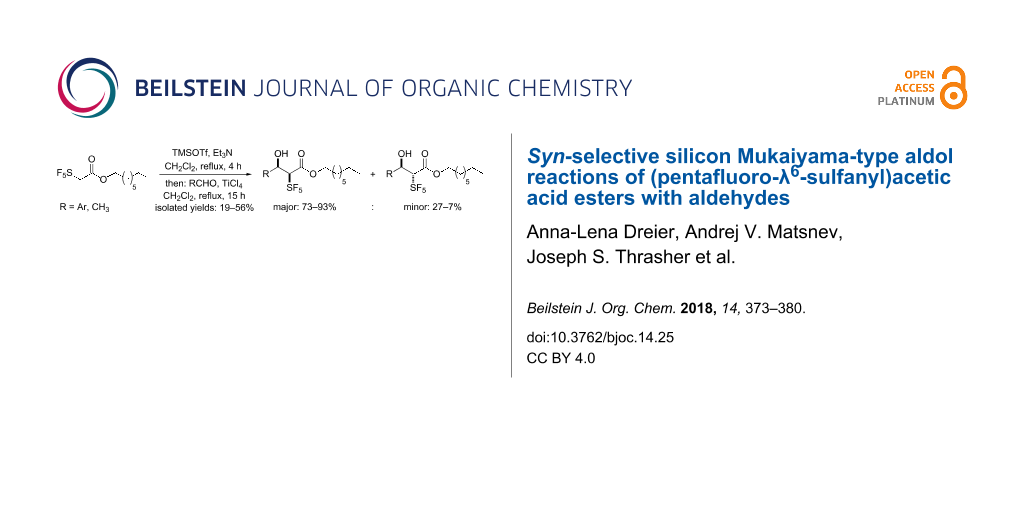
Abstract
Aldol reactions belong to the most frequently used C–C bond forming transformations utilized particularly for the construction of complex structures. The selectivity of these reactions depends on the geometry of the intermediate enolates. Here, we have reacted octyl pentafluoro-λ6-sulfanylacetate with substituted benzaldehydes and acetaldehyde under the conditions of the silicon-mediated Mukaiyama aldol reaction. The transformations proceeded with high diastereoselectivity. In case of benzaldehydes with electron-withdrawing substituents in the para-position, syn-α-SF5-β-hydroxyalkanoic acid esters were produced. The reaction was also successful with meta-substituted benzaldehydes and o-fluorobenzaldehyde. In contrast, p-methyl-, p-methoxy-, and p-ethoxybenzaldehydes led selectively to aldol condensation products with (E)-configured double bonds in 30–40% yields. In preliminary experiments with an SF5-substituted acetic acid morpholide and p-nitrobenzaldehyde, a low amount of an aldol product was formed under similar conditions.

Graphical Abstract
Introduction
The classical acid- or base-catalyzed directed cross aldol reaction of an aldehyde and an enolizable second carbonyl compound is one of the most powerful and reliable carbon–carbon bond-forming transformations in organic synthesis applied most successfully for the construction of natural products and their analogs [1,2]. Later this type of reaction was extended to enolized carboxylic acid derivatives, particularly to silylated ketene acetals, as reaction partners for carbonyl active compounds [3-5]. Mild and highly selective reaction conditions could be developed in parallel with the progress in understanding the mechanism of these transformations [6-9]. Also, aldol reactions with fluorinated substrates, particularly with trifluoromethyl-containing ones, were investigated [10-13].
In recent years, the pentafluoro-λ6-sulfanyl (SF5) substituent has come into the focus of chemists because of the remarkable effects of this substituent on the physical and chemical properties of compounds, which are important for agricultural and medicinal chemistry as well as for materials sciences. While aromatic [14,15] and heteroaromatic [16,17] SF5 compounds have become readily available and many applications have been described [18,19], the chemistry of aliphatic analogs is still underdeveloped [20]. Generally, the incorporation of SF5 groups into aliphatic positions is based on radical addition of SF5X (X = Cl, Br, SF5) across π-bonds. The unconventional conditions usually required were overcome by Dolbier’s elegant triethylborane initiation [21]. Recently, the radical arylation of a SF5-substituted alkene was realized in order to gain access to SF5-containing dihydrobenzofurans and indolines [22]. There are not many transformations of aliphatic SF5 compounds described in the literature. Among them are the preparation and derivatization of SF5-aldehydes [23], Diels–Alder reactions [24-26], the “click reaction” of SF5-acetylenes with azides to form triazoles [27], and 1,3-dipolar cycloadditions of azomethine ylides with pentafluoro-λ6-sulfanyl-substituted acrylic esters and amides [28].
A couple of years ago, we became interested in SF5-substituted ester enolates as reaction intermediates. Thus, in 2016 we reported a highly anti-selective aldol addition of SF5-substituted acetic ester-based boron enolates to aromatic and aliphatic aldehydes to form anti-2-pentafluoro-λ6-sulfanyl-3-hydroxyalkyl-carboxylic acid esters [29]. Quite similar results were published independently by Carreira et al. a couple of weeks before us [30].
Recently, we discovered that silylated enolates can be formed as mixtures of (E)- and (Z)-isomers from SF5-substituted acetic acid esters of aliphatic terminal allylic alcohols at low temperature. At slightly elevated temperature, the latter diastereomers are transformed to γ,δ-unsaturated α-pentafluoro-λ6-sulfanyl alkanoic acids in an Ireland–Claisen rearrangement [31]. Under slightly modified conditions, this type of rearrangement was also successful for SF5-substituted acetic esters of cinnamyl alcohols [32].
Herein we describe our results [33] of highly syn-diastereoselective silicon Mukaiyama-type aldol reactions of SF5-acetic acid esters with different aldehydes. While our research was well underway, Ponomarenko and Röschenthaler et al. reported similar Ti(IV)-mediated aldol reactions proceeding via titanium enolates and some succeeding or competing reactions of intermediates or products [34].
Results and Discussion
As a model for our investigations we chose the reaction of the less volatile octyl 2-(pentafluoro-λ6-sulfanyl)acetate (1) with p-nitrobenzaldehyde. Analogously to a protocol used by Ishihara et al. [35] for an Evans aldol reaction of trifluoropropanoic amides, we refluxed 1 equiv of ester 1 with 1.5 equiv trimethylsilyl trifluoromethanesulfonate (TMSOTf) and 1.5 equiv triethylamine (Et3N) in dichloromethane for 4 hours. Then the mixture was cooled down to 0 °C and 1 equiv of p-nitrobenzaldehyde and 0.3 equiv of TiCl4 were added under stirring. Stirring at room temperature was continued for 15 hours. Then the reaction was quenched by the addition of ice-water. After work-up 22% of the aldol addition products were formed in a syn/anti-ratio of 97:3 as determined by 19F NMR spectroscopy (Scheme 1). Subsequently, the reaction conditions were optimized (Table 1).
![[1860-5397-14-25-i1]](/bjoc/content/inline/1860-5397-14-25-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Silicon-mediated Mukaiyama-type aldol reaction of octyl 2-(pentafluoro-λ6-sulfanyl)acetate (1) with p-nitrobenzaldehyde.
Scheme 1: Silicon-mediated Mukaiyama-type aldol reaction of octyl 2-(pentafluoro-λ6-sulfanyl)acetate (1) with ...
Table 1: Optimization of the reaction conditions for the silicon Mukaiyama-type aldol reaction of ester 1 with p-nitrobenzaldehyde.
| Entry |
TMSOTf
[equiv] |
Et3N
[equiv] |
Lewis acid
[equiv] |
Temp. |
Time
[h] |
Yield 2 + 3 [%]a
(2:3-ratio) |
| 1 | 1.5 | 1.5 | 0.3 TiCl4 | rt | 15 | 22 (93:7) |
| 2 | 1.5 | 1.5 | 0.3 TiCl4 | reflux | 15 | 53 (93:7) |
| 3 | 1.5 | 1.5 | 0.3 TiCl4 | reflux | 3 days | 40 (78:22) |
| 4 | 1.2 | 1.5 | 0.3 TiCl4 | reflux | 15 | 61 (73:27) |
| 5 | 1.5 | 3.0 | 0.3 TiCl4 | reflux | 15 | 0 |
| 6 | 1.5 | 1.5 | 1.0 TiCl4 | reflux | 15 | 67 (81:19) |
| 7 | 1.5 | 1.5 | 0.3 BF3·OEt2 | reflux | 15 | 0 |
| 8 | 1.5 | 1.5 | 1.0 BF3·OEt2 | reflux | 15 | 0 |
| 9 | 1.5 | 1.5 | 1.3 BF3·OEt2 | reflux | 15 | 0 |
aDetermined by 19F NMR spectroscopy of the crude product mixture.
Elevation of the reaction temperature (15 h reflux) led to an increase of the yield, while the syn/anti-ratio was not changed (Table 1, entry 2). Elongation of the reaction time resulted in the formation of more side products, drop of aldol products’ yield and selectivity (Table 1, entry 3). A lower amount of TMSOTf (1.2 equiv) led to increased yield but lower selectivity (Table 1, entry 4). Application of excess Et3N delivered a complex mixture of products including fluorine-free ones. Only traces of the aldol products were detected (Table 1, entry 5). Increasing the amount of TiCl4 to 1.0 equiv resulted in an increased yield (67%), but lower selectivity (81:19, Table 1, entry 6). All attempts to catalyze the reaction with BF3·OEt2 as a Lewis acid failed. Aldol products were not found (Table 1, entries 7–9). Thus, the conditions of entry 2 presented the best compromise regarding the yield and selectivity. Therefore, these conditions were used for the reactions of other aromatic and aliphatic aldehydes (Table 2).
Table 2: Results of reactions of ester 1 with different aldehydes.
![[Graphic 1]](/bjoc/content/inline/1860-5397-14-25-i7.svg?max-width=637&scale=1.0)
|
||||
| Entry | Compounds | R | Yield 2 + 3 [%]a | 2 : 3 ratiob |
| 1 | a | p-NO2-C6H4 | 53 (40) | 93:7 (99:1) |
| 2 | b | C6H5 | 44 (30) | 86:14 (93:7) |
| 3 | c | p-F-C6H4 | 44 (37) | 86:14 (91:9) |
| 4 | d | p-Cl-C6H4 | 38 (19) | 81:19 (87:13) |
| 5 | e | p-Br-C6H4 | 39 (19) | 83:17 (86:14) |
| 6 | f | p-SF5-C6H4 | 42 (22) | 95:5 (95:5) |
| 7 | g | p-CH3-C6H4 | 0 | – |
| 8 | h | p-CH3O-C6H4 | 0 | – |
| 9 | i | p-C2H5O-C6H4 | 0 | – |
| 10 | j | m-NO2-C6H4 | 61 (44) | 90:10 (90:10) |
| 11 | k | m-CH3-C6H4 | 57 (41) | 73:27 (84:16) |
| 12 | l | m-CH3O-C6H4 | 35 (26) | 88:12 (99:1) |
| 13 | m | o-F-C6H4 | 69 (56) | 83:17 (90:10) |
| 14 | n | o-Br-C6H4 | 0 | – |
| 15 | o | 2,6-Cl2-C6H3 | 0 | – |
| 16 | p | 2,4-(NO2)2-C6H3 | 0 | – |
| 17 | q | CH3 | 58 (37) | 87:13 (89:11) |
| 18 | r | cyclo-C6H11 | 0 | – |
| 19 | s | CH2=CH | 0 | – |
| 20 | t | CH3-CH=CH | 0 | – |
aCombined yield determined by 19F NMR spectroscopy of the crude mixture, isolated yield after repeated chromatography in parentheses; bsyn/anti-ratio of the crude product, ratio after chromatographic purification in parentheses determined by 19F NMR spectroscopy.
Table 2 shows that benzaldehyde and five of its derivatives substituted with electron withdrawing groups in para-position gave the desired aldol addition products in fair yields and with diastereoselectivities between 81:19 and 95:5 in favor of the syn-products (Table 2, entries 1–6). In contrast, from the reactions of 1 with benzaldehydes bearing electron-donating substituents (methyl-, methoxy-, ethoxy-) no aldol addition products could be isolated (Table 2, entries 7–9), but the aldol condensation products 4g–i were isolated in fair yields (see below). On the other hand, meta-substituted benzaldehydes gave mainly the syn-aldolates 2j–l and small amounts of the anti-isomers 3j–l regardless of the electronic nature of the aryl substituent (Table 2, entries 10–12). 2-Fluorobenzaldehyde gave the highest yield (69%) and 83:17 diastereoselectivity (Table 2, entry 13), while 2-bromo-, 2,6-dichloro- and 2,4-dinitrobenzaldehydes failed to give any aldol products (Table 2, entries 14–16). Besides the starting materials, only minor amounts of SF5-containing side products of unknown structure were detected in the 19F NMR spectra of the crude product mixtures. Among the saturated and unsaturated aliphatic aldehydes, only acetaldehyde gave the expected aldolates (58% yield, ratio 87:13, Table 2, entry 17), while cyclohexane carbaldehyde, acrolein, and crotonaldehyde did not give any aldol products (Table 2, entries 18–20).
During the aldol addition, two stereocenters were formed giving either the syn-2 or the anti-isomers 3. In all reactions one of the diastereomers was formed in large excess. In most cases, this isomer was isolated in an almost pure form after repeated chromatography, which explains the low isolated yields.
Unfortunately, all products are oils and could not be crystallized. Thus, in order to determine the relative stereochemistry, we analyzed the vicinal coupling constants of the protons attached to the stereocenters. In the syn-isomers 2 these protons are anti to each other, while in the anti-isomers 3 these protons are in a syn-arrangement (Figure 1). Such an arrangement was found in the crystalline state of an anti-aldol product we obtained as a result of a boron-mediated aldol reaction of the benzyl ester analog of compound 1 with benzaldehyde [29]. This product and a couple of its aryl-substituted analogs exhibited coupling constants of 2.4–4.0 Hz. The products 3 produced as minor compounds in the present study had coupling constants between 3.3 and 4.0 Hz, which is typical for a gauche arrangement of the coupling nuclei according to the Karplus equation. Also the other NMR data agree with those found for authentic anti-aldol addition products. Thus, the minor products of the silicon Mukaiyama aldol reactions are the anti-isomers 3.
![[1860-5397-14-25-1]](/bjoc/content/figures/1860-5397-14-25-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Newman projections of the syn- and the anti-diastereomeric aldol addition products.
Figure 1: Newman projections of the syn- and the anti-diastereomeric aldol addition products.
On the other hand, the vicinal coupling constants of the major products were found between 8.5 Hz for the p-nitro derivative 2a to about 9.4 Hz for derivatives 2b–e with less electron-withdrawing substituents in para- and meta-positions, showing that these protons are in anti-position to each other as shown for compounds 2 in Figure 1. Thus, the major products are syn-isomers. Almost identical coupling constants and chemical shifts were also found for the aldol addition products of methyl SF5-acetate with benzaldehyde, p-nitro-, and p-methoxybenzaldehyde as described recently by Ponomarenko and Röschenthaler et al. [34].
Considering our earlier results [31] on TMSOTf-mediated Claisen-type rearrangements of SF5-acetates of allyl alcohols, we favor the initial formation of (Z)-enolates (ketene silylacetals) 5 in the aldol reactions (Scheme 2).
![[1860-5397-14-25-i2]](/bjoc/content/inline/1860-5397-14-25-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Mechanism of the formation of aldol addition products.
Scheme 2: Mechanism of the formation of aldol addition products.
From this enolate, two transition states A and B can be formed for the aldol reactions. B should be less favored due to the steric (and may be also electronic) repulsion of the aryl and the SF5 groups. Consequently, the syn-products resulting from transition state A are the major products of the aldol addition reactions.
As mentioned above, aldol addition products were not isolated from the reaction of 1 with electron rich p-methyl-, p-methoxy-, and p-ethoxybenzaldehydes. Here, aldol condensation products 4g–i were obtained as single isomers after chromatography (Scheme 3).
![[1860-5397-14-25-i3]](/bjoc/content/inline/1860-5397-14-25-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Formation of (E)-configured aldol condensation products.
Scheme 3: Formation of (E)-configured aldol condensation products.
In order to ascertain the configuration of our products, we performed a heteronuclear Overhauser effect (1H,19F-correlation spectrum (gHOESY), see Supporting Information File 1 for a copy of the spectrum). From this spectrum it becomes clear that the four equatorial fluorine atoms do interact with the vinylic proton. Thus, these atoms must be located in spatial proximity. This is possible only in the (E)-configuration of the double bond. A second proof for this configuration is the signal of the vinylic proton at δ = 7.41 ppm, which is a singlet. This was also found in the spectra of the analogous methyl esters, while for the corresponding (Z)-products a multiplet was identified, which is formed by 4JH,F coupling with the four equatorial fluorine atoms of the SF5 group [34].
The formation of the condensation products might occur via two alternative mechanisms. Both variants depend on the initial formation of aldol addition products. These products could undergo acid catalyzed dehydration during aqueous work-up. Due to the presence of Lewis acids, which will be hydrolyzed, water would most probably eliminate via an intermediate benzyl cation, which would be stabilized by the electron-donating substituents in p-position. Consequently, the formation of a mixture of E/Z-isomers would be expected. Therefore, we were in favor of an alternative mechanism involving elimination subsequent to the addition step. According to Denmark’s mechanism [5] for the silicon Mukaiyama aldol reaction (see above), the nucleophilic attack of the silicon enolate (in our case the ketene silylacetal) at the TiCl4-activated aldehyde results in the formation of intermediate 6. Under the influence of the electron-donating substituent, the elimination of titanium oxide dichloride (Ti(O)Cl2) is favored under liberation of a chloride. For the formed oxonium ion, two conformers A and B are possible due to free rotation around the single bond neighboring the SF5 group and the former benzylic carbon atom (Scheme 4).
![[1860-5397-14-25-i4]](/bjoc/content/inline/1860-5397-14-25-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Anticipated mechanism of formation of aldol condensation products.
Scheme 4: Anticipated mechanism of formation of aldol condensation products.
Due to the possible repulsive interaction of the SF5 group with the quinoid ring in conformer A, conformer B should be favored. Assisted by the chloride, this species is deprotonated forming the condensation product 4h with an (E)-configured double bond. Obviously, this formal dehydration is possible only in the presence of electron-donating para-substituents in the benzaldehyde. In order to evaluate this assumption, we performed the following NMR experiment: the ester 1 was dissolved in CD2Cl2 and TMSOTf and Et3N were successively added at room temperature. Then the mixture was refluxed for 4 hours, cooled down to ambient temperature, and p-anisaldehyde and TiCl4 were added. This mixture was heated to 40 °C in a sealed tube overnight and directly investigated by 1H and 19F NMR spectroscopy showing the formation of product 4h. Thus, a concerted mechanism seems to be responsible for the exclusive formation of the (E)-configured products.
Finally, we attempted to incorporate SF5-substituted acetamides into aldol additions with the intension of applying amides in Evans-type substrate directed asymmetric C–C-bond forming reactions. Therefore, 2-(pentafluoro-λ6-sulfanyl)acetic morpholide (8) was prepared by reaction of SF5-CH2C(O)Cl (7) [36,37] with morpholine (Scheme 5).
![[1860-5397-14-25-i5]](/bjoc/content/inline/1860-5397-14-25-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of SF5-substituted acetmorpholide 8.
Scheme 5: Synthesis of SF5-substituted acetmorpholide 8.
According to the general protocol, the morpholide 8 was treated with TMSOTf and Et3N and refluxed in CH2Cl2 for 4 hours. Then this mixture was treated with p-nitrobenzaldehyde and TiCl4 and refluxed in CH2Cl2 in a sealed tube for 15 hours. However, after aqueous work-up, the starting amide 8 and p-nitrobenzaldehyde were mainly recovered. A new SF5-containing compound was detected (19F NMR) in the crude product mixture, but we were unable to isolate this product. The following 19F NMR data were found δ = 57.7 ppm (dm, 2JF,F = 147.6 Hz, 4F) and δ = 74.7 ppm (qn, 2JF,F = 147.6 Hz, 1F), which are different from the starting material and from the corresponding carboxylic acid [36]. Therefore, we repeated the reaction in an NMR tube and treated the morpholide 8 with TMSOTf and Et3N in CD2Cl2 at room temperature. After a short period of time, the 1H and 19F NMR spectra showed the exclusive formation of the (Z)-ketene aminal. Signals of 8 were not found any more (Scheme 6).
![[1860-5397-14-25-i6]](/bjoc/content/inline/1860-5397-14-25-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Intermediate formation of the (Z)-ketene aminal from morpholide 8 with TMSOTf/ Et3N and subsequent transformation to an aldol addition product 9 with p-nitrobenzaldehyde.
Scheme 6: Intermediate formation of the (Z)-ketene aminal from morpholide 8 with TMSOTf/ Et3N and subsequent ...
The structure of this intermediate became evident from its NMR data (see Supporting Information File 1). In particular a quintet (3JH,F = 7.0 Hz) at δ = 5.15 ppm in the 1H NMR spectrum is indicative of a vinylic proton. A quintet (2JC,F = 19.4 Hz) at δ = 106.1 ppm in the 13C NMR spectrum can be assigned to the SF5-substituted carbon atom, and for the second carbon atom of the double bond a quintet (3JC,F = 4.6 Hz) appears at δ = 156.9 ppm. From the poorly resolved multiplets at 91.6 ppm and 74.1 ppm in the 19F NMR spectrum one can reason that the SF5 group is attached to a double bond. Its (Z)-configuration was deduced from a 1H,19F-correlation spectrum (gHOESY) showing an interaction of the equatorial fluorine atoms of the SF5 group and protons of the TMS group (see Supporting Information File 1 for a copy of the spectrum). Consequently, these two substituents must be located at the same side of the double bond.
Subsequently, the reaction mixture was treated with p-nitrobenzaldehyde and TiCl4 and allowed to remain at room temperature for 3 days while new peaks appeared in NMR spectra. Besides signals of p-nitrobenzaldehyde, triethylamine hydrochloride, and several silicon compounds, the aforementioned additional signals were found. From two new doublets for two protons each of a 1,4-disubstituted benzene ring (δ = 8.32 and δ = 7.96), a multiplet for one proton at δ = 4.62 ppm, a broad singlet of one proton at δ = 6.29 ppm, and the two multiplets of the morpholine ring for four protons each at δ = 3.37 ppm and δ = 3.28 ppm, one can reason the formation of an aldol addition product 9. This assignment is supported by a multiplet for one axial fluorine atom at δ = 75.39 ppm (J = 147 Hz) and a doublet (of multiplets) for four fluorine atoms at δ = 57.6 ppm (J = 147 Hz) in the 19F NMR spectra (overlapping with a doublet of another compound). Unfortunately, after work-up, we have not yet been able to isolate this product. Starting amide 8 was a major component of the crude reaction product.
In summary, the formation of the intermediate ketene aminal occurred very fast and was highly (Z)-selective. Unfortunately, the aldol product, which was formed after treatment with p-nitrobenzaldehyde, could not yet be isolated. Anyway, the preliminary results using morpholide 8 are promising. We expect that optimization of the reaction conditions and application of enantiopure Lewis acids or SF5-substituted acetamides bearing a chiral auxiliary will result in asymmetric aldol addition reactions in the future.
Conclusion
From octyl SF5-acetate and TMSOTf/Et3N, a (Z)-enolate (ketene silylacetal) was preferentially formed as has already been shown in our earlier investigations. The C–C bond forming step proceeded preferably via a transition state with a syn-arrangement of the SF5 and OTMS groups resulting in the formation of syn-aldol products in case of aldehydes with electron-withdrawing substituents in the para-position or any substituents in the meta-position. In contrast, aldols derived from aldehydes with electron-donating substituents in the para-position were not stable under the reaction conditions. A formal Lewis acid (TiCl4)-assisted elimination of water produced the (E)-configured aldol condensation products. Preliminary results of formation of an aldol addition product from the reaction of an SF5-substituted acetmorpholide and p-nitrobenzaldehyde are promising, and successful asymmetric reactions may be expected.
Supporting Information
| Supporting Information File 1: General procedure, synthesis of the aldol products, spectroscopic data, and copies of 1H, 13C, and 19F NMR spectra. | ||
| Format: PDF | Size: 7.5 MB | Download |
References
-
Mukaiyama, T. Org. React. 1982, 28, 203–331. doi:10.1002/0471264180.or028.03
Return to citation in text: [1] -
Mahrwald, R., Ed. Modern Aldol Reactions; Wiley-VCH: Weinheim, 2004; Vol. 1 and 2.
Return to citation in text: [1] -
Saigo, K.; Osaki, M.; Mukaiyama, T. Chem. Lett. 1975, 4, 989–990. doi:10.1246/cl.1975.989
Return to citation in text: [1] -
Reetz, M. T.; Kunisch, F.; Heitmann, P. Tetrahedron Lett. 1986, 27, 4721–4724. doi:10.1016/S0040-4039(00)85047-9
Return to citation in text: [1] -
Denmark, S. E.; Beutner, G. L.; Wynn, T.; Eastgate, M. D. J. Am. Chem. Soc. 2005, 127, 3774–3789. doi:10.1021/ja047339w
Return to citation in text: [1] [2] -
Gawronski, J.; Wascinska, N.; Gajewy, J. Chem. Rev. 2008, 108, 5227–5252. doi:10.1021/cr800421c
Return to citation in text: [1] -
Beutner, G. L.; Denmark, S. E. Angew. Chem., Int. Ed. 2013, 52, 9086–9096. doi:10.1002/anie.201302084
Return to citation in text: [1] -
Kann, S. B. J.; Ng, K. K.-H.; Peterson, I. Angew. Chem., Int. Ed. 2013, 52, 9097–9108. doi:10.1002/anie.201303914
Return to citation in text: [1] -
Matsuo, J.-i.; Murakami, M. Angew. Chem., Int. Ed. 2013, 52, 9109–9118. doi:10.1002/anie.201303192
Return to citation in text: [1] -
Lefebvre, O.; Brigaud, T.; Portella, C. J. Org. Chem. 2001, 66, 1941–1946. doi:10.1021/jo001549j
Return to citation in text: [1] -
Sato, K.; Sekiguchi, T.; Ishihara, T.; Konno, T.; Yamanaka, H. Chem. Lett. 2004, 33, 154–155. doi:10.1246/cl.2004.154
Return to citation in text: [1] -
Itoh, Y.; Yamanaka, M.; Mikami, K. J. Am. Chem. Soc. 2004, 126, 13174–13175. doi:10.1021/ja046518a
Return to citation in text: [1] -
Ramachandran, P. V.; Parthasarathy, G.; Gagare, P. D. Org. Lett. 2010, 12, 4474–4477. doi:10.1021/ol1016178
Return to citation in text: [1] -
Umemoto, T.; Garrick, L. M.; Saito, N. Beilstein J. Org. Chem. 2012, 8, 461–471. doi:10.3762/bjoc.8.53
Return to citation in text: [1] -
Kirsch, P. The pentafluorosulfanyl group and related structures. Modern Fluoroorganic Chemistry, 2nd ed.; Wiley-VCH: Weinheim, 2013; pp 179–191. doi:10.1002/9783527651351
Return to citation in text: [1] -
Kosobokov, M.; Cui, B.; Balia, A.; Matsuzaki, K.; Tokunaga, E.; Saito, N.; Shibata, N. Angew. Chem., Int. Ed. 2016, 55, 10781–10785. doi:10.1002/anie.201605008
Return to citation in text: [1] -
Das, P.; Tokunaga, E.; Shibata, N. Tetrahedron Lett. 2017, 58, 4803–4815. doi:10.1016/j.tetlet.2017.11.015
Return to citation in text: [1] -
Altomonte, S.; Zanda, M. J. Fluorine Chem. 2012, 143, 57–93. doi:10.1016/j.jfluchem.2012.06.030
Return to citation in text: [1] -
Savoie, P. R.; Welch, J. T. Chem. Rev. 2015, 115, 1130–1190. doi:10.1021/cr500336u
Return to citation in text: [1] -
Kirsch, P.; Röschenthaler, G.-V. Functional Compounds Based on Hypervalent Sulfur Fluorides. In Current Fluoroorganic Chemistry. New Synthetic Directions, Technologies, Materials, and Biological Applications; Soloshonok, V. A.; Mikami, K.; Yamazaki, T.; Welch, J. T.; Honek, J. F., Eds.; ACS Symposium Series, Vol. 949; American Chemical Society: Washington, DC, 2007; pp 221–243. doi:10.1021/bk-2007-0949.ch013
Return to citation in text: [1] -
Aït-Mohand, S.; Dolbier, W. R., Jr. Org. Lett. 2002, 4, 3013–3015. doi:10.1021/ol026483o
Return to citation in text: [1] -
Desroches, J.; Gilbert, A.; Houle, C.; Paquin, J.-F. Synthesis 2017, 49, 4827–4844. doi:10.1055/s-0036-1589514
Return to citation in text: [1] -
Ngo, S. C.; Lin, J.-H.; Savoie, P. R.; Hines, E. M.; Pugliese, K. M.; Welch, J. T. Eur. J. Org. Chem. 2012, 4902–4905. doi:10.1002/ejoc.201200763
Return to citation in text: [1] -
Dolbier, W. R., Jr.; Mitani, A.; Xu, W.; Ghiviriga, I. Org. Lett. 2006, 8, 5573–5575. doi:10.1021/ol0622662
Return to citation in text: [1] -
Brel, V. K. Synthesis 2006, 339–343. doi:10.1055/s-2005-918508
Return to citation in text: [1] -
Duda, B.; Lentz, D. Org. Biomol. Chem. 2015, 13, 5625–5628. doi:10.1039/C5OB00610D
Return to citation in text: [1] -
Huang, Y.; Gard, G. L.; Shreeve, J. M. Tetrahedron Lett. 2010, 51, 6951–6954. doi:10.1016/j.tetlet.2010.10.149
Return to citation in text: [1] -
Falkowska, E.; Tognetti, V.; Joubert, L.; Jubault, P.; Bouillon, J.-P.; Pannecoucke, X. RSC Adv. 2015, 5, 6864–6868. doi:10.1039/C4RA14075C
Return to citation in text: [1] -
Friese, F. W.; Dreier, A.-L.; Matsnev, A. V.; Daniliuc, C. G.; Thrasher, J. S.; Haufe, G. Org. Lett. 2016, 18, 1012–1015. doi:10.1021/acs.orglett.6b00136
Return to citation in text: [1] [2] -
Joilton, A.; Plancher, J.-M.; Carreira, E. M. Angew. Chem., Int. Ed. 2016, 55, 2113–2117. doi:10.1002/anie.201510380
Return to citation in text: [1] -
Dreier, A.-L.; Matsnev, A. V.; Thrasher, J. S.; Haufe, G. J. Fluorine Chem. 2014, 167, 84–90. doi:10.1016/j.jfluchem.2014.05.006
Return to citation in text: [1] [2] -
Dreier, A.-L.; Beutel, B.; Mück-Lichtenfeld, C.; Matsnev, A. V.; Thrasher, J. S.; Haufe, G. J. Org. Chem. 2017, 82, 1638–1648. doi:10.1021/acs.joc.6b02805
Return to citation in text: [1] -
Dreier, A.-L. Darstellung von α-(Pentafluorsulfanyl)-substituierten Carbonylverbindungen mittels Ireland-Claisen-Umlagerungen sowie Mukaiyama-Aldolreaktionen. Ph.D. Thesis, University of Münster, Münster, Germany, 2015.
Return to citation in text: [1] -
Ponomarenko, M. V.; Grabowsky, S.; Pal, R.; Röschenthaler, G.-V.; Fokin, A. A. J. Org. Chem. 2016, 81, 6783–6791. doi:10.1021/acs.joc.6b00946
Return to citation in text: [1] [2] [3] -
Shimada, T.; Yoshioka, M.; Konno, T.; Ishihara, T. Org. Lett. 2006, 8, 1129–1131. doi:10.1021/ol0531435
Return to citation in text: [1] -
Kleemann, G.; Seppelt, K. Chem. Ber. 1979, 112, 1140–1146. doi:10.1002/cber.19791120409
Return to citation in text: [1] [2] -
Martinez, H.; Zheng, Z.; Dolbier, W. R., Jr. J. Fluorine Chem. 2012, 143, 112–122. doi:10.1016/j.jfluchem.2012.03.010
Return to citation in text: [1]
| 36. | Kleemann, G.; Seppelt, K. Chem. Ber. 1979, 112, 1140–1146. doi:10.1002/cber.19791120409 |
| 1. | Mukaiyama, T. Org. React. 1982, 28, 203–331. doi:10.1002/0471264180.or028.03 |
| 2. | Mahrwald, R., Ed. Modern Aldol Reactions; Wiley-VCH: Weinheim, 2004; Vol. 1 and 2. |
| 14. | Umemoto, T.; Garrick, L. M.; Saito, N. Beilstein J. Org. Chem. 2012, 8, 461–471. doi:10.3762/bjoc.8.53 |
| 15. | Kirsch, P. The pentafluorosulfanyl group and related structures. Modern Fluoroorganic Chemistry, 2nd ed.; Wiley-VCH: Weinheim, 2013; pp 179–191. doi:10.1002/9783527651351 |
| 29. | Friese, F. W.; Dreier, A.-L.; Matsnev, A. V.; Daniliuc, C. G.; Thrasher, J. S.; Haufe, G. Org. Lett. 2016, 18, 1012–1015. doi:10.1021/acs.orglett.6b00136 |
| 10. | Lefebvre, O.; Brigaud, T.; Portella, C. J. Org. Chem. 2001, 66, 1941–1946. doi:10.1021/jo001549j |
| 11. | Sato, K.; Sekiguchi, T.; Ishihara, T.; Konno, T.; Yamanaka, H. Chem. Lett. 2004, 33, 154–155. doi:10.1246/cl.2004.154 |
| 12. | Itoh, Y.; Yamanaka, M.; Mikami, K. J. Am. Chem. Soc. 2004, 126, 13174–13175. doi:10.1021/ja046518a |
| 13. | Ramachandran, P. V.; Parthasarathy, G.; Gagare, P. D. Org. Lett. 2010, 12, 4474–4477. doi:10.1021/ol1016178 |
| 30. | Joilton, A.; Plancher, J.-M.; Carreira, E. M. Angew. Chem., Int. Ed. 2016, 55, 2113–2117. doi:10.1002/anie.201510380 |
| 6. | Gawronski, J.; Wascinska, N.; Gajewy, J. Chem. Rev. 2008, 108, 5227–5252. doi:10.1021/cr800421c |
| 7. | Beutner, G. L.; Denmark, S. E. Angew. Chem., Int. Ed. 2013, 52, 9086–9096. doi:10.1002/anie.201302084 |
| 8. | Kann, S. B. J.; Ng, K. K.-H.; Peterson, I. Angew. Chem., Int. Ed. 2013, 52, 9097–9108. doi:10.1002/anie.201303914 |
| 9. | Matsuo, J.-i.; Murakami, M. Angew. Chem., Int. Ed. 2013, 52, 9109–9118. doi:10.1002/anie.201303192 |
| 27. | Huang, Y.; Gard, G. L.; Shreeve, J. M. Tetrahedron Lett. 2010, 51, 6951–6954. doi:10.1016/j.tetlet.2010.10.149 |
| 3. | Saigo, K.; Osaki, M.; Mukaiyama, T. Chem. Lett. 1975, 4, 989–990. doi:10.1246/cl.1975.989 |
| 4. | Reetz, M. T.; Kunisch, F.; Heitmann, P. Tetrahedron Lett. 1986, 27, 4721–4724. doi:10.1016/S0040-4039(00)85047-9 |
| 5. | Denmark, S. E.; Beutner, G. L.; Wynn, T.; Eastgate, M. D. J. Am. Chem. Soc. 2005, 127, 3774–3789. doi:10.1021/ja047339w |
| 28. | Falkowska, E.; Tognetti, V.; Joubert, L.; Jubault, P.; Bouillon, J.-P.; Pannecoucke, X. RSC Adv. 2015, 5, 6864–6868. doi:10.1039/C4RA14075C |
| 21. | Aït-Mohand, S.; Dolbier, W. R., Jr. Org. Lett. 2002, 4, 3013–3015. doi:10.1021/ol026483o |
| 23. | Ngo, S. C.; Lin, J.-H.; Savoie, P. R.; Hines, E. M.; Pugliese, K. M.; Welch, J. T. Eur. J. Org. Chem. 2012, 4902–4905. doi:10.1002/ejoc.201200763 |
| 20. | Kirsch, P.; Röschenthaler, G.-V. Functional Compounds Based on Hypervalent Sulfur Fluorides. In Current Fluoroorganic Chemistry. New Synthetic Directions, Technologies, Materials, and Biological Applications; Soloshonok, V. A.; Mikami, K.; Yamazaki, T.; Welch, J. T.; Honek, J. F., Eds.; ACS Symposium Series, Vol. 949; American Chemical Society: Washington, DC, 2007; pp 221–243. doi:10.1021/bk-2007-0949.ch013 |
| 24. | Dolbier, W. R., Jr.; Mitani, A.; Xu, W.; Ghiviriga, I. Org. Lett. 2006, 8, 5573–5575. doi:10.1021/ol0622662 |
| 25. | Brel, V. K. Synthesis 2006, 339–343. doi:10.1055/s-2005-918508 |
| 26. | Duda, B.; Lentz, D. Org. Biomol. Chem. 2015, 13, 5625–5628. doi:10.1039/C5OB00610D |
| 18. | Altomonte, S.; Zanda, M. J. Fluorine Chem. 2012, 143, 57–93. doi:10.1016/j.jfluchem.2012.06.030 |
| 19. | Savoie, P. R.; Welch, J. T. Chem. Rev. 2015, 115, 1130–1190. doi:10.1021/cr500336u |
| 16. | Kosobokov, M.; Cui, B.; Balia, A.; Matsuzaki, K.; Tokunaga, E.; Saito, N.; Shibata, N. Angew. Chem., Int. Ed. 2016, 55, 10781–10785. doi:10.1002/anie.201605008 |
| 17. | Das, P.; Tokunaga, E.; Shibata, N. Tetrahedron Lett. 2017, 58, 4803–4815. doi:10.1016/j.tetlet.2017.11.015 |
| 22. | Desroches, J.; Gilbert, A.; Houle, C.; Paquin, J.-F. Synthesis 2017, 49, 4827–4844. doi:10.1055/s-0036-1589514 |
| 33. | Dreier, A.-L. Darstellung von α-(Pentafluorsulfanyl)-substituierten Carbonylverbindungen mittels Ireland-Claisen-Umlagerungen sowie Mukaiyama-Aldolreaktionen. Ph.D. Thesis, University of Münster, Münster, Germany, 2015. |
| 31. | Dreier, A.-L.; Matsnev, A. V.; Thrasher, J. S.; Haufe, G. J. Fluorine Chem. 2014, 167, 84–90. doi:10.1016/j.jfluchem.2014.05.006 |
| 32. | Dreier, A.-L.; Beutel, B.; Mück-Lichtenfeld, C.; Matsnev, A. V.; Thrasher, J. S.; Haufe, G. J. Org. Chem. 2017, 82, 1638–1648. doi:10.1021/acs.joc.6b02805 |
| 5. | Denmark, S. E.; Beutner, G. L.; Wynn, T.; Eastgate, M. D. J. Am. Chem. Soc. 2005, 127, 3774–3789. doi:10.1021/ja047339w |
| 36. | Kleemann, G.; Seppelt, K. Chem. Ber. 1979, 112, 1140–1146. doi:10.1002/cber.19791120409 |
| 37. | Martinez, H.; Zheng, Z.; Dolbier, W. R., Jr. J. Fluorine Chem. 2012, 143, 112–122. doi:10.1016/j.jfluchem.2012.03.010 |
| 31. | Dreier, A.-L.; Matsnev, A. V.; Thrasher, J. S.; Haufe, G. J. Fluorine Chem. 2014, 167, 84–90. doi:10.1016/j.jfluchem.2014.05.006 |
| 34. | Ponomarenko, M. V.; Grabowsky, S.; Pal, R.; Röschenthaler, G.-V.; Fokin, A. A. J. Org. Chem. 2016, 81, 6783–6791. doi:10.1021/acs.joc.6b00946 |
| 29. | Friese, F. W.; Dreier, A.-L.; Matsnev, A. V.; Daniliuc, C. G.; Thrasher, J. S.; Haufe, G. Org. Lett. 2016, 18, 1012–1015. doi:10.1021/acs.orglett.6b00136 |
| 34. | Ponomarenko, M. V.; Grabowsky, S.; Pal, R.; Röschenthaler, G.-V.; Fokin, A. A. J. Org. Chem. 2016, 81, 6783–6791. doi:10.1021/acs.joc.6b00946 |
| 34. | Ponomarenko, M. V.; Grabowsky, S.; Pal, R.; Röschenthaler, G.-V.; Fokin, A. A. J. Org. Chem. 2016, 81, 6783–6791. doi:10.1021/acs.joc.6b00946 |
| 35. | Shimada, T.; Yoshioka, M.; Konno, T.; Ishihara, T. Org. Lett. 2006, 8, 1129–1131. doi:10.1021/ol0531435 |
© 2018 Dreier et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)