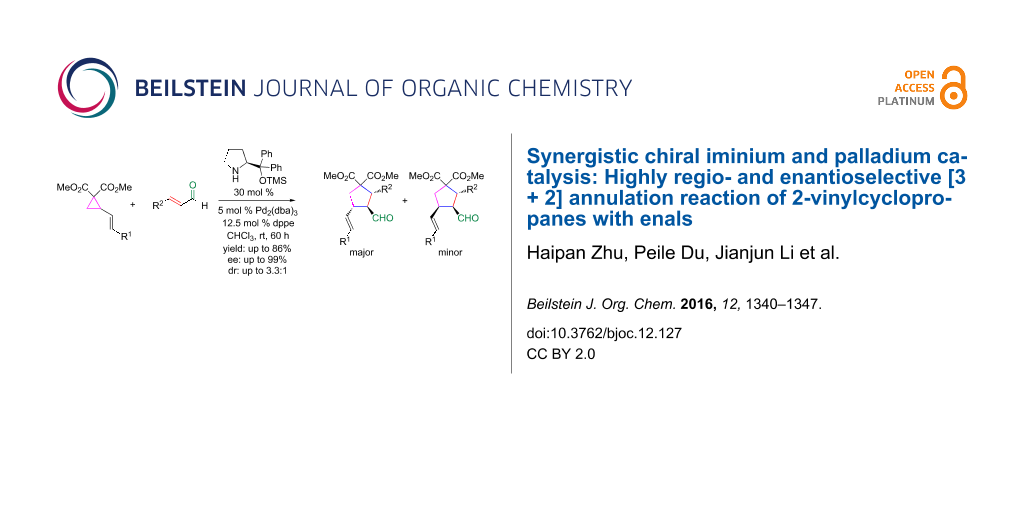
Abstract
A cooperative catalytic strategy of chiral iminium catalysis by regioselective activation of the C=C bond in enals and a transition metal promoting to open the 2-vinylcyclopropanes for highly regio- and enantioselective [3 + 2] cycloaddition reaction of 2-vinylcyclopropanes with α,β-unsaturated aldehydes has been developed.

Graphical Abstract
Introduction
The power of “donor–acceptor” (D–A) cyclopropanes as versatile 1,3-dipolar components is fuelled by its capacity of serving a complementary approach to a wide array of 5-membered ring structures, which are difficult or impossible to access by classic [3 + 2] cycloaddition reactions [1-34]. In recent years, significant efforts have been devoted to developing a catalytic enantioselective version of the processes. In this context, the D–A cyclopropanes have been applied for the reaction with highly active dipolarophiles, such as electrophilic C=O [35], e.g., aldehydes [36-38], ketones [38,39], and imines [40], and nucleophilic enol ethers [38,41], enamides [42], and indoles [43]. Nonetheless, the reactions with the α,β-unsaturated aldehydes and ketones face important challenges. To the best of our knowledge, so far merely two catalyst manifolds have been realized to effect the transformations with C=C double bonds instead of C=O in the α,β-unsaturated systems. Tsuji described the first organometallic promoted non-asymmetric reaction between D–A cyclopropanes and methyl vinyl ketone and α,β-unsaturated esters [44]. Trost and co-workers orchestrated the only example of the enantioselective reaction of D–A cyclopropanes with C=C double bonds with Meldrum’s acid and alkylidenes or azlactone alkylidenes, catalyzed by the chiral Trost Pd(0)-complexes [45]. However, it is difficult to apply the catalytic system for the regio-controlled reaction with C=C bonds in α,β-unsaturated carbonyl compounds, particularly enals. The highly active aldehyde functionality reacts more favorably with the D–A cyclopropane resulting 1,3-dipoles, as elegantly demonstrated by Johnson and Waser for the formation of chiral tetrahydrofurans (Scheme 1, reaction 1) [36,38]. Achieving a regioselective control at the C=C bond rather than at C=O in enals represents a challenge and has not been reported.
![[1860-5397-12-127-i1]](/bjoc/content/inline/1860-5397-12-127-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Catalytic regio- and enantioselective [3 + 2] annulation reactions of 2-vinylcyclopropanes with enals.
Scheme 1: Catalytic regio- and enantioselective [3 + 2] annulation reactions of 2-vinylcyclopropanes with ena...
Synergistic catalysis is a very important and useful strategy in organic synthesis by offering power for improving reaction efficiency and/or realizing impossible processes [46-55]. Recently, we developed an enantioselective addition of aldehydes to vinylpyridines and vinylarenes catalyzed by synergistic catalysis of iminium catalyst and Brønsted acid [56]. Herein we wish to disclose the first synergistic catalytic enantioselective [3 + 2] annulation reaction between 2-vinylcyclopropanes and enals via 1,4-addition (Scheme 1, reaction 2). The process proceeds highly regio- and enantioselectively with C=C bonds in enals. Notably, a synergistic catalytic system is implemented and makes this previously inaccessible [3 + 2] annulation transformation possible.
Results and Discussion
To render the [3 + 2] annulation reaction to selectively act on the C=C double bond rather than on the aldehyde in enals 1, we proposed a new cooperative iminium and Lewis acid (LA) catalysis strategy (Scheme 1, reaction 2) [49,50,57-76]. The iminium catalysis plays an important dual role in the process. The formed iminium ion 4 derived from aldehyde 1 and an amine catalyst activates the C=C bond and sterically blocks the attack of the C=N iminium ion functionality posed by the bulky amine catalyst. In parallel, a LA promotes to open the D–A cyclopropanes 2. The cooperative activation of two independent substrates by respective iminium and Lewis acid catalysis may enable an unprecedented catalytic regio- and enantioselective [3 + 2] annulation process, which offers a new approach to synthetically important heavily functionalized chiral cyclopentane structures 3, bearing at least 3 stereogenic centers in this one-pot operation [77,78].
To test the feasibility of the designed [3 + 2] annulation process [79-94], we started our investigation by carrying out the reaction between the commonly used D–A system dimethyl 2-vinylcyclopropane-1,1-dicarboxylate (1a) and trans-cinnamaldehyde (2a) catalyzed in the presence of a LA and chiral amine I in CH2Cl2 at rt for 48 h (Table 1). A series of Lewis acids were initially screened. FeCl3 and Cu(OTf)2 gave the 1,2-cycloaddition product tetrahydrofuran 4a (Table 1, entries 1 and 2). It is also disappointing that others Lewis acids, such as CuCl2, MgI2, ZnBr2, ZnCl2 and FeCl2 failed to promote these processes (Table 1, entries 3–7). Inspired by Trost’s work of Pd(0)-catalyzed annulations of D–A cyclopropanes with C=C double bonds with Meldrum’s acid and alkylidenes or azlactone alkylidenes [45], we probed the Pd2(dba)3-dppe complex for the 1,4-addition cycloaddition reaction (Table 1, entry 8). It was found that the reaction took place to afford the desired cyclopentane 3a.
Table 1: Screening of lewis acids.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-127-i4.svg?max-width=637&scale=1.0)
|
|||
| Entry | LA | Yield (%)b, 3a | Yield (%)b, 4a |
|---|---|---|---|
| 1 | FeCl3 | 0 | 53 |
| 2 | Cu(OTf)2 | 0 | 47 |
| 3 | CuCl2 | 0 | 0 |
| 4 | MgI2 | 0 | 0 |
| 5 | ZnBr2 | 0 | 0 |
| 6 | ZnCl2 | 0 | 0 |
| 7 | FeCl2 | 0 | 0 |
| 8c | Pd2(dba)3 | 48 | 0 |
aThe reaction was carried out with 1a (36.8 mg, 0.2 mmol) and 2a (26.4 mg, 0.2 mmol) in the presence of 10 mol % LA and 30 mol % amine I in 0.8 mL of CH2Cl2 at rt for 48 h. bIsolated yields; c5 mol % Pd2(dba)3 and 12.5 mol % dppe was used.
Encouraged by this result, we carried out further investigations of the co-catalysts promoted process (Table 2). First, we determined the diastereo- and enantioselectivity of the reaction. The 1H NMR of the reaction crude mixture showed three diastereoisomers. The two major diastereoisomers were determined to be (2S,3S,4S)-3a’ and (2S,3S,4R)-3a’’ in 2:1 ratio (Table 2, entry 1) based on single X-ray crystallographic analysis (see Scheme 2). Unfortunately, the third diastereoisomer 3a’’’ was too hard to be separated to determine its stereochemistry. The enantioselectivities of two major diastereoisomers are even more encouraging (80 and 76% ee). Further investigations of solvents revealed the medium-dependent effect (Table 2, entries 1–8). No reaction happened in toluene (Table 2, entry 2). Disappointing outcomes were also received in DCE, ether, CH3CN and EtOAc (Table 2, entries 3–6). Gratifyingly, in CHCl3 this reaction proceeded smoothly to furnish the desired cyclopentanes in 63% yield with 99% ee for major 3a’ and 83% ee for minor 3a’’ with a dr ratio of 1.7:1 (Table 2, entry 7). The reaction performed in THF was interesting: No reaction occurred at rt (Table 2, entry 8), but at 50 °C, 54% yield with high enantioselectivity for both isomers while 3a’’ as the major product (dr: 3a’’:3a’ = 5:1, Table 2, entry 9) was obtained. We decided to further optimize the reaction in CHCl3 accordingly (Table 2, entries 10–13). A longer reaction time helped to increase the reaction yield (60 h, 76% yield entry 10). More steric hindered amine catalysts with bigger TES and TBDMS groups, II and III, were then probed and gave rise to the slight drop of enantioseletivity (Table 2, entries 11 and 12). A further optimization of reaction conditions found that the addition of additional 0.5 equiv 1a into the reaction mixture in 4 portions significantly improved the reaction yield (83%, Table 2, entry 13). In order to improve the diastereoselectivity of this reaction, other cyclopentanes used in Trost’s system were also tested in this reaction [45]. Unfortunately, the reactions proceeded slowly to afford the cycloaddition products in less than 10% yield.
Table 2: The optimization of reaction conditions.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-127-i5.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Amine cat. | Solvent | Yield (%)b | ee (3a’, 3a’’)c | dr (3a’:3a’’)d |
|---|---|---|---|---|---|
| 1 | I | CH2Cl2 | 48 | 80, 76 | 2:1 |
| 2 | I | toluene | – | – | – |
| 3 | I | DCE | < 20 | – | – |
| 4 | I | ether | < 20 | – | – |
| 5 | I | CH3CN | < 20 | – | – |
| 6 | I | EtOAc | < 20 | – | – |
| 7 | I | CHCl3 | 63 | 99, 83 | 1.7:1 |
| 8 | I | THF | – | – | – |
| 9e | I | THF | 54 | 90, 90 | 1:5 |
| 10f | I | CHCl3 | 76 | 99, 83 | 1.7:1 |
| 11f | II | CHCl3 | 76 | 96, 80 | 1.7:1 |
| 12f | III | CHCl3 | 61 | 97, 82 | 1.7:1 |
| 13f,g | I | CHCl3 | 83 | 99, 83 | 1.7:1 |
aThe reaction was carried out with 1a (36.8 mg, 0.2 mmol) and 2a (26.4 mg, 0.2 mmol) in the presence of 5 mol % Pd2(dba)3, 12.5 mol % dppe and 30 mol % organic catalyst in 0.8 mL of solvent at rt for 48 h. bIsolated yields. cDetermined by HPLC analysis. dDetermined by 1H NMR spectroscopy of the crude mixture. eThe reaction was run at 50 °C. fThe reaction was stirred for 60 h. gAdditional 0.5 equiv 1a in 0.4 mL of CHCl3 was added into the reaction mixture in 4 portions every 12 h.
![[1860-5397-12-127-i2]](/bjoc/content/inline/1860-5397-12-127-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Single X-ray crystal structures of 7h’ and 7h’’.
Scheme 2: Single X-ray crystal structures of 7h’ and 7h’’.
We then selected the use of co-catalysts of Pd2(dba)3 and organocatalyst I in CHCl3 at room temperature to evaluate the generality of this [3 + 2] annulation process by the variation of vinylcyclopropanes and enals (Table 3). The results exhibit that the synergistic catalyzed enantioselective [3 + 2] annulation process serves as a general approach to structurally chiral cyclopentanes bearing 3-consecutive stereogenic centers with high regio- and enantioselectivities. It was found that a wide range of aromatic α,β-unsaturated aldehydes can effectively participate in the process (Table 3, entries 1–12). The aromatic α,β-unsaturated aldehydes tethering electron-neutral, -withdrawing, and -donating substituents at the para-position of the phenyl ring gave good to high yields and excellent enantioselectivities for major isomer 3’ and minor 3’’’ products, while the electronic effect on enantioselectivity is more pronounced for minor 3’’ (Table 3, entries 1–5). A similar trend is observed with the aromatic α,β-unsaturated aldehydes with electron-withdrawing at meta-position (Table 3, entries 6 and 7). Those with electron-withdrawing, and -donating groups at ortho-position furnished excellent enantioselectivities for both 3’ and 3’’ products in cases studied (Table 3, entries 8–11). Moreover, the heteroaromatic furanyl α,β-unsaturated aldehyde 2l can also be tolerated with good yield and 97% ee for the major product (Table 3, entry 12). More significantly, the more steric demanding D–A cyclopropane bearing a phenyl ring instead of H can effectively participate in the process to deliver the desired product with achieving an excellent level of enantioselectivity albeit a relatively low yield (Table 3, 46%, entry 13). It is noteworthy that although aliphatic enals also can engage in this [3 + 2] annulation reaction. Unfortunately, we could not separate them on chiral HPLC column for the determination of the enantioselectivity by all means we have attempted (data not shown).
Table 3: Scope of the [3 + 2] annulation reaction of D–A cyclopropanes with enals.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-127-i6.svg?max-width=637&scale=1.0)
|
||||
| Entry | R1, R2, 3 | Yield (%)b | ee (3a’, 3a’’,3a’’’)c | dr (3a’:3a’’,3a’’’)d |
|---|---|---|---|---|
| 1 | H, Ph, 3a | 83 | 99, 83, 99 | 1.7:1:0.7 |
| 2 | H, 4-ClC6H4, 3b | 86 | 97, 77, 99 | 2:1:0.4 |
| 3 | H, 4-BrC6H4, 3c | 84 | 94, 70, 99 | 2.5:1:0.5 |
| 4 | H, 4-MeC6H4, 3d | 70 | 99, 85, 99 | 2:1:0.7 |
| 5 | H, 4-MeOC6H4, 3e | 71 | 99, 99, 86 | 2:1:0.5 |
| 6 | H, 3-FC6H4, 3f | 85 | 97, 99, 76 | 2.5:1:0.6 |
| 7 | H, 3-CF3C6H4, 3g | 61 | 90, 66, 99 | 3.3:1:0.5 |
| 8 | H, 2-ClC6H4, 3h | 65 | 98, 88, – | 1.7:1:0.6 |
| 9 | H, 2-BrC6H4 3i | 77 | 99, 90, – | 2.5:1:0.8 |
| 10 | H, 2-MeC6H4, 3j | 72 | 99, 91, 99 | 3.3:1:0.4 |
| 11 | H, 2-MeOC6H4, 3k | 81 | 99, 99, 92 | 2:1:0.8 |
| 12 | H, 2-furanyl, 3l | 70 | 99, 97, 72 | 1.3:1:0.7 |
| 13e | Ph, 4-ClC6H4, 3m | 46 | 92, 86, 99 | 1.7:1:0.2 |
aUnless specified, see experimental section for details. bIsolated yields. cDetermined by HPLC analysis. dDetermined by 1H NMR spectroscopy of crude product. eThe reaction was run at 50 °C for 120 h.
The absolute configuration of cyclopentanes 3’ and 3’’ were determined based on the derivatives 7h’ and 7h’’ of 3h (Scheme 2) [95].
We proposed two possible transition states (TS) 8a’ and 8a” to rationalize the observed configurations (Scheme 3). The trans-C=C double bond in iminium ion 4 dictates the R group at pseudo axial position in the cyclic 5-membered ring TS 8a’ and 8a”. This orientation avoids the A[1,3] strain induced by the catalyst-derived enamine. The Pd(II)-π3 complex moiety at pseudo axial and equatorial positions leads to respective TS 8a’ and 8a”, while 8a’ is more stable due to the minimization of the A[1,3] interaction. Therefore, it is observed 3a’ produced from corresponding 8a’ as the major diastereomer whereas 3a” as minor one.
![[1860-5397-12-127-i3]](/bjoc/content/inline/1860-5397-12-127-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The proposed transition states.
Scheme 3: The proposed transition states.
Conclusion
We have developed a cooperative catalytic strategy for highly regio- and enantioselective [3 + 2] cycloaddition reactions of vinylcyclopropanes with α,β-unsaturated aldehydes for the first time. The combination of a chiral iminium catalyst, which activates the C=C bond and blocks the C=O bond in enals, and a Lewis acid promoting to open the vinylcyclopropanes enables the annulation process to proceed with the challenging C=C bond. A high level of enantioselectivity could be achieved here. This previously unattainable [3 + 2] annulation transformation serves as a general approach to the preparation of new densely functionalized chiral cyclopentanes. This synergistic catalysis strategy holds great potentials for further exploration of new cycloaddition reactions involving enals and other D–A systems. The endeavor is being pursued in our laboratories.
Experimental
General procedure for the [3 + 2] annulation
A mixture of 1a (0.2 mmol, 36.8 mg), 2a (0.2 mmol, 26.4 mg), Pd2(dba)3 (0.01 mmol, 9.2 mg), dppe (0.025 mmol, 10 mg) and I (0.06 mmol, 18.5 mg) in 0.8 mL CHCl3 was stirred for 60 h at rt. During this period, 1a (0.1 mmol, 18.4 mg) in 0.4 mL CHCl3 was added into the solution for total 4 times every 12 h, the mixture was purified by column chromatography on silica gel, eluted by petroleum ether/EtOAc = 20:1 to 10:1 to give the desired product 3a in 83% yield as a colorless oil.
Supporting Information
| Supporting Information File 1: Experimental and analytical data. | ||
| Format: PDF | Size: 3.4 MB | Download |
Acknowledgements
Financial support of this research from the program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (No. 201226, H. L.), the National Science Foundation of China (No. 21372073, W. W.), the Fundamental Research Funds for the Central Universities and East China University of Science and Technology (start-up funds, H. L. and W. W.), and the China 111 Project (Grant B07023, H. L. and W.W.) is gratefully acknowledged.
References
-
Danishefsky, S. Acc. Chem. Res. 1979, 12, 66–72. doi:10.1021/ar50134a004
Return to citation in text: [1] [2] [3] -
Verhé, R.; de Kimpe, N. Synthesis and Reactivity of Electrophilic Cyclopropanes. In The Carbonyl Group; Rappoport, Z., Ed.; PATAI'S Chemistry of Functional Groups, Vol. 1 and 2; John Wiley & Sons: Chichester, United Kingdom, 1987; pp 445–564. doi:10.1002/0470023449.ch9
Return to citation in text: [1] -
Reissig, H.-U. Organic Synthesis via Cyclopropanes: Principles and Applications. In Cyclopropyl Group; Rappoport, Z., Ed.; PATAI'S Chemistry of Functional Groups, Vol. 1 and 2; John Wiley & Sons: Chichester, United Kingdom, 1987; pp 375–443. doi:10.1002/0470023449.ch8
Return to citation in text: [1] [2] [3] -
Salaün, J. Cyclopropane Derivatives and their Diverse Biological Activities. In Small Ring Compounds in Organic Synthesis VI; de Meijere, A., Ed.; Topics in Current Chemistry, Vol. 207; Springer: Berlin, Germany, 2000; pp 1–67. doi:10.1007/3-540-48255-5_1
Return to citation in text: [1] -
Reissig, H.-U.; Zimmer, R. Chem. Rev. 2003, 103, 1151–1196. doi:10.1021/cr010016n
Return to citation in text: [1] -
Yu, M.; Pagenkopf, B. L. Tetrahedron 2005, 61, 321–347. doi:10.1016/j.tet.2004.10.077
Return to citation in text: [1] -
Carson, C. A.; Kerr, M. A. Chem. Soc. Rev. 2009, 38, 3051–3060. doi:10.1039/b901245c
Return to citation in text: [1] -
Lebold, T. P.; Kerr, M. A. Pure Appl. Chem. 2010, 82, 1797–1812. doi:10.1351/PAC-CON-09-09-28
Return to citation in text: [1] -
Mel’nikov, M. Ya.; Budynina, E. M.; Ivanova, O. A.; Trushkov, I. V. Mendeleev Commun. 2011, 21, 293–301. doi:10.1016/j.mencom.2011.11.001
Return to citation in text: [1] -
Wang, Z. Synlett 2012, 23, 2311–2327. doi:10.1055/s-0032-1317082
Return to citation in text: [1] -
Tang, P.; Qin, Y. Synthesis 2012, 44, 2969–2984. doi:10.1055/s-0032-1317011
Return to citation in text: [1] -
Cavitt, M. A.; Phun, L. H.; France, S. Chem. Soc. Rev. 2014, 43, 804–818. doi:10.1039/C3CS60238A
Return to citation in text: [1] -
Schneider, T. F.; Kaschel, J.; Werz, D. B. Angew. Chem., Int. Ed. 2014, 53, 5504–5523. doi:10.1002/anie.201309886
Return to citation in text: [1] -
Stork, G.; Gregson, M. J. Am. Chem. Soc. 1969, 91, 2373–2374. doi:10.1021/ja01037a032
Return to citation in text: [1] -
Stork, G.; Grieco, P. A. J. Am. Chem. Soc. 1969, 91, 2407–2408. doi:10.1021/ja01037a059
Return to citation in text: [1] -
Stork, G.; Marx, M. J. Am. Chem. Soc. 1969, 91, 2371–2373. doi:10.1021/ja01037a031
Return to citation in text: [1] -
Stork, G.; Grieco, P. A. Tetrahedron Lett. 1971, 1807–1810. doi:10.1016/S0040-4039(01)87467-0
Return to citation in text: [1] -
Corey, E. J.; Balanson, R. D. Tetrahedron Lett. 1973, 14, 3153–3156. doi:10.1016/S0040-4039(00)79797-8
Return to citation in text: [1] -
Danishefsky, S.; Dynak, J.; Hatch, E.; Yamamoto, M. J. Am. Chem. Soc. 1974, 96, 1256–1259. doi:10.1021/ja00811a068
Return to citation in text: [1] -
Danishefsky, S.; Tsai, M. Y.; Dynak, J. J. Chem. Soc., Chem. Commun. 1975, 7–8. doi:10.1039/c39750000007
Return to citation in text: [1] -
Danishefsky, S.; McKee, R.; Singh, R. K. J. Am. Chem. Soc. 1977, 99, 7711–7713. doi:10.1021/ja00465a054
Return to citation in text: [1] -
de Meijere, A. Angew. Chem., Int. Ed. Engl. 1979, 18, 809–826. doi:10.1002/anie.197908093
Return to citation in text: [1] -
Wenkert, E.; Alonso, M. E.; Buckwalter, B. L.; Chou, K. J. J. Am. Chem. Soc. 1977, 99, 4778–4782. doi:10.1021/ja00456a040
Return to citation in text: [1] -
Piers, E.; Reissig, H.-U. Angew. Chem., Int. Ed. Engl. 1979, 18, 791–792. doi:10.1002/anie.197907911
Return to citation in text: [1] -
Wenkert, E. Acc. Chem. Res. 1980, 13, 27–31. doi:10.1021/ar50145a005
Return to citation in text: [1] -
Reissig, H.-U.; Hirsch, E. Angew. Chem., Int. Ed. Engl. 1980, 19, 813–814. doi:10.1002/anie.198008131
Return to citation in text: [1] -
Reissig, H.-U. Tetrahedron Lett. 1981, 22, 2981–2984. doi:10.1016/S0040-4039(01)81805-0
Return to citation in text: [1] -
Brückner, C.; Reissig, H.-U. J. Chem. Soc., Chem. Commun. 1985, 1512–1513. doi:10.1039/C39850001512
Return to citation in text: [1] -
Brückner, C.; Reissig, H.-U. Angew. Chem., Int. Ed. Engl. 1985, 24, 588–589. doi:10.1002/anie.198505881
Return to citation in text: [1] -
Grimm, E. L.; Zschiesche, R.; Reissig, H. U. J. Org. Chem. 1985, 50, 5543–5545. doi:10.1021/jo00350a022
Return to citation in text: [1] -
Reißig, H.-U.; Reichelt, I.; Lorey, H. Liebigs Ann. Chem. 1986, 1986, 1924–1939. doi:10.1002/jlac.198619861113
Return to citation in text: [1] -
Brueckner, C.; Reissig, H. U. J. Org. Chem. 1988, 53, 2440–2450. doi:10.1021/jo00246a010
Return to citation in text: [1] -
Reissig, H.-U.; Holzinger, H.; Glomsda, G. Tetrahedron 1989, 45, 3139–3150. doi:10.1016/S0040-4020(01)80140-X
Return to citation in text: [1] -
Reissig, H.-U. Donor-acceptor-substituted cyclopropanes: Versatile building blocks in organic synthesis. Small ring compounds in organic synthesis III; Top. Curr. Chem., Vol. 144; Springer: Berlin, Germany, 1988; pp 73–135. doi:10.1007/BFb0111229
Return to citation in text: [1] -
Campbell, M. J.; Johnson, J. S.; Parsons, A. T.; Pohlhaus, P. D.; Sanders, S. D. J. Org. Chem. 2010, 75, 6317–6325. doi:10.1021/jo1010735
Return to citation in text: [1] -
Parsons, A. T.; Johnson, J. S. J. Am. Chem. Soc. 2009, 131, 3122–3123. doi:10.1021/ja809873u
Return to citation in text: [1] [2] -
Benfatti, F.; de Nanteuil, F.; Waser, J. Org. Lett. 2012, 14, 386–389. doi:10.1021/ol203144v
Return to citation in text: [1] -
de Nanteuil, F.; Serrano, E.; Perrotta, D.; Waser, J. J. Am. Chem. Soc. 2014, 136, 6239–6242. doi:10.1021/ja5024578
Return to citation in text: [1] [2] [3] [4] -
Benfatti, F.; de Nanteuil, F.; Waser, J. Chem. – Eur. J. 2012, 18, 4844–4849. doi:10.1002/chem.201103971
Return to citation in text: [1] -
Parsons, A. T.; Smith, A. G.; Neel, A. N.; Johnson, J. S. J. Am. Chem. Soc. 2010, 132, 9688–9692. doi:10.1021/ja1032277
Return to citation in text: [1] -
Xu, H.; Qu, J.-P.; Liao, S.; Xiong, H.; Tang, Y. Angew. Chem., Int. Ed. 2013, 52, 4004–4007. doi:10.1002/anie.201300032
Return to citation in text: [1] -
de Nanteuil, F.; Waser, J. Angew. Chem., Int. Ed. 2011, 50, 12075–12079. doi:10.1002/anie.201106255
Return to citation in text: [1] -
Xiong, H.; Xu, H.; Liao, S.; Xie, Z.; Tang, Y. J. Am. Chem. Soc. 2013, 135, 7851–7854. doi:10.1021/ja4042127
Return to citation in text: [1] -
Shimizu, I.; Ohashi, Y.; Tsuji, J. Tetrahedron Lett. 1985, 23, 3825–3828. doi:10.1016/S0040-4039(00)89261-8
Return to citation in text: [1] -
Trost, B. M.; Morris, P. J.; Sprague, S. J. J. Am. Chem. Soc. 2012, 134, 17823–17831. doi:10.1021/ja309003x
Return to citation in text: [1] [2] [3] -
Sträter, N.; Lipscomb, W. N.; Klabunde, T.; Krebs, B. Angew. Chem., Int. Ed. Engl. 1996, 35, 2024–2055. doi:10.1002/anie.199620241
Return to citation in text: [1] -
Sawamura, M.; Sudoh, M.; Ito, Y. J. Am. Chem. Soc. 1996, 118, 3309–3310. doi:10.1021/ja954223e
Return to citation in text: [1] -
Xu, H.; Zuend, S. J.; Woll, M. G.; Tao, Y.; Jacobsen, E. N. Science 2010, 327, 986–990. doi:10.1126/science.1182826
Return to citation in text: [1] -
Krautwald, S.; Sarlah, D.; Schafroth, M. A.; Carreira, E. M. Science 2013, 340, 1065–1068. doi:10.1126/science.1237068
Return to citation in text: [1] [2] -
Allen, A. E.; MacMillan, D. W. C. Chem. Sci. 2012, 3, 633–658. doi:10.1039/c2sc00907b
Return to citation in text: [1] [2] -
Ahire, M. M.; Mhaske, S. B. Angew. Chem., Int. Ed. 2014, 53, 7038–7042. doi:10.1002/anie.201400623
Return to citation in text: [1] -
Meazza, M.; Ceban, V.; Pitak, M. B.; Coles, S. J.; Rios, R. Chem. – Eur. J. 2014, 20, 16853–16857. doi:10.1002/chem.201404565
Return to citation in text: [1] -
Logan, K. M.; Smith, K. B.; Brown, M. K. Angew. Chem., Int. Ed. 2015, 54, 5228–5231. doi:10.1002/anie.201500396
Return to citation in text: [1] -
Zhu, S.; Zhang, J.; Chen, K.; Jiang, H. Angew. Chem., Int. Ed. 2015, 54, 9414–9418. doi:10.1002/anie.201504964
Return to citation in text: [1] -
Peng, H.; Akhmedov, N. G.; Liang, Y.-F.; Jiao, N.; Shi, X. J. Am. Chem. Soc. 2015, 137, 8912–8915. doi:10.1021/jacs.5b05415
Return to citation in text: [1] -
Wang, S.; Li, X.; Liu, H.; Xu, L.; Zhang, J.; Li, J.; Li, H.; Wang, W. J. Am. Chem. Soc. 2015, 137, 2303–2310. doi:10.1021/ja511143b
Return to citation in text: [1] -
Shao, Z.; Zhang, H. Chem. Soc. Rev. 2009, 38, 2745–2755. doi:10.1039/b901258n
Return to citation in text: [1] -
de Armas, P.; Tejedor, D.; García-Tellado, F. Angew. Chem., Int. Ed. 2010, 49, 1013–1016. doi:10.1002/anie.200906018
Return to citation in text: [1] -
Zhong, C.; Shi, X. Eur. J. Org. Chem. 2010, 2999–3025. doi:10.1002/ejoc.201000004
Return to citation in text: [1] -
Rueping, M.; Koenigs, R. M.; Atodiresei, I. Chem. – Eur. J. 2010, 16, 9350–9365. doi:10.1002/chem.201001140
Return to citation in text: [1] -
Zhou, J. Chem. – Asian J. 2010, 5, 422–434. doi:10.1002/asia.200900458
Return to citation in text: [1] -
Loh, C. C. J.; Enders, D. Chem. – Eur. J. 2012, 18, 10212–10225. doi:10.1002/chem.201200287
Return to citation in text: [1] -
Patil, N. T.; Shinde, V. S.; Gajula, B. Org. Biomol. Chem. 2012, 10, 211–224. doi:10.1039/C1OB06432K
Return to citation in text: [1] -
Pellissier, H. Tetrahedron 2013, 69, 7171–7210. doi:10.1016/j.tet.2013.06.020
Return to citation in text: [1] -
Du, Z.; Shao, Z. Chem. Soc. Rev. 2013, 42, 1337–1378. doi:10.1039/C2CS35258C
Return to citation in text: [1] -
Ibrahem, I.; Córdova, A. Angew. Chem., Int. Ed. 2006, 45, 1952–1956. doi:10.1002/anie.200504021
Return to citation in text: [1] -
Bihelovic, F.; Matovic, R.; Vulovic, B.; Saicic, R. N. Org. Lett. 2007, 9, 5063–5066. doi:10.1021/ol7023554
Return to citation in text: [1] -
Usui, I.; Schmidt, S.; Breit, B. Org. Lett. 2009, 11, 1453–1456. doi:10.1021/ol9001812
Return to citation in text: [1] -
Capdevila, M. G.; Benfatti, F.; Zoli, L.; Stenta, M.; Cozzi, P. G. Chem. – Eur. J. 2010, 16, 11237–11241. doi:10.1002/chem.201001693
Return to citation in text: [1] -
Ikeda, M.; Miyake, Y.; Nishibayashi, Y. Angew. Chem., Int. Ed. 2010, 49, 7289–7293. doi:10.1002/anie.201002591
Return to citation in text: [1] -
Yoshida, A.; Ikeda, M.; Hattori, G.; Miyake, Y.; Nishibayashi, Y. Org. Lett. 2011, 13, 592–595. doi:10.1021/ol1027865
Return to citation in text: [1] -
Motoyama, K.; Ikeda, M.; Miyake, Y.; Nishibayashi, Y. Eur. J. Org. Chem. 2011, 2239–2246. doi:10.1002/ejoc.201100044
Return to citation in text: [1] -
Sinisi, R.; Vita, M. V.; Gualandi, A.; Emer, E.; Cozzi, P. G. Chem. – Eur. J. 2011, 17, 7404–7408. doi:10.1002/chem.201100729
Return to citation in text: [1] -
Afewerki, S.; Ibrahem, I.; Rydfjord, J.; Breistein, P.; Córdova, A. Chem. – Eur. J. 2012, 18, 2972–2977. doi:10.1002/chem.201103366
Return to citation in text: [1] -
Ma, G.; Afewerki, S.; Deiana, L.; Palo-Nieto, C.; Liu, L.; Sun, J.; Ibrahem, I.; Córdova, A. Angew. Chem., Int. Ed. 2013, 52, 6050–6054. doi:10.1002/anie.201300559
Return to citation in text: [1] -
Krautwald, S.; Schafroth, M. A.; Sarlah, D.; Carreira, E. M. J. Am. Chem. Soc. 2014, 136, 3020–3023. doi:10.1021/ja5003247
Return to citation in text: [1] -
Wang, H.; Yang, W.; Liu, H.; Wang, W.; Li, H. Org. Biomol. Chem. 2012, 10, 5032–5035. doi:10.1039/c2ob25682g
Return to citation in text: [1] -
Sathishkannan, G.; Srinivasan, K. Org. Lett. 2011, 13, 6002–6005. doi:10.1021/ol2024423
Return to citation in text: [1] -
Laugeois, M.; Ponra, S.; Ratovelomanana-Vidal, V.; Michelet, V.; Vitale, M. R. Chem. Commun. 2016, 52, 5332–5335. doi:10.1039/C6CC01775D
Return to citation in text: [1] -
Goldberg, A. F. G.; O’Connor, N. R.; Craig, R. A.; Stoltz, B. M. Org. Lett. 2012, 14, 5314–5317. doi:10.1021/ol302494n
Return to citation in text: [1] -
Zu, L.; Li, H.; Xie, H.; Wang, J.; Jiang, W.; Tang, T.; Wang, W. Angew. Chem., Int. Ed. 2007, 46, 3732–3734. doi:10.1002/anie.200700485
Return to citation in text: [1] -
Wang, J.; Li, H.; Xie, H.; Zu, L.; Shen, X.; Wang, W. Angew. Chem., Int. Ed. 2007, 46, 9050–9053. doi:10.1002/anie.200703163
Return to citation in text: [1] -
Vignola, N.; List, B. J. Am. Chem. Soc. 2004, 126, 450–451. doi:10.1021/ja0392566
Return to citation in text: [1] -
Enders, D.; Wang, C.; Bats, J. W. Angew. Chem., Int. Ed. 2008, 47, 7539–7542. doi:10.1002/anie.200802532
Return to citation in text: [1] -
Tan, B.; Shi, Z.; Chua, P. J.; Zhong, G. Org. Lett. 2008, 10, 3425–3428. doi:10.1021/ol801246m
Return to citation in text: [1] -
Tan, B.; Chua, P. J.; Zeng, X.; Lu, M.; Zhong, G. Org. Lett. 2008, 10, 3489–3492. doi:10.1021/ol801273x
Return to citation in text: [1] -
Hong, B.-C.; Nimje, R. Y.; Lin, C.-W.; Liao, J.-H. Org. Lett. 2011, 13, 1278–1281. doi:10.1021/ol1030487
Return to citation in text: [1] -
Hong, B.-C.; Dange, N. S.; Hsu, C.-S.; Liao, J.-H.; Lee, G.-H. Org. Lett. 2011, 13, 1338–1341. doi:10.1021/ol200006e
Return to citation in text: [1] -
Tan, B.; Candeias, N. R.; Barbas, C. F., III. Nat. Chem. 2011, 3, 473–477. doi:10.1038/nchem.1039
Return to citation in text: [1] -
Albertshofer, K.; Tan, B.; Barbas, C. F., III. Org. Lett. 2012, 14, 1834–1837. doi:10.1021/ol300441z
Return to citation in text: [1] -
Zhao, G.-L.; Ibrahem, I.; Dziedzic, P.; Sun, J.; Bonneau, C.; Córdova, A. Chem. – Eur. J. 2008, 14, 10007–10011. doi:10.1002/chem.200801082
Return to citation in text: [1] -
Sun, W.; Zhu, G.; Wu, C.; Hong, L.; Wang, R. Chem. – Eur. J. 2012, 18, 6737–6741. doi:10.1002/chem.201200478
Return to citation in text: [1] -
Remeš, M.; Veselý, J. Eur. J. Org. Chem. 2012, 3747–3752. doi:10.1002/ejoc.201200334
Return to citation in text: [1] -
Liu, G.; Shirley, M. E.; Van, K. N.; McFarlin, R. L.; Romo, D. Nat. Chem. 2013, 5, 1049–1057. doi:10.1038/nchem.1788
Return to citation in text: [1] -
CCDC-1032267 and -1032268 for compounds 7h’ and 7h”, respectively contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk.
Return to citation in text: [1]
| 95. | CCDC-1032267 and -1032268 for compounds 7h’ and 7h”, respectively contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk. |
| 45. | Trost, B. M.; Morris, P. J.; Sprague, S. J. J. Am. Chem. Soc. 2012, 134, 17823–17831. doi:10.1021/ja309003x |
| 45. | Trost, B. M.; Morris, P. J.; Sprague, S. J. J. Am. Chem. Soc. 2012, 134, 17823–17831. doi:10.1021/ja309003x |
| 1. | Danishefsky, S. Acc. Chem. Res. 1979, 12, 66–72. doi:10.1021/ar50134a004 |
| 2. | Verhé, R.; de Kimpe, N. Synthesis and Reactivity of Electrophilic Cyclopropanes. In The Carbonyl Group; Rappoport, Z., Ed.; PATAI'S Chemistry of Functional Groups, Vol. 1 and 2; John Wiley & Sons: Chichester, United Kingdom, 1987; pp 445–564. doi:10.1002/0470023449.ch9 |
| 3. | Reissig, H.-U. Organic Synthesis via Cyclopropanes: Principles and Applications. In Cyclopropyl Group; Rappoport, Z., Ed.; PATAI'S Chemistry of Functional Groups, Vol. 1 and 2; John Wiley & Sons: Chichester, United Kingdom, 1987; pp 375–443. doi:10.1002/0470023449.ch8 |
| 4. | Salaün, J. Cyclopropane Derivatives and their Diverse Biological Activities. In Small Ring Compounds in Organic Synthesis VI; de Meijere, A., Ed.; Topics in Current Chemistry, Vol. 207; Springer: Berlin, Germany, 2000; pp 1–67. doi:10.1007/3-540-48255-5_1 |
| 5. | Reissig, H.-U.; Zimmer, R. Chem. Rev. 2003, 103, 1151–1196. doi:10.1021/cr010016n |
| 6. | Yu, M.; Pagenkopf, B. L. Tetrahedron 2005, 61, 321–347. doi:10.1016/j.tet.2004.10.077 |
| 7. | Carson, C. A.; Kerr, M. A. Chem. Soc. Rev. 2009, 38, 3051–3060. doi:10.1039/b901245c |
| 8. | Lebold, T. P.; Kerr, M. A. Pure Appl. Chem. 2010, 82, 1797–1812. doi:10.1351/PAC-CON-09-09-28 |
| 9. | Mel’nikov, M. Ya.; Budynina, E. M.; Ivanova, O. A.; Trushkov, I. V. Mendeleev Commun. 2011, 21, 293–301. doi:10.1016/j.mencom.2011.11.001 |
| 10. | Wang, Z. Synlett 2012, 23, 2311–2327. doi:10.1055/s-0032-1317082 |
| 11. | Tang, P.; Qin, Y. Synthesis 2012, 44, 2969–2984. doi:10.1055/s-0032-1317011 |
| 12. | Cavitt, M. A.; Phun, L. H.; France, S. Chem. Soc. Rev. 2014, 43, 804–818. doi:10.1039/C3CS60238A |
| 13. | Schneider, T. F.; Kaschel, J.; Werz, D. B. Angew. Chem., Int. Ed. 2014, 53, 5504–5523. doi:10.1002/anie.201309886 |
| 14. | Stork, G.; Gregson, M. J. Am. Chem. Soc. 1969, 91, 2373–2374. doi:10.1021/ja01037a032 |
| 15. | Stork, G.; Grieco, P. A. J. Am. Chem. Soc. 1969, 91, 2407–2408. doi:10.1021/ja01037a059 |
| 16. | Stork, G.; Marx, M. J. Am. Chem. Soc. 1969, 91, 2371–2373. doi:10.1021/ja01037a031 |
| 17. | Stork, G.; Grieco, P. A. Tetrahedron Lett. 1971, 1807–1810. doi:10.1016/S0040-4039(01)87467-0 |
| 18. | Corey, E. J.; Balanson, R. D. Tetrahedron Lett. 1973, 14, 3153–3156. doi:10.1016/S0040-4039(00)79797-8 |
| 19. | Danishefsky, S.; Dynak, J.; Hatch, E.; Yamamoto, M. J. Am. Chem. Soc. 1974, 96, 1256–1259. doi:10.1021/ja00811a068 |
| 20. | Danishefsky, S.; Tsai, M. Y.; Dynak, J. J. Chem. Soc., Chem. Commun. 1975, 7–8. doi:10.1039/c39750000007 |
| 21. | Danishefsky, S.; McKee, R.; Singh, R. K. J. Am. Chem. Soc. 1977, 99, 7711–7713. doi:10.1021/ja00465a054 |
| 22. | de Meijere, A. Angew. Chem., Int. Ed. Engl. 1979, 18, 809–826. doi:10.1002/anie.197908093 |
| 23. | Wenkert, E.; Alonso, M. E.; Buckwalter, B. L.; Chou, K. J. J. Am. Chem. Soc. 1977, 99, 4778–4782. doi:10.1021/ja00456a040 |
| 24. | Piers, E.; Reissig, H.-U. Angew. Chem., Int. Ed. Engl. 1979, 18, 791–792. doi:10.1002/anie.197907911 |
| 25. | Wenkert, E. Acc. Chem. Res. 1980, 13, 27–31. doi:10.1021/ar50145a005 |
| 26. | Reissig, H.-U.; Hirsch, E. Angew. Chem., Int. Ed. Engl. 1980, 19, 813–814. doi:10.1002/anie.198008131 |
| 27. | Reissig, H.-U. Tetrahedron Lett. 1981, 22, 2981–2984. doi:10.1016/S0040-4039(01)81805-0 |
| 28. | Brückner, C.; Reissig, H.-U. J. Chem. Soc., Chem. Commun. 1985, 1512–1513. doi:10.1039/C39850001512 |
| 29. | Brückner, C.; Reissig, H.-U. Angew. Chem., Int. Ed. Engl. 1985, 24, 588–589. doi:10.1002/anie.198505881 |
| 30. | Grimm, E. L.; Zschiesche, R.; Reissig, H. U. J. Org. Chem. 1985, 50, 5543–5545. doi:10.1021/jo00350a022 |
| 31. | Reißig, H.-U.; Reichelt, I.; Lorey, H. Liebigs Ann. Chem. 1986, 1986, 1924–1939. doi:10.1002/jlac.198619861113 |
| 32. | Brueckner, C.; Reissig, H. U. J. Org. Chem. 1988, 53, 2440–2450. doi:10.1021/jo00246a010 |
| 33. | Reissig, H.-U.; Holzinger, H.; Glomsda, G. Tetrahedron 1989, 45, 3139–3150. doi:10.1016/S0040-4020(01)80140-X |
| 34. | Reissig, H.-U. Donor-acceptor-substituted cyclopropanes: Versatile building blocks in organic synthesis. Small ring compounds in organic synthesis III; Top. Curr. Chem., Vol. 144; Springer: Berlin, Germany, 1988; pp 73–135. doi:10.1007/BFb0111229 |
| 40. | Parsons, A. T.; Smith, A. G.; Neel, A. N.; Johnson, J. S. J. Am. Chem. Soc. 2010, 132, 9688–9692. doi:10.1021/ja1032277 |
| 77. | Wang, H.; Yang, W.; Liu, H.; Wang, W.; Li, H. Org. Biomol. Chem. 2012, 10, 5032–5035. doi:10.1039/c2ob25682g |
| 78. | Sathishkannan, G.; Srinivasan, K. Org. Lett. 2011, 13, 6002–6005. doi:10.1021/ol2024423 |
| 38. | de Nanteuil, F.; Serrano, E.; Perrotta, D.; Waser, J. J. Am. Chem. Soc. 2014, 136, 6239–6242. doi:10.1021/ja5024578 |
| 39. | Benfatti, F.; de Nanteuil, F.; Waser, J. Chem. – Eur. J. 2012, 18, 4844–4849. doi:10.1002/chem.201103971 |
| 79. | Laugeois, M.; Ponra, S.; Ratovelomanana-Vidal, V.; Michelet, V.; Vitale, M. R. Chem. Commun. 2016, 52, 5332–5335. doi:10.1039/C6CC01775D |
| 80. | Goldberg, A. F. G.; O’Connor, N. R.; Craig, R. A.; Stoltz, B. M. Org. Lett. 2012, 14, 5314–5317. doi:10.1021/ol302494n |
| 81. | Zu, L.; Li, H.; Xie, H.; Wang, J.; Jiang, W.; Tang, T.; Wang, W. Angew. Chem., Int. Ed. 2007, 46, 3732–3734. doi:10.1002/anie.200700485 |
| 82. | Wang, J.; Li, H.; Xie, H.; Zu, L.; Shen, X.; Wang, W. Angew. Chem., Int. Ed. 2007, 46, 9050–9053. doi:10.1002/anie.200703163 |
| 83. | Vignola, N.; List, B. J. Am. Chem. Soc. 2004, 126, 450–451. doi:10.1021/ja0392566 |
| 84. | Enders, D.; Wang, C.; Bats, J. W. Angew. Chem., Int. Ed. 2008, 47, 7539–7542. doi:10.1002/anie.200802532 |
| 85. | Tan, B.; Shi, Z.; Chua, P. J.; Zhong, G. Org. Lett. 2008, 10, 3425–3428. doi:10.1021/ol801246m |
| 86. | Tan, B.; Chua, P. J.; Zeng, X.; Lu, M.; Zhong, G. Org. Lett. 2008, 10, 3489–3492. doi:10.1021/ol801273x |
| 87. | Hong, B.-C.; Nimje, R. Y.; Lin, C.-W.; Liao, J.-H. Org. Lett. 2011, 13, 1278–1281. doi:10.1021/ol1030487 |
| 88. | Hong, B.-C.; Dange, N. S.; Hsu, C.-S.; Liao, J.-H.; Lee, G.-H. Org. Lett. 2011, 13, 1338–1341. doi:10.1021/ol200006e |
| 89. | Tan, B.; Candeias, N. R.; Barbas, C. F., III. Nat. Chem. 2011, 3, 473–477. doi:10.1038/nchem.1039 |
| 90. | Albertshofer, K.; Tan, B.; Barbas, C. F., III. Org. Lett. 2012, 14, 1834–1837. doi:10.1021/ol300441z |
| 91. | Zhao, G.-L.; Ibrahem, I.; Dziedzic, P.; Sun, J.; Bonneau, C.; Córdova, A. Chem. – Eur. J. 2008, 14, 10007–10011. doi:10.1002/chem.200801082 |
| 92. | Sun, W.; Zhu, G.; Wu, C.; Hong, L.; Wang, R. Chem. – Eur. J. 2012, 18, 6737–6741. doi:10.1002/chem.201200478 |
| 93. | Remeš, M.; Veselý, J. Eur. J. Org. Chem. 2012, 3747–3752. doi:10.1002/ejoc.201200334 |
| 94. | Liu, G.; Shirley, M. E.; Van, K. N.; McFarlin, R. L.; Romo, D. Nat. Chem. 2013, 5, 1049–1057. doi:10.1038/nchem.1788 |
| 36. | Parsons, A. T.; Johnson, J. S. J. Am. Chem. Soc. 2009, 131, 3122–3123. doi:10.1021/ja809873u |
| 37. | Benfatti, F.; de Nanteuil, F.; Waser, J. Org. Lett. 2012, 14, 386–389. doi:10.1021/ol203144v |
| 38. | de Nanteuil, F.; Serrano, E.; Perrotta, D.; Waser, J. J. Am. Chem. Soc. 2014, 136, 6239–6242. doi:10.1021/ja5024578 |
| 56. | Wang, S.; Li, X.; Liu, H.; Xu, L.; Zhang, J.; Li, J.; Li, H.; Wang, W. J. Am. Chem. Soc. 2015, 137, 2303–2310. doi:10.1021/ja511143b |
| 35. | Campbell, M. J.; Johnson, J. S.; Parsons, A. T.; Pohlhaus, P. D.; Sanders, S. D. J. Org. Chem. 2010, 75, 6317–6325. doi:10.1021/jo1010735 |
| 49. | Krautwald, S.; Sarlah, D.; Schafroth, M. A.; Carreira, E. M. Science 2013, 340, 1065–1068. doi:10.1126/science.1237068 |
| 50. | Allen, A. E.; MacMillan, D. W. C. Chem. Sci. 2012, 3, 633–658. doi:10.1039/c2sc00907b |
| 57. | Shao, Z.; Zhang, H. Chem. Soc. Rev. 2009, 38, 2745–2755. doi:10.1039/b901258n |
| 58. | de Armas, P.; Tejedor, D.; García-Tellado, F. Angew. Chem., Int. Ed. 2010, 49, 1013–1016. doi:10.1002/anie.200906018 |
| 59. | Zhong, C.; Shi, X. Eur. J. Org. Chem. 2010, 2999–3025. doi:10.1002/ejoc.201000004 |
| 60. | Rueping, M.; Koenigs, R. M.; Atodiresei, I. Chem. – Eur. J. 2010, 16, 9350–9365. doi:10.1002/chem.201001140 |
| 61. | Zhou, J. Chem. – Asian J. 2010, 5, 422–434. doi:10.1002/asia.200900458 |
| 62. | Loh, C. C. J.; Enders, D. Chem. – Eur. J. 2012, 18, 10212–10225. doi:10.1002/chem.201200287 |
| 63. | Patil, N. T.; Shinde, V. S.; Gajula, B. Org. Biomol. Chem. 2012, 10, 211–224. doi:10.1039/C1OB06432K |
| 64. | Pellissier, H. Tetrahedron 2013, 69, 7171–7210. doi:10.1016/j.tet.2013.06.020 |
| 65. | Du, Z.; Shao, Z. Chem. Soc. Rev. 2013, 42, 1337–1378. doi:10.1039/C2CS35258C |
| 66. | Ibrahem, I.; Córdova, A. Angew. Chem., Int. Ed. 2006, 45, 1952–1956. doi:10.1002/anie.200504021 |
| 67. | Bihelovic, F.; Matovic, R.; Vulovic, B.; Saicic, R. N. Org. Lett. 2007, 9, 5063–5066. doi:10.1021/ol7023554 |
| 68. | Usui, I.; Schmidt, S.; Breit, B. Org. Lett. 2009, 11, 1453–1456. doi:10.1021/ol9001812 |
| 69. | Capdevila, M. G.; Benfatti, F.; Zoli, L.; Stenta, M.; Cozzi, P. G. Chem. – Eur. J. 2010, 16, 11237–11241. doi:10.1002/chem.201001693 |
| 70. | Ikeda, M.; Miyake, Y.; Nishibayashi, Y. Angew. Chem., Int. Ed. 2010, 49, 7289–7293. doi:10.1002/anie.201002591 |
| 71. | Yoshida, A.; Ikeda, M.; Hattori, G.; Miyake, Y.; Nishibayashi, Y. Org. Lett. 2011, 13, 592–595. doi:10.1021/ol1027865 |
| 72. | Motoyama, K.; Ikeda, M.; Miyake, Y.; Nishibayashi, Y. Eur. J. Org. Chem. 2011, 2239–2246. doi:10.1002/ejoc.201100044 |
| 73. | Sinisi, R.; Vita, M. V.; Gualandi, A.; Emer, E.; Cozzi, P. G. Chem. – Eur. J. 2011, 17, 7404–7408. doi:10.1002/chem.201100729 |
| 74. | Afewerki, S.; Ibrahem, I.; Rydfjord, J.; Breistein, P.; Córdova, A. Chem. – Eur. J. 2012, 18, 2972–2977. doi:10.1002/chem.201103366 |
| 75. | Ma, G.; Afewerki, S.; Deiana, L.; Palo-Nieto, C.; Liu, L.; Sun, J.; Ibrahem, I.; Córdova, A. Angew. Chem., Int. Ed. 2013, 52, 6050–6054. doi:10.1002/anie.201300559 |
| 76. | Krautwald, S.; Schafroth, M. A.; Sarlah, D.; Carreira, E. M. J. Am. Chem. Soc. 2014, 136, 3020–3023. doi:10.1021/ja5003247 |
| 44. | Shimizu, I.; Ohashi, Y.; Tsuji, J. Tetrahedron Lett. 1985, 23, 3825–3828. doi:10.1016/S0040-4039(00)89261-8 |
| 36. | Parsons, A. T.; Johnson, J. S. J. Am. Chem. Soc. 2009, 131, 3122–3123. doi:10.1021/ja809873u |
| 38. | de Nanteuil, F.; Serrano, E.; Perrotta, D.; Waser, J. J. Am. Chem. Soc. 2014, 136, 6239–6242. doi:10.1021/ja5024578 |
| 43. | Xiong, H.; Xu, H.; Liao, S.; Xie, Z.; Tang, Y. J. Am. Chem. Soc. 2013, 135, 7851–7854. doi:10.1021/ja4042127 |
| 46. | Sträter, N.; Lipscomb, W. N.; Klabunde, T.; Krebs, B. Angew. Chem., Int. Ed. Engl. 1996, 35, 2024–2055. doi:10.1002/anie.199620241 |
| 47. | Sawamura, M.; Sudoh, M.; Ito, Y. J. Am. Chem. Soc. 1996, 118, 3309–3310. doi:10.1021/ja954223e |
| 48. | Xu, H.; Zuend, S. J.; Woll, M. G.; Tao, Y.; Jacobsen, E. N. Science 2010, 327, 986–990. doi:10.1126/science.1182826 |
| 49. | Krautwald, S.; Sarlah, D.; Schafroth, M. A.; Carreira, E. M. Science 2013, 340, 1065–1068. doi:10.1126/science.1237068 |
| 50. | Allen, A. E.; MacMillan, D. W. C. Chem. Sci. 2012, 3, 633–658. doi:10.1039/c2sc00907b |
| 51. | Ahire, M. M.; Mhaske, S. B. Angew. Chem., Int. Ed. 2014, 53, 7038–7042. doi:10.1002/anie.201400623 |
| 52. | Meazza, M.; Ceban, V.; Pitak, M. B.; Coles, S. J.; Rios, R. Chem. – Eur. J. 2014, 20, 16853–16857. doi:10.1002/chem.201404565 |
| 53. | Logan, K. M.; Smith, K. B.; Brown, M. K. Angew. Chem., Int. Ed. 2015, 54, 5228–5231. doi:10.1002/anie.201500396 |
| 54. | Zhu, S.; Zhang, J.; Chen, K.; Jiang, H. Angew. Chem., Int. Ed. 2015, 54, 9414–9418. doi:10.1002/anie.201504964 |
| 55. | Peng, H.; Akhmedov, N. G.; Liang, Y.-F.; Jiao, N.; Shi, X. J. Am. Chem. Soc. 2015, 137, 8912–8915. doi:10.1021/jacs.5b05415 |
| 42. | de Nanteuil, F.; Waser, J. Angew. Chem., Int. Ed. 2011, 50, 12075–12079. doi:10.1002/anie.201106255 |
| 1. | Danishefsky, S. Acc. Chem. Res. 1979, 12, 66–72. doi:10.1021/ar50134a004 |
| 3. | Reissig, H.-U. Organic Synthesis via Cyclopropanes: Principles and Applications. In Cyclopropyl Group; Rappoport, Z., Ed.; PATAI'S Chemistry of Functional Groups, Vol. 1 and 2; John Wiley & Sons: Chichester, United Kingdom, 1987; pp 375–443. doi:10.1002/0470023449.ch8 |
| 38. | de Nanteuil, F.; Serrano, E.; Perrotta, D.; Waser, J. J. Am. Chem. Soc. 2014, 136, 6239–6242. doi:10.1021/ja5024578 |
| 41. | Xu, H.; Qu, J.-P.; Liao, S.; Xiong, H.; Tang, Y. Angew. Chem., Int. Ed. 2013, 52, 4004–4007. doi:10.1002/anie.201300032 |
| 45. | Trost, B. M.; Morris, P. J.; Sprague, S. J. J. Am. Chem. Soc. 2012, 134, 17823–17831. doi:10.1021/ja309003x |
| 1. | Danishefsky, S. Acc. Chem. Res. 1979, 12, 66–72. doi:10.1021/ar50134a004 |
| 3. | Reissig, H.-U. Organic Synthesis via Cyclopropanes: Principles and Applications. In Cyclopropyl Group; Rappoport, Z., Ed.; PATAI'S Chemistry of Functional Groups, Vol. 1 and 2; John Wiley & Sons: Chichester, United Kingdom, 1987; pp 375–443. doi:10.1002/0470023449.ch8 |
© 2016 Zhu et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)