary addition product to generate the central double bond of 16.
![[1860-5397-12-118-i7]](/bjoc/content/inline/1860-5397-12-118-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Palladium-catalyzed cyclization of p-methoxy-substituted aryl iodide 6a/6b to compound 16.
Scheme 7: Palladium-catalyzed cyclization of p-methoxy-substituted aryl iodide 6a/6b to compound 16.
Discussion
The nucleophilic addition of organometallics such as lithium, magnesium or zinc organic compounds to carbonyl groups leading to alcohols is an important standard operation in organic synthesis providing high stereoselectivities in many cases. The in situ generation of the nucleophilic species from the corresponding organic halides in the presence of the carbonyl compound is also known, e.g., the Barbier reaction employing magnesium. The related transformation described in this report very likely involves an arylpalladium species as nucleophile that is in situ generated from the aryl iodide moiety. Similar palladium-catalyzed processes (palladium Barbier reactions) are relatively rare (for a review, see [18]). Early studies were reported by Y. Yamamoto et al. [19] and this group also published examples involving an alkyne palladation step to vinyl palladium intermediates that are able to undergo additions to carbonyl groups [20,21]. Very extensive investigations with a variety of o-haloaniline derivatives as precursors have been reported by the group of Solé, Bonjoch and Fernández [22,23]. They also analyzed this reaction and the competing enolate arylation by computational studies [24,25] (for a review, see [26]). Singular contributions employing different systems were contributed by other groups [27-29]. Scheme 8 shows a typical example of Solé et al. [23], related to our systems, furnishing a tricyclic compound in good yield with moderate diastereoselectivity.
![[1860-5397-12-118-i8]](/bjoc/content/inline/1860-5397-12-118-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Typical palladium-catalyzed cyclization of an o-iodoaniline derivative to a tricyclic tertiary alcohol as reported by Solé et al. [23].
Scheme 8: Typical palladium-catalyzed cyclization of an o-iodoaniline derivative to a tricyclic tertiary alco...
The transformations described in the present report are highly stereoselective (we hesitate to use the term stereospecific here since the mass balances are often too low to rigorously exclude the formation of other diastereomers) since the configuration of the cyclic precursor ketones 3–5 is transferred to that of the cyclization products as shown in Schemes 4–6. Surprisingly, the presence of excess of the base triethylamine and the fairly long reaction times do not lead to noticeable epimerization of the precursor γ-ketoesters that would lead to an erosion of the observed stereoselectivity. In all examples the methoxycarbonyl group and the hydroxy group are arranged trans to each other irrespective of the configuration of the third stereogenic center at the bridgehead. A transition-state model that rationalizes this observation is depicted in Scheme 9. We propose a four-center interaction of the carbonyl moiety with the carbon–palladium bond in the transition state (TS) and due to this highly ordered arrangement only a boat-like transition state with a pseudo-equatorial position of the methoxycarbonyl group seems to be possible. The rigid benzene backbone further restricts the flexibility of the system. This model explains the observed trans-arrangement of the methoxycarbonyl group and the hydroxy group leading to diastereomers a, if the hydrogen at C-1’ is in a pseudo-equatorial position and diastereomers b if this atom occupies a pseudo-axial position. This transition state is in accordance with the model that has been proposed on the basis of DFT calculations by Solé and Fernández [24-26].
![[1860-5397-12-118-i9]](/bjoc/content/inline/1860-5397-12-118-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Proposed transition state (TS) explaining the stereoselective formation of cyclization products.
Scheme 9: Proposed transition state (TS) explaining the stereoselective formation of cyclization products.
The intermediate arylpalladium species is certainly generated by the standard oxidative insertion of palladium(0) into the C–I bond of the aryl iodide moiety. After the C–C bond formation leading to the bi- or tricyclic products, palladium(II) has to be reduced back to a palladium(0) species in order to allow a catalytic use of the metal. It is well known that several reagents (alkenes or alcohols [19]) are able to achieve this reduction. We therefore assume that triethylamine is the reducing reagent under the conditions employed in this study [30]. Possible mechanisms for this process leading to the formation of an iminium intermediate or a dehydrogenation of the amine are depicted in Scheme 10.
![[1860-5397-12-118-i10]](/bjoc/content/inline/1860-5397-12-118-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Possible mechanism of the reduction of palladium(II) to palladium(0) by triethylamine (additional ligands at palladium are not depicted for clarity of the presentation).
Scheme 10: Possible mechanism of the reduction of palladium(II) to palladium(0) by triethylamine (additional l...
Conclusion
We have found new examples of intramolecular palladium-catalyzed nucleophilic additions of aryl iodides to alkyl ketones. These additions proceed in the presence of only 2–5 mol % Pd(PPh3)4 and afford bi- and tricyclic compounds with excellent stereoselectivity and in moderate to very good efficacy. The low mass balance observed in several cases may be due to subsequent reactions such as simple de-iodination of the precursor compounds or elimination of water in the products. However, in general none of these byproducts has been isolated. For compound 2 the bulky isopropyl group slows down the addition to the carbonyl group and an enolate arylation was observed instead as major reaction pathway. Although the scope of the discovered aryl iodide addition to carbonyl groups may be limited it is attractive since only low catalyst loadings are required and interesting products are formed with high stereoselectivity.
Supporting Information
| Supporting Information File 1: Characterization data and copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 1.3 MB | Download |
References
-
Berndt, M.; Gross, S.; Hölemann, A.; Reissig, H.-U. Synlett 2004, 422–438. doi:10.1055/s-2004-815429
Return to citation in text: [1] -
Saadi, J.; Reissig, H.-U. Synlett 2009, 2089–2092. doi:10.1055/s-0029-1217520
Return to citation in text: [1] -
Saadi, J.; Lentz, D.; Reissig, H.-U. Org. Lett. 2009, 11, 3334–3337. doi:10.1021/ol901183h
Return to citation in text: [1] -
Saadi, J.; Brüdgam, I.; Reissig, H.-U. Beilstein J. Org. Chem. 2010, 6, 1229–1245. doi:10.3762/bjoc.6.141
Return to citation in text: [1] -
Saadi, J. SmI2-Induced Cyclizations of Alkenyl- and Alkynyl-Substituted γ-, δ- and ε-Ketoesters – An Approach to Medium-Sized Carbocycles. Ph.D. Thesis, Freie Universität Berlin, Germany, 2009.
Return to citation in text: [1] -
Reissig, H.-U. Top. Curr. Chem. 1988, 144, 73–135.
Return to citation in text: [1] -
Reissig, H.-U.; Zimmer, R. Chem. Rev. 2003, 103, 1151–1196. doi:10.1021/cr010016n
Return to citation in text: [1] -
Kunkel, E.; Reichelt, I.; Reissig, H.-U. Liebigs Ann. Chem. 1984, 512–530. doi:10.1002/jlac.198419840311
Return to citation in text: [1] -
Reichelt, I.; Reissig, H.-U. Liebigs Ann. Chem. 1984, 531–551. doi:10.1002/jlac.198419840312
Return to citation in text: [1] -
Kunkel, E.; Reichelt, I.; Reissig, H.-U. Liebigs Ann. Chem. 1984, 802–819. doi:10.1002/jlac.198419840416
Return to citation in text: [1] -
Khan, F. A.; Czerwonka, R.; Reissig, H.-U. Synlett 1996, 533–535. doi:10.1055/s-1996-5483
Return to citation in text: [1] -
Khan, F. A.; Czerwonka, R.; Reissig, H.-U. Eur. J. Org. Chem. 2000, 3607–3617. doi:10.1002/1099-0690(200011)2000:21<3607::AID-EJOC3607>3.0.CO;2-N
Return to citation in text: [1] -
CCDC-1469195 (for 12a) contains the supplementary crystallographic data. These data can be obtained free of charge form The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Farrugia, L. J. J. Appl. Crystallogr. 1997, 30, 565. doi:10.1107/S0021889897003117
Return to citation in text: [1] [2] [3] [4] -
CCDC-1469196 (for 14a) contains the supplementary crystallographic data. These data can be obtained free of charge form The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
CCDC-1469197 (for 15a) contains the supplementary crystallographic data. These data can be obtained free of charge form The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
CCDC-1469198 (for 15b) contains the supplementary crystallographic data. These data can be obtained free of charge form The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1] -
Yamamoto, Y.; Nakamura, I. Top. Organomet. Chem. 2005, 14, 211–239. doi:10.1007/b104136
Return to citation in text: [1] -
Quan, L. G.; Lamrani, M.; Yamamoto, Y. J. Am. Chem. Soc. 2000, 122, 4827–4828. doi:10.1021/ja000415k
Return to citation in text: [1] [2] -
Gevorgyan, V.; Quan, L. G.; Yamamoto, Y. Tetrahedron Lett. 1999, 40, 4089–4092. doi:10.1016/S0040-4039(99)00656-5
Return to citation in text: [1] -
Quan, L. G.; Gevorgyan, V.; Yamamoto, Y. J. Am. Chem. Soc. 1999, 121, 3545–3546. doi:10.1021/ja983645w
Return to citation in text: [1] -
Solé, D.; Vallverdú, L.; Peidró, E.; Bonjoch, J. Chem. Commun. 2001, 1888–1889. doi:10.1039/b105686g
Return to citation in text: [1] -
Solé, D.; Vallverdú, L.; Solans, X.; Font-Bardía, M.; Bonjoch, J. J. Am. Chem. Soc. 2003, 125, 1587–1594. doi:10.1021/ja029114w
Return to citation in text: [1] [2] [3] -
Solé, D.; Mariani, F.; Fernández, I.; Sierra, M. A. J. Org. Chem. 2012, 77, 10272–10284. doi:10.1021/jo301924e
Return to citation in text: [1] [2] -
Solé, D.; Fernández, I.; Sierra, M. A. Chem. – Eur. J. 2012, 18, 6950–6958. doi:10.1002/chem.201102811
Return to citation in text: [1] [2] -
Solé, D.; Fernández, I. Acc. Chem. Res. 2014, 47, 168–179. doi:10.1021/ar400104j
Return to citation in text: [1] [2] -
Zhao, Y.-B.; Mariampillai, B.; Candito, D. A.; Laleu, B.; Li, M.; Lautens, M. Angew. Chem. 2009, 121, 1881–1884. doi:10.1002/ange.200805780
Angew. Chem., Int. Ed. 2009, 48, 1849–1852. doi:10.1002/anie.200805780
Return to citation in text: [1] -
Jia, Y.-X.; Katayev, D.; Kündig, E. P. Chem. Commun. 2010, 46, 130–132. doi:10.1039/B917958E
Return to citation in text: [1] -
Giorgi, G.; Maiti, S.; López-Alvarado, P.; Menéndez, J. C. Org. Biomol. Chem. 2011, 9, 2722–2730. doi:10.1039/c0ob00526f
Return to citation in text: [1] -
Coquerel, Y.; Brémond, P.; Rodriguez, J. J. Organomet. Chem. 2007, 692, 4805–4808. doi:10.1016/j.jorganchem.2007.05.053
Return to citation in text: [1]
| 1. | Berndt, M.; Gross, S.; Hölemann, A.; Reissig, H.-U. Synlett 2004, 422–438. doi:10.1055/s-2004-815429 |
| 2. | Saadi, J.; Reissig, H.-U. Synlett 2009, 2089–2092. doi:10.1055/s-0029-1217520 |
| 3. | Saadi, J.; Lentz, D.; Reissig, H.-U. Org. Lett. 2009, 11, 3334–3337. doi:10.1021/ol901183h |
| 4. | Saadi, J.; Brüdgam, I.; Reissig, H.-U. Beilstein J. Org. Chem. 2010, 6, 1229–1245. doi:10.3762/bjoc.6.141 |
| 10. | Kunkel, E.; Reichelt, I.; Reissig, H.-U. Liebigs Ann. Chem. 1984, 802–819. doi:10.1002/jlac.198419840416 |
| 19. | Quan, L. G.; Lamrani, M.; Yamamoto, Y. J. Am. Chem. Soc. 2000, 122, 4827–4828. doi:10.1021/ja000415k |
| 8. | Kunkel, E.; Reichelt, I.; Reissig, H.-U. Liebigs Ann. Chem. 1984, 512–530. doi:10.1002/jlac.198419840311 |
| 9. | Reichelt, I.; Reissig, H.-U. Liebigs Ann. Chem. 1984, 531–551. doi:10.1002/jlac.198419840312 |
| 20. | Gevorgyan, V.; Quan, L. G.; Yamamoto, Y. Tetrahedron Lett. 1999, 40, 4089–4092. doi:10.1016/S0040-4039(99)00656-5 |
| 21. | Quan, L. G.; Gevorgyan, V.; Yamamoto, Y. J. Am. Chem. Soc. 1999, 121, 3545–3546. doi:10.1021/ja983645w |
| 6. | Reissig, H.-U. Top. Curr. Chem. 1988, 144, 73–135. |
| 7. | Reissig, H.-U.; Zimmer, R. Chem. Rev. 2003, 103, 1151–1196. doi:10.1021/cr010016n |
| 14. | Farrugia, L. J. J. Appl. Crystallogr. 1997, 30, 565. doi:10.1107/S0021889897003117 |
| 5. | Saadi, J. SmI2-Induced Cyclizations of Alkenyl- and Alkynyl-Substituted γ-, δ- and ε-Ketoesters – An Approach to Medium-Sized Carbocycles. Ph.D. Thesis, Freie Universität Berlin, Germany, 2009. |
| 18. | Yamamoto, Y.; Nakamura, I. Top. Organomet. Chem. 2005, 14, 211–239. doi:10.1007/b104136 |
| 15. | CCDC-1469196 (for 14a) contains the supplementary crystallographic data. These data can be obtained free of charge form The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 16. | CCDC-1469197 (for 15a) contains the supplementary crystallographic data. These data can be obtained free of charge form The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 17. | CCDC-1469198 (for 15b) contains the supplementary crystallographic data. These data can be obtained free of charge form The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 14. | Farrugia, L. J. J. Appl. Crystallogr. 1997, 30, 565. doi:10.1107/S0021889897003117 |
| 14. | Farrugia, L. J. J. Appl. Crystallogr. 1997, 30, 565. doi:10.1107/S0021889897003117 |
| 13. | CCDC-1469195 (for 12a) contains the supplementary crystallographic data. These data can be obtained free of charge form The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 11. | Khan, F. A.; Czerwonka, R.; Reissig, H.-U. Synlett 1996, 533–535. doi:10.1055/s-1996-5483 |
| 12. | Khan, F. A.; Czerwonka, R.; Reissig, H.-U. Eur. J. Org. Chem. 2000, 3607–3617. doi:10.1002/1099-0690(200011)2000:21<3607::AID-EJOC3607>3.0.CO;2-N |
| 14. | Farrugia, L. J. J. Appl. Crystallogr. 1997, 30, 565. doi:10.1107/S0021889897003117 |
| 26. | Solé, D.; Fernández, I. Acc. Chem. Res. 2014, 47, 168–179. doi:10.1021/ar400104j |
| 22. | Solé, D.; Vallverdú, L.; Peidró, E.; Bonjoch, J. Chem. Commun. 2001, 1888–1889. doi:10.1039/b105686g |
| 23. | Solé, D.; Vallverdú, L.; Solans, X.; Font-Bardía, M.; Bonjoch, J. J. Am. Chem. Soc. 2003, 125, 1587–1594. doi:10.1021/ja029114w |
| 24. | Solé, D.; Mariani, F.; Fernández, I.; Sierra, M. A. J. Org. Chem. 2012, 77, 10272–10284. doi:10.1021/jo301924e |
| 25. | Solé, D.; Fernández, I.; Sierra, M. A. Chem. – Eur. J. 2012, 18, 6950–6958. doi:10.1002/chem.201102811 |
| 19. | Quan, L. G.; Lamrani, M.; Yamamoto, Y. J. Am. Chem. Soc. 2000, 122, 4827–4828. doi:10.1021/ja000415k |
| 30. | Coquerel, Y.; Brémond, P.; Rodriguez, J. J. Organomet. Chem. 2007, 692, 4805–4808. doi:10.1016/j.jorganchem.2007.05.053 |
| 23. | Solé, D.; Vallverdú, L.; Solans, X.; Font-Bardía, M.; Bonjoch, J. J. Am. Chem. Soc. 2003, 125, 1587–1594. doi:10.1021/ja029114w |
| 24. | Solé, D.; Mariani, F.; Fernández, I.; Sierra, M. A. J. Org. Chem. 2012, 77, 10272–10284. doi:10.1021/jo301924e |
| 25. | Solé, D.; Fernández, I.; Sierra, M. A. Chem. – Eur. J. 2012, 18, 6950–6958. doi:10.1002/chem.201102811 |
| 26. | Solé, D.; Fernández, I. Acc. Chem. Res. 2014, 47, 168–179. doi:10.1021/ar400104j |
| 27. |
Zhao, Y.-B.; Mariampillai, B.; Candito, D. A.; Laleu, B.; Li, M.; Lautens, M. Angew. Chem. 2009, 121, 1881–1884. doi:10.1002/ange.200805780
Angew. Chem., Int. Ed. 2009, 48, 1849–1852. doi:10.1002/anie.200805780 |
| 28. | Jia, Y.-X.; Katayev, D.; Kündig, E. P. Chem. Commun. 2010, 46, 130–132. doi:10.1039/B917958E |
| 29. | Giorgi, G.; Maiti, S.; López-Alvarado, P.; Menéndez, J. C. Org. Biomol. Chem. 2011, 9, 2722–2730. doi:10.1039/c0ob00526f |
| 23. | Solé, D.; Vallverdú, L.; Solans, X.; Font-Bardía, M.; Bonjoch, J. J. Am. Chem. Soc. 2003, 125, 1587–1594. doi:10.1021/ja029114w |
© 2016 Saadi et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)