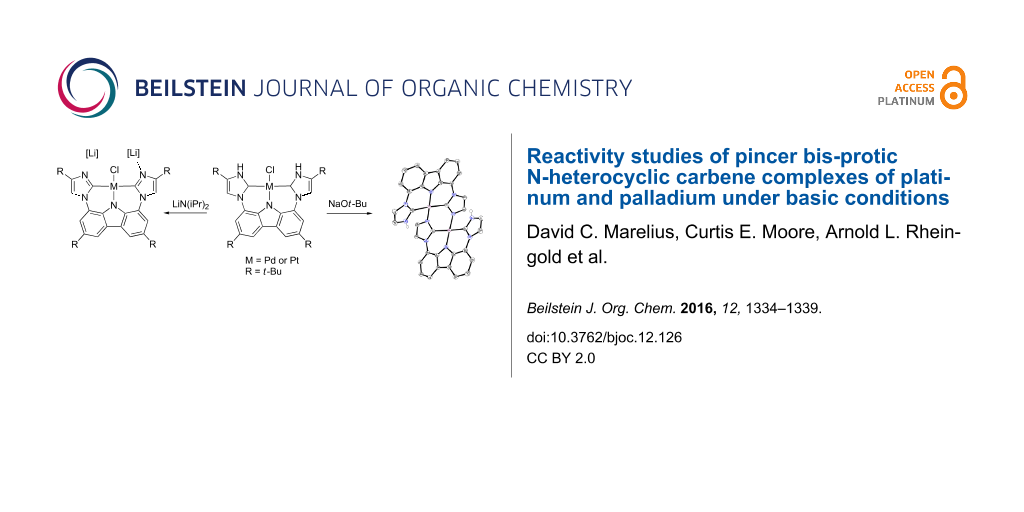
Abstract
Bis-protic N-heterocyclic carbene complexes of platinum and palladium (4) yield dimeric structures 6 when treated with sodium tert-butoxide in CH2Cl2. The use of a more polar solvent (THF) and a strong base (LiN(iPr)2) gave the lithium chloride adducts monobasic complex 7 or analogous dibasic complex 8.

Graphical Abstract
Introduction
N-Heterocyclic carbenes (NHCs) have been extensively researched for a number of purposes since 1991 when Arduengo first isolated free NHCs [1-3]. NHCs as ligands have been known even longer. In 1968, Wanzlick and Öfele separately synthesized mercury(II) and chromium(0) imidazol-2-ylidene complexes [3]. Nearly 50 years of NHC ligand research have demonstrated the importance of the electronic and steric effects that can be modified by altering the alkyl or aryl groups on each nitrogen atom. Less common are protic imidazol-2-ylidene (PNHC) ligands with a hydrogen atom on one or both of the stabilizing nitrogens. The synthesis of PNHC complexes has proven to be a challenge, which has limited studies of their reactivity [4-8].
Protic imidazol-2-ylidene ligands (e.g., 1) have been shown to form an imidazolyl ligand (e.g., 2) after deprotonation with a basic proton-accepting nitrogen (Figure 1). We are unaware of reports on an experimentally determined pKa value of a PNHC imidazolidene complex, but looking at related derivatives, Isobe showed that a 2-palladated pyridine was 3.57 pKa units more basic than pyridine [9,10]. Considering reactions other than simple proton transfer, imidazol-2-yl complexes have recently been used to bind to a second transition metal [11]. Additionally, Cp*Ir complexes from our group [12] demonstrated heterolysis of the H–H bond of H2 and of the C–H bond of acetylene. The same ligand in CpRu complexes 2 and 3 showed heterolysis of dihydrogen [13]. 1 had a much faster ligand exchange rate after ionization as compared to the Cp*Ir analog (ethylene bound in 5 min at rt (CpRu) instead of 16 h at 70 °C (Cp*Ir)). Species 1 could be converted in situ to the hydride and isolated, or generated in situ and used as a transfer hydrogenation catalyst. Interestingly, the ligand substitution rate of ethylene and the heterolysis of dihydrogen was much greater for 3 than for 2. With only a few papers exploring the utility of these imidazol-2-yl complexes, we aim to extend this to our recently reported pincer bis PNHC complexes 4-PdCl and 4-PtCl and their triflato analogs [14]. The design of these complexes was inspired by studies of Kunz et al. on aprotic analogs [15,16].
![[1860-5397-12-126-1]](/bjoc/content/figures/1860-5397-12-126-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Previously reported PNHC complexes of interest for this work, along with the targeted complex 5.
Figure 1: Previously reported PNHC complexes of interest for this work, along with the targeted complex 5.
Results and Discussion
The loss of one NH proton from the bis-PNHC complex 4 could lead to structure 5, a complex concurrently containing a PNHC proton donor and a bond activating imidazol-2-yl unit. In an attempt to form 5, 4-PdCl was dissolved in CD2Cl2, and the solution was saturated with ethylene, followed by the addition of sodium tert-butoxide. After 2 h at room temperature, an NMR spectrum was acquired that showed a new, unsymmetrical species, as expected for 5. Crystals were grown by vapor diffusion of pentanes into benzene and analyzed. Surprisingly, the data showed that the dimer 6-Pd had formed such that the open site was not filled with ethylene, but rather was occupied by an imidazolyl nitrogen from a second complex (Figure 2). The palladium and platinum dimer complexes, 6-Pd and 6-Pt, could be formed by addition of sodium tert-butoxide to the chloride analogs (Scheme 1), and isolated in 50–56% yields.
![[1860-5397-12-126-2]](/bjoc/content/figures/1860-5397-12-126-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Crystal structure of 6-Pd. The atoms of the tert-butyl groups, C–H bonds, and the solvent have been omitted for clarity.
Figure 2: Crystal structure of 6-Pd. The atoms of the tert-butyl groups, C–H bonds, and the solvent have been...
![[1860-5397-12-126-i1]](/bjoc/content/inline/1860-5397-12-126-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Formation of dimers 6-Pd and 6-Pt by addition of NaOt-Bu.
Scheme 1: Formation of dimers 6-Pd and 6-Pt by addition of NaOt-Bu.
The examination of the dimer crystal structure (see Figure 2 for 6-Pd) shows strain in the Pd1–N1’ (and Pd1’–N1) bond. This is due to the metal that remains in the plane defined by the three coordinating atoms of the tridentate ligand (i.e., C1, N3, and C4). The fourth donor atom from the other has to bend out of the plane with the N1’ imidazole ring because of the adjacent sterics of the tert-butyl groups on the imidazole. The strain can be quantified by examining how far the metal is from the N1 (or N1’)-bound imidazole plane (C1–C2–C3–N1–N2 plane and the symmetry-equivalent atoms): for 6-Pd, 1.241 Å and, for 6-Pt, 1.094 Å (Table 1). Further evidence is given by the dihedral angles: Pd1–C1–N1–Pd1’ = −40.4(4)° and 36.5(3)° for 6-Pd and 6-Pt, respectively, C1–Pd1–N1’–C1’ = 72.4(3)° and 67.3(2)° for 6-Pd and 6-Pt, respectively.
Table 1: Key bond lengths (Å) and angles (°) of dimers 6 compared to parent compounds 4.
| 4-PdCl | 6-Pd | 4-PtCl | 6-Pt | |
|---|---|---|---|---|
| M–N1 | – | 2.092(3) | – | 2.079(3) |
| M–N3 | 1.961(3) | 1.962(3) | 1.9627(19) | 1.969(3) |
| M–C4 | 2.006(4), 1.998(4) | 2.034(4) | 2.007(2), 2.014(2) | 2.014(3) |
| M–C1 | – | 1.984(4) | – | 1.996(3) |
| N3–M–X | 178.09(10) | 176.3(1) | 179.85(6) | 175.6(1) |
| C1–M–C4 | 175.43(17) | 167.9(2) | 176.29(9) | 168.3(1) |
| M out planea | – | 1.241 | – | 1.094 |
aThe metal-to-plane distance defined by the five corresponding N-coordinated imidazole atoms; this value would be near zero in the absence of strain.
The NMR results are completely consistent with persistence of the dimers in solution. For monomeric species such as 4-PdCl and 4-PtCl, the NH proton resonance is typically downfield shifted with a chemical shift of ca. 11 ppm, whereas this signal is strongly shifted upfield to 8.03 (6-Pd) or 8.19 ppm (6-Pt). The crystal structures for both 6-Pd and 6-Pt show that the NH is located above the pi system of one imidazole ring of the other half of the dimer, which would be expected to shield the NH and cause a significant upfield chemical shift. Moreover, a ROESY experiment on 6-Pt (Figure S6, Supporting Information File 1) confirms that the NH (N5’, Figure 2) has a through-space interaction with the proton on the imidazole ring (C3, Figure 2), a situation that would not be possible for a monomeric structure.
Attempts to synthesize 5 using sodium alkoxide bases led to the formation of dimer structures 6 with presumed loss of NaCl. Therefore, lithium chloride adducts 7 were targeted because LiCl adduct 3 was isolable yet highly reactive. As demonstrated by NMR spectroscopy, the dissolution of 4-PdCl in a mixture of THF (0.7 mL) and C6D6 (0.1 mL) followed by the addition of one equivalent of LiN(iPr)2 deprotonates one of the PNHC complexes. This gives 7-Pd, without evidence of dimer formation (Scheme 2). The addition of a second equivalent of LiN(iPr)2 deprotonates the second PNHC complex, giving 8-Pd. The 1H NMR spectrum for compound 7-Pt consists of a single NH peak at 10.90 ppm and six aromatic peaks, which all integrate to one proton. The asymmetry is also observed in the 13C NMR spectrum, which consists of 18 peaks between 100 and 170 ppm. As for 8-Pt, the 1H NMR spectrum has no peak where the NH peak typically is located, and in the aromatic region there are three peaks. The 13C NMR spectrum thus consists of 9 peaks between 100 and 170 ppm, showing the reappearance of symmetry.
![[1860-5397-12-126-i2]](/bjoc/content/inline/1860-5397-12-126-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Formation of 7 and 8 by addition of LiN(iPr)2 (1 or 2 equiv) (R = tert-butyl).
Scheme 2: Formation of 7 and 8 by addition of LiN(iPr)2 (1 or 2 equiv) (R = tert-butyl).
15N chemical shift data give structural insight (Table 2), as exemplified by 1–3 [10]. The Δx (difference in 15N shifts for compound x) is near zero for a PNHC (1), maximum for the imidazolyl conjugate base 2, and slightly less for an imidazolyl lithium chloride adduct 3. The δN for the aprotic nitrogen incapable of acid base chemistry (N2) hardly changes, whereas for the protic (N1), the changes depend on its environment. In the following discussion, 1 and 4-PtCl are considered the reference starting material complexes because they are both neutral PNHC species with M–Cl moieties. Their ∆x values are named as ∆ref. For 1, ∆ref = 1.5, whereas for 4-PdCl, ∆ref = −13.1. This difference is likely due to a variety of factors related to the electronics of the nonprotic substituent of the PNHC and/or the ring size of the chelates, which is beyond the scope of this paper. For a given metal center and mono- or bis-NHC framework, we want to diagnose the effects of chemical changes at N1. For this purpose, we introduce the quantity ∆∆ = ∆x − ∆ref to account for the change in Δx that accompanies a chemical change from the reference compound to the new species x. Looking at complex 6-Pt, ∆∆ for the NH nitrogen is only 0.3 ppm and only −4.4 ppm for the NM nitrogen. The ∆∆ values for both nitrogens are relatively unchanged as the carbene character of the ligand is still intact. Interestingly, 6-Pd shows a similar small ∆∆ value for the NH (2.6) but a greater ∆∆ for the NM of 17.4 ppm. This is possibly a result of the differing ring strain in the Pt case (M out of plane 1.094 Å for 6-Pt vs 1.241 for 6-Pd) and/or smaller M–N distance (2.079(3) Å for 6-Pt vs 2.092(3) Å for 6-Pd, Table 1). Turning now to 7-Pt and 8-Pt, we note that the deprotonated nitrogens of 7-Pt and 8-Pt show large values of ∆∆ = 80.5 ppm and 77.8 ppm, respectively. This resembles the results for the CpRu species 2 and 3 (∆∆ = 88.0 ppm and 73.2 ppm, respectively). The values of ∆∆ for the deprotonated nitrogens (that bear a Pt or Pd) in the crystallographically characterized dimers 6-Pt and 6-Pd are much smaller (17.4, −4.4). The large ∆∆ values for the deprotonated nitrogens of 7-Pt and 8-Pt could be due to either structural formulation as a LiCl adduct or free imidazolyl species. However, the dependence of the stability of 7 and 8 on the presence of chloride (see below) argues strongly for a chloride ligand on the central metal. This leads us to assign structures 7-Pt and 8-Pt as LiCl-imidazolyl adducts. All attempts at characterization by crystallography have been unsuccessful due to the very water-sensitive nature of 7 and 8. Therefore, the environment of the lithium and chloride is unknown but it is assumed to be similar to that of 3, which is formed under very similar conditions. The 15N chemical shift data give structural insight that is unavailable by any other means regarding the absence of solid-state structures or meaningfully diagnostic 7Li chemical shifts.
Table 2: 15N chemical shift values (ppm) of complexes in this paper.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-126-i3.svg?max-width=637&scale=1.0)
|
||||
| Complex | N1b | N2c | ∆xd | ∆∆e |
|---|---|---|---|---|
| 1 | −197.1 | −198.6 | 1.5 | 0 (defined) |
| 2 | −110.4 | −199.9 | 89.5 | 88.0 |
| 3 | −122.1 | −196.8 | 74.7 | 73.2 |
| 4-PtCl | −211.0 | −197.9 | −13.1 | 0 (defined) |
| 7-Pt NH | −212.7 | −198.4 | −14.4 | −1.3 |
| 7-Pt Li | −129.9 | −197.3 | 67.4 | 80.5 |
| 8-Pt | −127.4 | −192.0 | 64.7 | 77.8 |
| 6-Pt NH | −209.9 | −197.0 | −12.9 | 0.3 |
| 6-Pt NPd | −215.3 | −197.8 | −17.5 | −4.4 |
| 6-Pd NH | −208.3 | −192.6 | −15.7 | 2.6 |
| 6-Pd NPt | −193.5 | −195.8 | 2.3 | 17.4 |
aDetermined using 1H,15N gHMBC on natural abundance material in THF (0.7 mL) and benzene-d6 (0.1 mL) (4-PtCl, 7, 8), or CD2Cl2 (6). bN1 = either NH or derivative of PNHC. cN2 = aprotic nitrogen incapable of acid base chemistry. d∆x = N1 − N2. e∆∆ = ∆x − ∆ref where ∆ref = −13.1 for type-1 compounds and 1.5 for type-4 compounds.
An indication of the reactivity of imidazolyl complexes was deduced after bubbling H2 through a solution of 7-Pt for 16 h in THF/C6D6, where only a small amount of 4-PtCl was regenerated, likely from adventitious water. The addition of AgOTf did not facilitate H2 heterolysis, but formed the dimer 6-Pt (Figure S7, Supporting Information File 1). Bubbling ethylene through a solution of 7-Pt gave some 4-PtCl, again likely from adventitious water (Figure S8, Supporting Information File 1). The rigorously dried substrate 1-heptene (20 equiv) was added to 8-Pt, where no reaction was observed even after heating at 70 °C for 4 h (Figure S9, Supporting Information File 1).
Unlike imidazolyl complex 3, 7-Pt showed no tendency to lose LiCl. One possible route to a more labile LiOTf adduct was to deprotonate 4-PtOTf, but the action of LiN(iPr)2 yielded mostly the dimer 6-Pt (Figure S10, Supporting Information File 1). Alternatively, the deprotonation of [4-Pt(CH3CN)]+ OTf− by LiN(iPr)2 (1 equiv) gave an analog of 5, where L = CH3CN. However in practice, this typically yielded a number of species (Figure S11, Supporting Information File 1). It appears that the LiCl-imidazolyl adduct complexes 7-Pt and 8-Pt have a much slower ligand exchange rate compared to complex 3. This was expected given the change from ruthenium to platinum [17].
To see if the faster ligand exchange would lead to LiCl loss with palladium, 7-Pd was synthesized. Unfortunately, similar results were observed with the platinum analog where 1-heptene did not react with 7-Pd (which was then converted to 8-Pd by addition of LiN(iPr)2). Then AgOTf was added to 8-Pd, which formed a deprotonated dimer complex. Even with palladium, the loss of the chloride ligand seemed to be too slow.
Conclusion
In conclusion, attempts at forming an imidazolyl complex from 4-MCl using sodium alkoxides led to strained dimers 6. However, 4-MCl could be deprotonated with either 1 or 2 equiv of LiN(iPr)2 to give 7, an intriguing species with one PNHC ligand and one Li-imidazolyl adduct, or 8, a bis imidazolyl complex. Unfortunately, substrates could not displace the chloride ligand without formation of dimer 6, and the deprotonated complexes were water sensitive. The attempts at deprotonating the more labile triflate complex 4-PtOTf led to the formation of dimer 6-Pt. To increase the lability of the chloride ligand, species 4-PdCl and 4-PdOTf were examined but gave dimer 6-Pd. In summary, the reactivity of bis-PNHC complexes 4 and bases appears to be dominated by the formation of the dimeric structures. Studies to reduce dimer formation by various means, such as increasing steric hindrance at the imidazolyl nitrogens, will be reported in due course.
Supporting Information
The Supporting Information contains details on syntheses of 6-Pd and 6-Pt, NMR data for 4-PtCl, 6-Pd, 6-Pt, 7-Pt, and 8-Pt and Figues S6–S12.
| Supporting Information File 1: Experimental information and NMR spectroscopy figures. | ||
| Format: PDF | Size: 1.1 MB | Download |
References
-
Hermann, W. A. Angew. Chem., Int. Ed. 2002, 41, 1290. doi:10.1002/1521-3773(20020415)41:8<1290::AID-ANIE1290>3.0.CO;2-Y
Return to citation in text: [1] -
Díez-González, S.; Marion, N.; Nolan, S. P. Chem. Rev. 2009, 109, 3612. doi:10.1021/cr900074m
Return to citation in text: [1] -
Hopkinson, M. N.; Richter, C.; Schedler, M.; Glorius, F. Nature 2014, 510, 485. doi:10.1038/nature13384
Return to citation in text: [1] [2] -
Kuwata, S.; Ikariya, T. Chem. – Eur. J. 2011, 17, 3542. doi:10.1002/chem.201003296
Return to citation in text: [1] -
Hahn, F. E. ChemCatChem 2013, 5, 419. doi:10.1002/cctc.201200567
Return to citation in text: [1] -
Zhao, B.; Han, Z.; Ding, K. Angew. Chem., Int. Ed. 2013, 52, 4744. doi:10.1002/anie.201204921
Return to citation in text: [1] -
Kuwata, S.; Ikariya, T. Chem. Commun. 2014, 50, 14290. doi:10.1039/C4CC04457F
Return to citation in text: [1] -
Jahnke, M. C.; Hahn, F. E. Coord. Chem. Rev. 2015, 293–294, 95. doi:10.1016/j.ccr.2015.01.014
Return to citation in text: [1] -
Isobe, K.; Kai, E.; Nakamura, Y.; Nishimoto, K.; Miwa, T.; Kawaguchi, S.; Kinoshita, K.; Nakatsu, K. J. Am. Chem. Soc. 1980, 102, 2475. doi:10.1021/ja00527a065
Return to citation in text: [1] -
Isobe, K.; Kawaguchi, S. Heterocycles 1981, 16, 1603. doi:10.3987/R-1981-09-1603
Return to citation in text: [1] [2] -
Flowers, S. E.; Cossairt, B. M. Organometallics 2014, 33, 4341. doi:10.1021/om500592u
Return to citation in text: [1] -
Miranda-Soto, V.; Grotjahn, D. B.; DiPasquale, A. G.; Rheingold, A. L. J. Am. Chem. Soc. 2008, 130, 13200. doi:10.1021/ja804713u
Return to citation in text: [1] -
Miranda-Soto, V.; Grotjahn, D. B.; Cooksy, A. L.; Golen, J. A.; Moore, C. E.; Rheingold, A. L. Angew. Chem., Int. Ed. 2011, 50, 631. doi:10.1002/anie.201005100
Return to citation in text: [1] -
Marelius, D. C.; Darrow, E. H.; Moore, C. E.; Golen, J. A.; Rheingold, A. L.; Grotjahn, D. B. Chem. – Eur. J. 2015, 21, 10988. doi:10.1002/chem.201501945
Return to citation in text: [1] -
Seyboldt, A.; Wucher, B.; Hohnstein, S.; Eichele, K.; Rominger, F.; Törnroos, K. W.; Kunz, D. Organometallics 2015, 34, 2717. doi:10.1021/om500836m
Return to citation in text: [1] -
Moser, M.; Wucher, B.; Kunz, D.; Rominger, F. Organometallics 2007, 26, 1024. doi:10.1021/om060952z
Return to citation in text: [1] -
Helm, L.; Merbach, A. E. Chem. Rev. 2005, 105, 1923. doi:10.1021/cr030726o
Return to citation in text: [1]
| 1. | Hermann, W. A. Angew. Chem., Int. Ed. 2002, 41, 1290. doi:10.1002/1521-3773(20020415)41:8<1290::AID-ANIE1290>3.0.CO;2-Y |
| 2. | Díez-González, S.; Marion, N.; Nolan, S. P. Chem. Rev. 2009, 109, 3612. doi:10.1021/cr900074m |
| 3. | Hopkinson, M. N.; Richter, C.; Schedler, M.; Glorius, F. Nature 2014, 510, 485. doi:10.1038/nature13384 |
| 11. | Flowers, S. E.; Cossairt, B. M. Organometallics 2014, 33, 4341. doi:10.1021/om500592u |
| 9. | Isobe, K.; Kai, E.; Nakamura, Y.; Nishimoto, K.; Miwa, T.; Kawaguchi, S.; Kinoshita, K.; Nakatsu, K. J. Am. Chem. Soc. 1980, 102, 2475. doi:10.1021/ja00527a065 |
| 10. | Isobe, K.; Kawaguchi, S. Heterocycles 1981, 16, 1603. doi:10.3987/R-1981-09-1603 |
| 4. | Kuwata, S.; Ikariya, T. Chem. – Eur. J. 2011, 17, 3542. doi:10.1002/chem.201003296 |
| 5. | Hahn, F. E. ChemCatChem 2013, 5, 419. doi:10.1002/cctc.201200567 |
| 6. | Zhao, B.; Han, Z.; Ding, K. Angew. Chem., Int. Ed. 2013, 52, 4744. doi:10.1002/anie.201204921 |
| 7. | Kuwata, S.; Ikariya, T. Chem. Commun. 2014, 50, 14290. doi:10.1039/C4CC04457F |
| 8. | Jahnke, M. C.; Hahn, F. E. Coord. Chem. Rev. 2015, 293–294, 95. doi:10.1016/j.ccr.2015.01.014 |
| 3. | Hopkinson, M. N.; Richter, C.; Schedler, M.; Glorius, F. Nature 2014, 510, 485. doi:10.1038/nature13384 |
| 15. | Seyboldt, A.; Wucher, B.; Hohnstein, S.; Eichele, K.; Rominger, F.; Törnroos, K. W.; Kunz, D. Organometallics 2015, 34, 2717. doi:10.1021/om500836m |
| 16. | Moser, M.; Wucher, B.; Kunz, D.; Rominger, F. Organometallics 2007, 26, 1024. doi:10.1021/om060952z |
| 14. | Marelius, D. C.; Darrow, E. H.; Moore, C. E.; Golen, J. A.; Rheingold, A. L.; Grotjahn, D. B. Chem. – Eur. J. 2015, 21, 10988. doi:10.1002/chem.201501945 |
| 13. | Miranda-Soto, V.; Grotjahn, D. B.; Cooksy, A. L.; Golen, J. A.; Moore, C. E.; Rheingold, A. L. Angew. Chem., Int. Ed. 2011, 50, 631. doi:10.1002/anie.201005100 |
| 12. | Miranda-Soto, V.; Grotjahn, D. B.; DiPasquale, A. G.; Rheingold, A. L. J. Am. Chem. Soc. 2008, 130, 13200. doi:10.1021/ja804713u |
| 10. | Isobe, K.; Kawaguchi, S. Heterocycles 1981, 16, 1603. doi:10.3987/R-1981-09-1603 |
© 2016 Marelius et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)