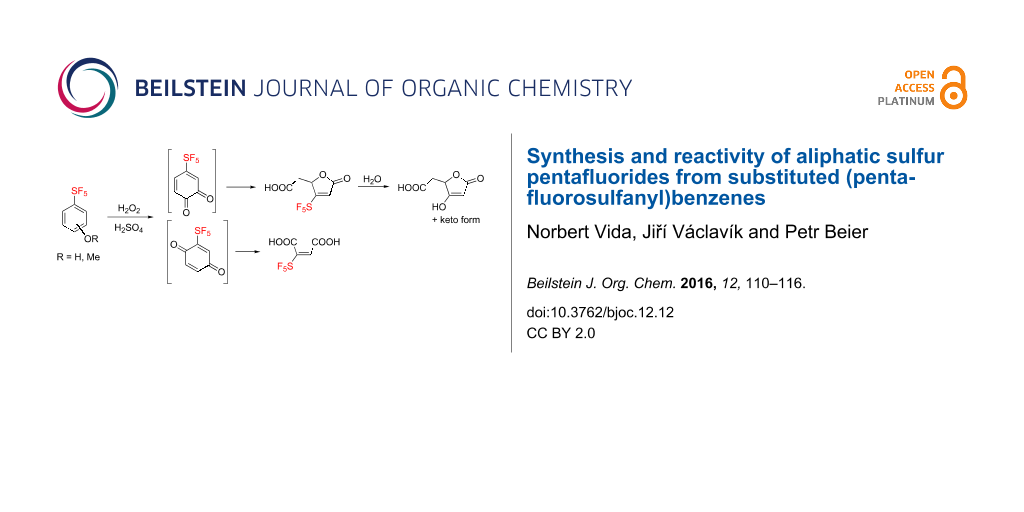
Abstract
Oxidation of 3- and 4-pentafluorosulfanyl-substituted anisoles and phenols with hydrogen peroxide and sulfuric acid provided a mixture of SF5-substituted muconolactone, maleic, and succinic acids. A plausible mechanism for the formation of the aliphatic SF5 compounds was presented and their chemical reactivity was investigated. SF5-substituted para-benzoquinone was synthesized; its oxidation led to an improved yield of 2-(pentafluorosulfanyl)maleic acid. The reaction of SF5-substituted maleic anhydride and para-benzoquinone with cyclopentadiene afforded the Diels–Alder adducts. Decomposition of 3-(pentafluorosulfanyl)muconolactone in acidic, neutral and basic aqueous media was investigated and the decarboxylation of 2-(pentafluorosulfanyl)maleic acid provided 3-(pentafluorosulfanyl)acrylic acid.

Graphical Abstract
Introduction
Fluorinated organic compounds have been one of the foci of chemical industry for the last several decades. The unique properties of fluorine atoms and fluorinated groups have been exploited in various applications ranging from advanced materials to bioactive compounds [1-3]. One such fluorinated functional group which has become the focus of systematic investigation only recently is the pentafluorosulfanyl (SF5) group [4,5]. The combination of high stability [6], lipophilicity [7] and strong electron-withdrawing character [8] – similar to but more extreme than the trifluoromethyl group – makes SF5 compounds promising candidates for agrochemicals and pharmaceuticals [9]. Recent studies showed several cases where SF5 analogues of established compounds showed improved potency [10-12]. Limiting factors for a more widespread use of SF5 compounds are low accessibility of basic building blocks and the lack of understanding of their chemical behavior. Lack of availability of SF5 compounds is particularly evident in aliphatics where radical addition of expensive and toxic SF5Cl to unsaturated compounds is practically the only synthetic option currently available [13,14]. We have recently described an alternative route to access SF5 aliphatic compounds, based on oxidation of much more readily available aromatic ones [15]. One of the methods we developed was based on the oxidation of 4-(pentafluorosulfanyl)anisole (1) or 4-(pentafluorosulfanyl)phenol (2) by a mixture of aqueous hydrogen peroxide and concentrated sulfuric acid (Scheme 1). The major product was muconolactone 3 while maleic acid 4 and succinic acid 5 were formed in small amounts. Herein we report a full account of this oxidation, alternative syntheses of maleic acid 4, and the results of chemical reactivity studies of acids 3 and 4.
![[1860-5397-12-12-i1]](/bjoc/content/inline/1860-5397-12-12-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Oxidation of SF5-anisole and phenol. 19F NMR yields are shown (isolated yields in parentheses).
Scheme 1: Oxidation of SF5-anisole and phenol. 19F NMR yields are shown (isolated yields in parentheses).
Results and Discussion
The proposed mechanism for the oxidation of 1 and 2 to SF5-containg products is shown in Scheme 2 [15]. The muconolactone derivative 3 is most probably the product of a series of transformations: electrophilic aromatic hydroxylation to the intermediate 6, oxidation to ortho-benzoquinone 7, Baeyer–Villiger (BV) oxidation, hydrolysis to muconic acid derivative 8, and finally intramolecular conjugate addition affording 3. Under certain conditions, small amounts of intermediates 6 and 8 were detected [15]. Interestingly, this reaction sequence is essentially identical to the aerobic microbial degradation of aromatic compounds, which involves hydroxylating oxygenase and ring-cleaving dioxygenase enzymes [16-19]. This sequence of reactions however, does not account for the presence of maleic acid derivative 4 in the product mixture. Control experiments in which 8 or 3 were reacted with H2O2/H2SO4 did not provide 4. This suggests that 4 may be formed by a second hydroxylation of 6 to 9. The second electrophilic aromatic hydroxylation is a favorable process because 6 is a more activated substrate than 1 or 2. Oxidation, BV oxidation and hydrolysis would afford the diacid 4 (Scheme 2). Succinic acid derivative 5 is another minor product and a control experiment showed that it was formed from 3. Mechanistic considerations of this process will be discussed later.
![[1860-5397-12-12-i2]](/bjoc/content/inline/1860-5397-12-12-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed mechanism for the formation of 3 and 4 from SF5 aromatics 1 and 2.
Scheme 2: Proposed mechanism for the formation of 3 and 4 from SF5 aromatics 1 and 2.
We hypothesized that the yield of 4 might be increased when starting from 3-(pentafluorosulfanyl)anisole (10) or 3-(pentafluorosulfanyl)phenol (11). In these substrates, the first hydroxylation is likely to occur in the para position to the methoxy or hydroxy groups facilitating the formation of the para-benzoquinone derivative 12. To test this hypothesis, both 10 and 11 were subjected to oxidation by H2O2/H2SO4 mixture. Indeed, the yield of 3 decreased considerably while the yield of 4 increased somewhat but still remains low (Scheme 3).
![[1860-5397-12-12-i3]](/bjoc/content/inline/1860-5397-12-12-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Oxidation of anisole 10 and phenol 11. 19F NMR yields are given.
Scheme 3: Oxidation of anisole 10 and phenol 11. 19F NMR yields are given.
The presumed intermediate 2-(pentafluorosulfanyl)-1,4-benzoquinone (12) in the pathway toward 4 was prepared independently (Scheme 4). Azo coupling [20] of phenol 11 with a diazonium salt prepared from sulfanilic acid afforded red azo product 13. The regioselectivity is governed by the strong para-directing hydroxy group despite the presence of the bulky SF5 group. Compounds with substituents ortho to the SF5 group are relatively uncommon [21]. Reduction with sodium dithionite [20] provided aminophenol 14 in good overall yield and oxidation with MnO2 [22] gave benzoquinone 12 also in a good yield. Further oxidation with H2O2/H2SO4 resulted in a mixture of four SF5-containing products. Maleic acid 4 was identified in the mixture (25% 19F NMR yield, Scheme 4) but chromatographic separation, crystallization or derivatization (CH2N2) attempts were not successful. Nevertheless, these results show that SF5-substituted para-benzoquinones are most likely intermediates in the formation of 4 by oxidation of aromatics.
![[1860-5397-12-12-i4]](/bjoc/content/inline/1860-5397-12-12-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of para-benzoquinone 12 and oxidation to maleic acid 4. 19F NMR yields are shown, in parentheses isolated yields.
Scheme 4: Synthesis of para-benzoquinone 12 and oxidation to maleic acid 4. 19F NMR yields are shown, in pare...
Benzoquinone 12 is a solid material with good stability at −20 °C. Several standard reactions were investigated. Reduction to hydroquinone 15 using catalytic palladium was very efficient (Scheme 5). The Diels–Alder reaction with cyclopentadiene proceeded in a high yield at ambient temperature with exclusive regiochemistry on the less substituted double bond of 12 and complete endo stereochemistry (Scheme 5).
![[1860-5397-12-12-i5]](/bjoc/content/inline/1860-5397-12-12-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Catalytic hydrogenation and Diels–Alder reaction of benzoquinone 12.
Scheme 5: Catalytic hydrogenation and Diels–Alder reaction of benzoquinone 12.
Employing density functional theory (DFT) with the B3LYP functional [23] and the aug-cc-pVDZ basis set [24,25], we calculated the reaction coordinates leading to four possible Diels–Alder products (Table 1). Single-point energies of the transition states suggested that pathway a was preferred over b (difference of ca. 10–20 kJ/mol). This was further supported by comparison of Gibbs free energy values. Boltzmann distribution was used to calculate theoretical endo/exo selectivity of 16a (98:2) from the differences of Gibbs energy of the transition states.
Table 1: Calculated reaction coordinates of Diels–Alder reaction of 12 with cyclopentadiene.a
| energy (kJ/mol) | pathway | ETSb | EPc |
|---|---|---|---|
single-point (Gibbs)
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-12-i11.svg?max-width=637&scale=1.0)
|
a-endo | 53.4 (116.9) | −57.4 (15.9) |
| a-exo | 64.1 (126.7) | −55.7 (20.1) | |
| b-endo | 74.6 (139.1) | −61.2 (14.8) | |
| b-exo | 75.6 (140.0) | −58.1 (20.2) | |
aComputed in Gaussian 09 Rev D.01 [26], rB3LYP/aug-cc-pVDZ, gas phase, 298.15 K; benergies of transition states relative to substrates; cenergies of products relative to substrates.
Geometries of the four transition states are depicted in Figure 1. The less abundant transition states (TS-endo-16b and TS-exo-16b) were asynchronous with the differences of bond lengths of 0.662 and 0.556 Å, respectively. Structures TS-endo-16a and TS-exo-16a also displayed a certain degree of asynchronicity with the bond length differences of 0.085 and 0.081 Å. The striking difference is probably caused by the steric demand of the SF5 group.
![[1860-5397-12-12-1]](/bjoc/content/figures/1860-5397-12-12-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Optimized geometries of transition states of Diels–Alder reaction of cyclopentadiene with 12. Selected bond lengths are given in Ångström.
Figure 1: Optimized geometries of transition states of Diels–Alder reaction of cyclopentadiene with 12. Selec...
The chemical behavior of muconolactone 3 was investigated next. This compound showed good stability at ambient temperature in a strongly acidic and oxidizing environment. However, lactone 3 was found to be unstable in water or in aqueous base. Monitoring a solution of 3 in D2O by 1H and 19F NMR spectroscopy showed a gradual disappearance of the SF5 signal. After 55 h at ambient temperature two sets of signals appeared in the 1H NMR spectrum; their integrals and coupling patterns suggested the presence of two structures of 17 in a 59:41 ratio. In DMSO-d6 only the enol form of 17 was observed. Decomposition of 3 is much faster in the presence of sodium bicarbonate. In a preparative experiment, 3 was dissolved in water and after 40 h at ambient temperature acid 17 was isolated in 60% yield confirming that 3 undergoes addition of water and elimination of HF and SF4 (Scheme 6). Nucleophile-induced elimination of SF5 groups in β-position of an electron-withdrawing group has been described [27].
![[1860-5397-12-12-i6]](/bjoc/content/inline/1860-5397-12-12-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
As already mentioned, compound 3 is much more stable in acidic than in neutral or basic aqueous medium. To investigate its reactivity in acids under various conditions, several screening experiments were carried out. In HCl (5 M), H2SO4 (5 M or 10 M) or 85% H3PO4 at ambient temperature the lactone 3 remained unaffected. At 80 °C the formation of levulinic acid derivative 18 was observed together with unidentified byproducts. Under elevated temperature (100 °C and above) the product of HF and SF4 elimination, (E)-4-oxopent-2-enoic acid (19) [28] formed (Scheme 7). In a preparative experiment using 85% H3PO4 at 100 °C, levulinic acid 18 was obtained in 14% yield. This result allowed us to propose that byproduct 5 observed during oxidation of SF5-anisoles and phenols could be formed from open-chain acids according to Scheme 7.
![[1860-5397-12-12-i7]](/bjoc/content/inline/1860-5397-12-12-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Formation of acids 5, 18 and 19 from lactone 3.
Scheme 7: Formation of acids 5, 18 and 19 from lactone 3.
Despite the lack of efficient synthetic protocols toward maleic acid 4, some basic chemical reactivity studies were performed. When heated to about 125 °C under reduced pressure, acid 4 eliminated water to afford anhydride 20 in a good yield. Anhydride 20 underwent a Diels–Alder reaction with cyclopentadiene at ambient temperature to provide a mixture of endo and exo products in an 8:1 ratio (Scheme 8). In comparison, it is known that maleic anhydride in analogous reactions affords exclusively the endo adduct [29].
![[1860-5397-12-12-i8]](/bjoc/content/inline/1860-5397-12-12-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of maleic anhydride 20 and Diels–Alder adducts 21.
Scheme 8: Synthesis of maleic anhydride 20 and Diels–Alder adducts 21.
Attempts to facilitate the isolation of very polar maleic acid 4 from the reaction mixture by derivatization to dimethyl ester using diazomethane were not successful. The dimethyl maleate derivative was formed but was accompanied by the [3 + 2] cycloaddition product 22. With excess of diazomethane, clean and efficient formation of 22 was observed (Scheme 9).
![[1860-5397-12-12-i9]](/bjoc/content/inline/1860-5397-12-12-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Reaction of maleic acid 4 with diazomethane.
Scheme 9: Reaction of maleic acid 4 with diazomethane.
Acid 4 was stable in water, methanol, isopropylalcohol, boiling benzene, or boiling aqueous sodium carbonate. However, it underwent decarboxylation in DMSO-d6. Monitoring by NMR showed decarboxylation even at room temperature. After 90 h at ambient temperature the starting compound 4 disappeared and 3-(pentafluorosulfanyl)prop-2-enoic acid (23) was formed. At 65 °C the starting material was no longer present after 1.5 h and the NMR yield of the acrylic acid derivative 23 was 60% (Scheme 10). Acrylic acid 23 was prepared previously by the addition of SF5Cl to allyl alcohol and subsequent elimination and oxidation [30]. We proposed that the decarboxylation reaction could be used for the synthesis of selectively deuterium labelled acrylic acid derivative 23. Indeed, when maleic acid 4 was evaporated twice with D2O to exchange acidic protons and heated in DMSO-d6, deuterated 23 was observed by NMR spectroscopy (Scheme 10).
![[1860-5397-12-12-i10]](/bjoc/content/inline/1860-5397-12-12-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Decarboxylation of maleic acid 4 to acrylic acid 23 in DMSO and the preparation of deuterium labelled 23.
Scheme 10: Decarboxylation of maleic acid 4 to acrylic acid 23 in DMSO and the preparation of deuterium labell...
Conclusion
In conclusion, similar to 4-(pentafluorosulfanyl)anisole and 4-(pentafluorosulfanyl)phenol the meta-derivatives 10 and 11 underwent oxidation with aqueous hydrogen peroxide in sulfuric acid to provide SF5-muconolactone and SF5-maleic acid as main products. Improved conversion of SF5-maleic acid was rationalized by preferential formation of SF5-substituted para-benzoquinone as an intermediate. This benzoquinone was independently prepared and its transformation to the maleic acid derivative by oxidation was verified. Reactions of SF5-maleinanhydride (formed by dehydration of SF5-maleic acid) and SF5-benzoquinone with cyclopentadiene provided the Diels–Alder adducts in good to excellent yields; the observed stereoselectivity of the latter reaction agreed with DFT calculations. SF5-muconolactone was found to be relatively stable in acidic media but eliminates the SF5 group in water or in aqueous base to form 2-(3,5-dioxotetrahydrofuran-2-yl)acetic acid, which in aqueous solution exists in equilibrium with its enol form. Further experiments are underway to find out whether aerobic microbial degradation of SF5-substituted aromatic compounds follows the same pathway as our chemical oxidation method.
Supporting Information
Synthesis and characterization of all new products, copies of 1H, 13C, and 19F NMR spectra of newly synthesized products, and computational data.
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 1.5 MB | Download |
References
-
Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2013. doi:10.1002/9783527651351
Return to citation in text: [1] -
Swallow, S. Fluorine-Containing Pharmaceuticals. In Fluorine in Pharmaceutical and Medicinal Chemistry; Gouverneur, V.; Müller, K., Eds.; Molecular Medicine and Medicinal Chemistry, Vol. 6; Imperial College Press: London, 2012; pp 141–174. doi:10.1142/9781848166363_0005
Return to citation in text: [1] -
Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c
Return to citation in text: [1] -
Altomonte, S.; Zanda, M. J. Fluorine Chem. 2012, 143, 57–93. doi:10.1016/j.jfluchem.2012.06.030
Return to citation in text: [1] -
Savoie, P. R.; Welch, J. T. Chem. Rev. 2015, 115, 1130–1190. doi:10.1021/cr500336u
Return to citation in text: [1] -
Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3064–3072. doi:10.1021/ja00875a006
Return to citation in text: [1] -
Hansch, C.; Leo, A.; Unger, S. H.; Kim, K. H.; Nikaitani, D.; Lien, E. J. J. Med. Chem. 1973, 16, 1207–1216. doi:10.1021/jm00269a003
Return to citation in text: [1] -
Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3072–3076. doi:10.1021/ja00875a007
Return to citation in text: [1] -
Crowley, P. J.; Mitchell, G.; Salmon, R.; Worthington, P. A. Chimia 2004, 58, 138–142. doi:10.2533/000942904777678172
Return to citation in text: [1] -
Lim, D. S.; Choi, J. S.; Pak, C. S.; Welch, J. T. J. Pestic. Sci. (Tokyo, Jpn.) 2007, 32, 255–259. doi:10.1584/jpestics.G06-50
Return to citation in text: [1] -
Welch, J. T.; Lim, D. S. Bioorg. Med. Chem. 2007, 15, 6659–6666. doi:10.1016/j.bmc.2007.08.012
Return to citation in text: [1] -
Wipf, P.; Mo, T.; Geib, S. J.; Caridha, D.; Dow, G. S.; Gerena, L.; Roncal, N.; Milner, E. E. Org. Biomol. Chem. 2009, 7, 4163–4165. doi:10.1039/b911483a
Return to citation in text: [1] -
Case, J. R.; Ray, N. H.; Roberts, H. L. J. Chem. Soc. 1961, 2066–2070. doi:10.1039/JR9610002066
Return to citation in text: [1] -
Aït-Mohand, S.; Dolbier, W. R., Jr. Org. Lett. 2002, 4, 3013–3015. doi:10.1021/ol026483o
Return to citation in text: [1] -
Vida, N.; Pastýříková, T.; Klepetářová, B.; Beier, P. J. Org. Chem. 2014, 79, 8906–8911. doi:10.1021/jo501562z
Return to citation in text: [1] [2] [3] -
Que, L., Jr.; Ho, R. Y. N. Chem. Rev. 1996, 96, 2607–2624. doi:10.1021/cr960039f
Return to citation in text: [1] -
Ullrich, R.; Hofrichter, M. Cell. Mol. Life Sci. 2007, 64, 271–293. doi:10.1007/s00018-007-6362-1
Return to citation in text: [1] -
Fuchs, G.; Boll, M.; Heider, J. Nat. Rev. Microbiol. 2011, 9, 803–816. doi:10.1038/nrmicro2652
Return to citation in text: [1] -
Diaz, E.; Jiménez, J. I.; Nogales, J. Curr. Opin. Biotechnol. 2013, 24, 431–442. doi:10.1016/j.copbio.2012.10.010
Return to citation in text: [1] -
Dieks, H.; Senge, M. O.; Kirste, B.; Kurreck, H. J. Org. Chem. 1997, 62, 8666–8680. doi:10.1021/jo970752k
Return to citation in text: [1] [2] -
Sipyagin, A. M.; Bateman, C. P.; Tan, Y.-T.; Thrasher, J. S. J. Fluorine Chem. 2001, 112, 287–295. doi:10.1016/s0022-1139(01)00514-0
Return to citation in text: [1] -
Boros, E. E.; Kaldor, I.; Turnbull, P. S. J. Heterocycl. Chem. 2011, 48, 733–736. doi:10.1002/jhet.571
Return to citation in text: [1] -
Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913
Return to citation in text: [1] -
Dunning, T. H., Jr. J. Chem. Phys. 1989, 90, 1007–1023. doi:10.1063/1.456153
Return to citation in text: [1] -
Kendall, R. A.; Dunning, T. H., Jr.; Harrison, R. J. J. Chem. Phys. 1992, 96, 6796–6806. doi:10.1063/1.462569
Return to citation in text: [1] -
Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013.
Return to citation in text: [1] -
Dolbier, W. R., Jr.; Zheng, Z. J. Org. Chem. 2009, 74, 5626–5628. doi:10.1021/jo9007699
Return to citation in text: [1] -
Seltzer, S.; Stevens, K. D. J. Org. Chem. 1968, 33, 2708–2711. doi:10.1021/jo01271a020
Return to citation in text: [1] -
Zhang, Z.; Peng, Z.-W.; Hao, M.-F.; Gao, J.-G. Synlett 2010, 2895–2898. doi:10.1055/s-0030-1259030
Return to citation in text: [1] -
Brel, V. K. Synthesis 2006, 339–343. doi:10.1055/s-2005-918508
Return to citation in text: [1]
| 24. | Dunning, T. H., Jr. J. Chem. Phys. 1989, 90, 1007–1023. doi:10.1063/1.456153 |
| 25. | Kendall, R. A.; Dunning, T. H., Jr.; Harrison, R. J. J. Chem. Phys. 1992, 96, 6796–6806. doi:10.1063/1.462569 |
| 1. | Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2013. doi:10.1002/9783527651351 |
| 2. | Swallow, S. Fluorine-Containing Pharmaceuticals. In Fluorine in Pharmaceutical and Medicinal Chemistry; Gouverneur, V.; Müller, K., Eds.; Molecular Medicine and Medicinal Chemistry, Vol. 6; Imperial College Press: London, 2012; pp 141–174. doi:10.1142/9781848166363_0005 |
| 3. | Purser, S.; Moore, P. R.; Swallow, S.; Gouverneur, V. Chem. Soc. Rev. 2008, 37, 320–330. doi:10.1039/b610213c |
| 20. | Dieks, H.; Senge, M. O.; Kirste, B.; Kurreck, H. J. Org. Chem. 1997, 62, 8666–8680. doi:10.1021/jo970752k |
| 7. | Hansch, C.; Leo, A.; Unger, S. H.; Kim, K. H.; Nikaitani, D.; Lien, E. J. J. Med. Chem. 1973, 16, 1207–1216. doi:10.1021/jm00269a003 |
| 22. | Boros, E. E.; Kaldor, I.; Turnbull, P. S. J. Heterocycl. Chem. 2011, 48, 733–736. doi:10.1002/jhet.571 |
| 20. | Dieks, H.; Senge, M. O.; Kirste, B.; Kurreck, H. J. Org. Chem. 1997, 62, 8666–8680. doi:10.1021/jo970752k |
| 4. | Altomonte, S.; Zanda, M. J. Fluorine Chem. 2012, 143, 57–93. doi:10.1016/j.jfluchem.2012.06.030 |
| 5. | Savoie, P. R.; Welch, J. T. Chem. Rev. 2015, 115, 1130–1190. doi:10.1021/cr500336u |
| 21. | Sipyagin, A. M.; Bateman, C. P.; Tan, Y.-T.; Thrasher, J. S. J. Fluorine Chem. 2001, 112, 287–295. doi:10.1016/s0022-1139(01)00514-0 |
| 15. | Vida, N.; Pastýříková, T.; Klepetářová, B.; Beier, P. J. Org. Chem. 2014, 79, 8906–8911. doi:10.1021/jo501562z |
| 15. | Vida, N.; Pastýříková, T.; Klepetářová, B.; Beier, P. J. Org. Chem. 2014, 79, 8906–8911. doi:10.1021/jo501562z |
| 29. | Zhang, Z.; Peng, Z.-W.; Hao, M.-F.; Gao, J.-G. Synlett 2010, 2895–2898. doi:10.1055/s-0030-1259030 |
| 13. | Case, J. R.; Ray, N. H.; Roberts, H. L. J. Chem. Soc. 1961, 2066–2070. doi:10.1039/JR9610002066 |
| 14. | Aït-Mohand, S.; Dolbier, W. R., Jr. Org. Lett. 2002, 4, 3013–3015. doi:10.1021/ol026483o |
| 16. | Que, L., Jr.; Ho, R. Y. N. Chem. Rev. 1996, 96, 2607–2624. doi:10.1021/cr960039f |
| 17. | Ullrich, R.; Hofrichter, M. Cell. Mol. Life Sci. 2007, 64, 271–293. doi:10.1007/s00018-007-6362-1 |
| 18. | Fuchs, G.; Boll, M.; Heider, J. Nat. Rev. Microbiol. 2011, 9, 803–816. doi:10.1038/nrmicro2652 |
| 19. | Diaz, E.; Jiménez, J. I.; Nogales, J. Curr. Opin. Biotechnol. 2013, 24, 431–442. doi:10.1016/j.copbio.2012.10.010 |
| 10. | Lim, D. S.; Choi, J. S.; Pak, C. S.; Welch, J. T. J. Pestic. Sci. (Tokyo, Jpn.) 2007, 32, 255–259. doi:10.1584/jpestics.G06-50 |
| 11. | Welch, J. T.; Lim, D. S. Bioorg. Med. Chem. 2007, 15, 6659–6666. doi:10.1016/j.bmc.2007.08.012 |
| 12. | Wipf, P.; Mo, T.; Geib, S. J.; Caridha, D.; Dow, G. S.; Gerena, L.; Roncal, N.; Milner, E. E. Org. Biomol. Chem. 2009, 7, 4163–4165. doi:10.1039/b911483a |
| 27. | Dolbier, W. R., Jr.; Zheng, Z. J. Org. Chem. 2009, 74, 5626–5628. doi:10.1021/jo9007699 |
| 9. | Crowley, P. J.; Mitchell, G.; Salmon, R.; Worthington, P. A. Chimia 2004, 58, 138–142. doi:10.2533/000942904777678172 |
| 15. | Vida, N.; Pastýříková, T.; Klepetářová, B.; Beier, P. J. Org. Chem. 2014, 79, 8906–8911. doi:10.1021/jo501562z |
| 28. | Seltzer, S.; Stevens, K. D. J. Org. Chem. 1968, 33, 2708–2711. doi:10.1021/jo01271a020 |
© 2016 Vida et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)