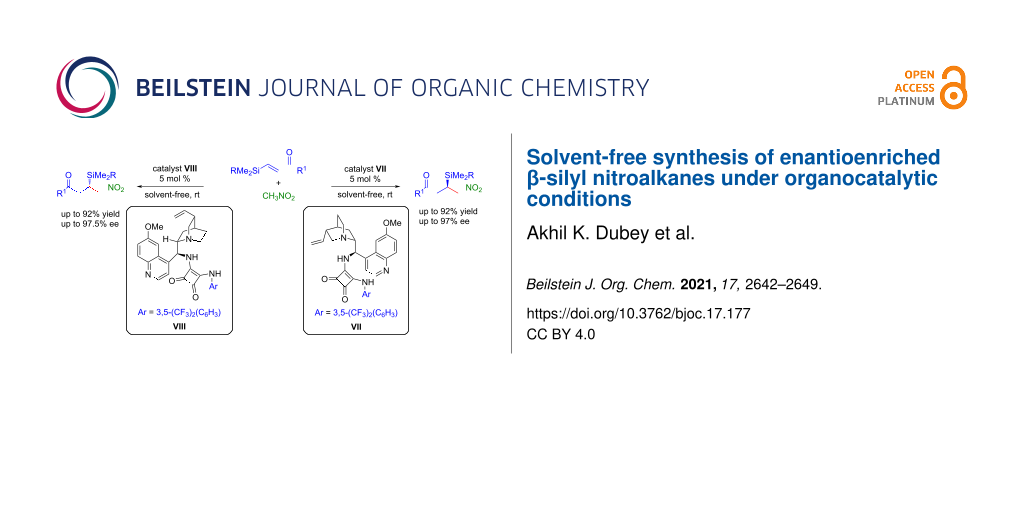
Abstract
An enantioselective 1,4-conjugate addition of nitromethane to β-silyl α,β-unsaturated carbonyl compounds catalyzed by bifunctional squaramide catalysts has been developed. This methodology offers both enantiomers of β-silyl nitroalkanes in good to excellent yields (up to 92%) and enantioselectivities (up to 97.5% ee) under solvent-free conditions at room temperature. Control experiments reveal that the presence of a β-silyl group in the enones is crucial for high reactivity under the optimized reaction conditions.

Graphical Abstract
Introduction
Enantioenriched organosilanes are attractive molecules in organic synthesis owing to their potential applications in stereoselective synthesis [1,2]. The unique sterical and electronical features of the C–Si bond can induce stereodifferentiation at the adjacent prostereogenic center in organic transformations [2]. In addition, the C–Si bond can be oxidized to a hydroxy group by Tamao–Fleming oxidation [3,4] or to an alkene unit via protodesilylation [5,6]. Many complex natural products, bioactive molecules, and drug molecules have been synthesized on exploitation of the above-mentioned properties of organosilanes [2,7-14]. A number of efficient catalytic enantioselective methods has been developed for the synthesis of chiral organosilanes [15-24]. Out of the chiral organosilanes, nitrosilanes are important synthetic targets as they are precursors of valuable β-aminosilanes [25-27]. Although there is huge success in the synthesis of enantioenriched organosilanes, catalytic routes to synthesize chiral β-nitrosilanes and in general nitrosilanes have not been well explored. Kobayashi and co-workers realized the synthesis of enantioenriched β-nitrosilanes through a Cu(II)–chiral bipyridine complex catalyzed enantioselective silyl transfer reaction to nitroalkenes using Suginome’s silylboron reagent (Scheme 1a) [28]. Recently, we have reported the synthesis of chiral β-nitrosilanes via an organocatalytic conjugate addition of nitromethane to β-silylmethylene malonates (Scheme 1b) [29]. As the catalytic enantioselective route is limited to accessible β-nitrosilanes, there is an urgent need to develop efficient catalytic protocols to deliver enantioenriched β-nitrosilanes from easily available starting materials.
Metal-catalyzed reaction of various nucleophiles to β-silyl α,β-unsaturated carbonyl compounds were documented as one of the straightforward and atom-economic approaches for the facile synthesis of chiral organosilanes (Scheme 1c–f) [30-33]. Recently, the aforementioned reaction under organocatalytic conditions has gained attention [34-36]. In this context, Huang, Fu and co-workers reported carbene-catalyzed enantioselective formal [4 + 2] annulation reactions of β-silyl enones with enals and with active acetic esters (Scheme 1g) for the preparation of chiral organosilanes [34-36]. Very recently, during the final stage of our work, the same group disclosed an organocatalyzed conjugate addition of thiols to β-silyl enones for the synthesis of chiral α-mercaptosilanes (Scheme 1g) [36].
![[1860-5397-17-177-i1]](/bjoc/content/inline/1860-5397-17-177-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Selected methods for the synthesis of enantioenriched β-silyl nitroalkanes, synthesis of chiral organosilanes from β-silyl α,β-unsaturated carbonyl compounds, and the present work.
Scheme 1: Selected methods for the synthesis of enantioenriched β-silyl nitroalkanes, synthesis of chiral org...
As a part of our ongoing program for the development of asymmetric catalytic approaches for the synthesis of enantioenriched organosilanes [29,37,38], we present herein an organocatalyzed conjugate addition reaction of nitromethane to β-silyl enones to afford chiral β-silyl nitroalkanes (Scheme 1). Notably, the developed method was not only carried out under solvent-free conditions at room temperature but was found to be tolerant to moisture and air. Therefore, this method offers an attractive and robust option for the preparation of chiral β-silyl nitroalkanes. In sharp contrast to the aforesaid reaction, organocatalytic conjugate addition reactions of nitroalkanes to enones have been well studied [39-43]. To the best of our knowledge, organocatalyzed or metal-catalyzed enantioselective conjugate additions of nitroalkanes to β-silyl enones are not yet known.
Results and Discussion
The optimization study began with the conjugate addition reaction between β-TMS enone 1a and nitromethane (2) as the model reaction. An uncatalyzed background reaction was not observed while performing the model reaction in toluene as a solvent at 30 °C for 24 h. To our delight, when the same reaction was carried out in presence of 5 mol % catalyst I in toluene at 30 °C for 48 h, the desired product 3a was obtained in 84% yield with 60% ee (Table 1, entry 1). Catalyst II was found to be unproductive as only 25% conversion of β-TMS enone 1a was observed (Table 1, entry 2). Gratifyingly, catalyst III furnished product ent-3a in 85% yield (Table 1, entry 3) with excellent enantioselectivity (94% ee). Whereas catalyst IV gave ent-3a in 85% yield with slightly lower enantioselectivity (91% ee) as compared to catalyst III (Table 1, entry 4). Catalyst V also led to product 3a in 66% yield and 78% ee (Table 1, entry 5). Catalyst VI, a pseudoenantiomer of catalyst V delivered ent-3a in 78% yield with 80% ee (Table 1, entry 6). The catalytic performance of the squaramide catalysts was also explored for the model reaction. Catalyst VII afforded the conjugate addition product 3a in 78% yield with excellent enantiopurity of 97% ee (Table 1, entry 7). A solvent survey (see Supporting Information File 1 for details) revealed that toluene is the most suitable solvent. Next, we targeted to make the reaction more time economical under mild conditions. For this purpose, the reaction was performed at different concentrations of the reaction mixture (Table 1, entries 8–11). It was observed that time required for completion of the reaction decreased with an increase of concentration of the reaction mixture while the enantiopurity of the product 3a remained unchanged (Table 1, entries 7–9). Next, the model reaction was performed using 10 equivalents of nitromethane (2) in the presence of 5 mol % catalyst VII under solvent-free conditions, and was complete within 24 h without affecting the enantioselectivity of product 3a (Table 1, entry 10). Reducing the loading of nitromethane (2) to 5 equivalents, a slight drop in yield (82%) of product 3a was observed whereas the enantioselectivity (97% ee) remained the same (Table 1, entry11). Upon further reduction in the loading of nitromethane (2) to 2.5 equivalents, the yield (82%), enantioselectivity (97% ee), and reaction time were not affected (Table 1, entry 12). Moreover, the reaction became sluggish when conducting the reaction with 2.5 mol % of the catalyst VII while keeping other parameters fixed (Table 1, entry 13). Performing the reaction with catalyst VIII, the pseudoenantiomeric catalyst of VII, furnished ent-3a in 80% yield and 94% ee (Table 1, entry 14). From the aforementioned studies, compromising slight lower yield of 3a, we set up the optimization conditions as: For 3a, 1a (0.2 mmol), 2 (0.5 mmol), 5 mol % of catalyst VII at 30–32 °C (Table 1, entry 12) and for ent-3a, 1a (0.2 mmol), 2 (0.5 mmol), 5 mol % of catalyst VIII at 30–32 °C (Table 1, entry 14).
Table 1: Catalysts screening and optimization of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-177-i6.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Cat. | Solvent (mL) | 2a (equiv) | Time (h) | Yield (%)b | ee (%)c |
| 1 | I | toluene (0.4) | 10 | 48 | 84 (98) | 60 |
| 2 | II | toluene (0.4) | 10 | 48 | ND (25) | ND |
| 3 | III | toluene (0.4) | 10 | 48 | 85 (>99) | −94d |
| 4 | IV | toluene (0.4) | 10 | 48 | 85 (>99) | −91d |
| 5 | V | toluene (0.4) | 10 | 48 | 66 (90) | 78 |
| 6 | VI | toluene (0.4) | 10 | 48 | 78 (97) | −80d |
| 7 | VII | toluene (0.4) | 10 | 48 | 78 (>99) | 97 |
| 8 | VII | toluene (0.2) | 10 | 42 | 78 (>99) | 97 |
| 9 | VII | toluene (0.1) | 10 | 24 | 80 (>99) | 97 |
| 10 | VII | – | 10 | 24 | 83 (>99) | 97 |
| 11 | VII | – | 5 | 24 | 82 (>99) | 97 |
| 12 | VII | – | 2.5 | 24 | 82 (>99) | 97 |
| 13e | VII | – | 10 | 24 | 56 (85) | 97 |
| 14 | VIII | – | 2.5 | 24 | 80 (>97) | −94d |
aReaction conditions: 1a (0.2 mmol), 2 (0.5–2.0 mmol), catalyst (0.01 mmol, 5 mol %) in toluene or neat at 30–32 °C. bIsolated yield after column chromatography, % of conversion of the starting material 1a is given in parentheses, determined by 1H NMR analysis of the crude reaction mixture. cDetermined by HPLC using a chiralpak OD-H column. dOpposite enantiomer. e2.5 mol % of the catalyst VII was used.
With the acceptable optimized reaction conditions in hand, we next investigated the generality and limitations of this enantioselective conjugate addition reaction. Under the optimized reaction conditions, the conjugate addition reaction of nitromethane (2) to a variety of β-silylenones 1 was carried out and the results are summarized in Scheme 2. β-Silylenones bearing electron-donating, electron-withdrawing groups and halogen substituents in the meta or para position of the phenyl ring reacted smoothly and furnished the desired products 3a–k in good to excellent yields (71.5–92%) and enantioselectivities (76–97.5% ee). The β-silylenone with a strong electron-withdrawing group (cyano) attached to the phenyl ring, was found to be most reactive as the reaction completed within 4 h and afforded the product 3e in good yield (88%) and enantioselectivity (95.5% ee). The β-silylenone with a naphthyl substituent also took part in the conjugate addition reaction and gave the corresponding product 3j in good yield (83%) and enantioselectivity (76% ee). The reaction also tolerated a 2-thienyl-substituted β-silylenone and the desired product 3k was obtained in good yield (88%) and enantioselectivity (97.5% ee). However, β-silylbutenone 1l failed to participate in the conjugate addition reaction with nitromethane under the optimized reaction conditions. Pleasingly, using 9-amino-9-deoxyepihydroquinidine (IX)–benzoic acid as organocatalyst system (see Supporting Information File 1 for details) promoted the addition reaction and product 3l was formed in good yield (79%) and excellent enantioselectivity (99% ee). The conjugate addition reaction between malononitrile and β-silylenone 1a was also investigated using 5 mol % of catalyst VII under the optimized reaction conditions. To our delight, the reaction completed within 4 h and the desired product 3m was isolated in excellent yield (97%) with moderate enantioselectivity (52% ee). β-Silylenone 2n bearing a o-chloro substituent in the aromatic ring remained unreactive under the optimized reaction conditions probably due to steric hindrance.
![[1860-5397-17-177-i2]](/bjoc/content/inline/1860-5397-17-177-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of substrates. Reaction conditions: 1 (0.2 mmol), 2 (0.5 mmol), catalyst VII (0.01 mmol, 5 mol %) at 30 °C. aIsolated yield of 3 after column chromatography. bConversion in % of the starting material 1 is given in parentheses, determined by 1H NMR analysis of the crude reaction mixture. cDetermined by HPLC using a chiral stationary phase. dEnantiomers could not be separated by AD-H, OD-H, OJ-H, and AS-H columns. eReaction conditions for 3l: 1l (0.2 mmol), 2 (2 mmol), catalyst IX (0.04 mmol, 20 mol %), benzoic acid (0.08 mmol, 40 mol %) in 0.9 mL toluene as the solvent (see Supporting Information File 1). fMalonitrile (0.6 mmol, 3 equiv) was used.
Scheme 2: Scope of substrates. Reaction conditions: 1 (0.2 mmol), 2 (0.5 mmol), catalyst VII (0.01 mmol, 5 mo...
The facile synthesis of both enantiomers of the targeted compounds is of paramount importance since biological activities are dictated by the absolute configuration of the products. To our delight, catalyst VIII, the pseudoenantiomeric catalyst of VII, allowed to synthesize the enantiomeric products ent-3 (Scheme 3) in high yields and enantioselectivities comparable to the corresponding enantiomers 3 under the optimized reaction conditions. The same set of β-silylenones was explored and an almost similar trend in reactivities, yields as well as enantioselectivities was observed.
![[1860-5397-17-177-i3]](/bjoc/content/inline/1860-5397-17-177-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of ent-3. Reaction conditions: 1 (0.2 mmol), 2 (0.5 mmol), catalyst VIII (0.01 mmol, 5 mol %) at 30 °C. aIsolated yield of ent-3 after column chromatography. bConversion in % of the starting material 1 is given in the parentheses, determined by 1H NMR analysis of the crude reaction mixture. cDetermined by HPLC using chiral stationary phase.
Scheme 3: Synthesis of ent-3. Reaction conditions: 1 (0.2 mmol), 2 (0.5 mmol), catalyst VIII (0.01 mmol, 5 mo...
To probe the role of the β-silyl group, the reaction of tert-butyl-substituted enone 3o and nitromethane (2) was conducted under the standard reaction conditions using catalyst VII or VIII, affording only trace amounts of products 4 or ent-4 even after stirring for 48 h [44]. When the same reaction was performed in the presence of 10 equivalents of nitromethane using catalyst VII, the product 4 was isolated in 26% yield and 89.5% ee after 96 h whereas the catalyst VIII led to ent-4 in 25% yield and 95% ee (Scheme 4). This observation confirmed that the presence of the β-silyl group in the enones played a key role in the high reactivity under the optimized reaction conditions.
![[1860-5397-17-177-i4]](/bjoc/content/inline/1860-5397-17-177-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Organocatalytic 1,4-conjuagte addition of nitromethane (2) to enone 3o.
Scheme 4: Organocatalytic 1,4-conjuagte addition of nitromethane (2) to enone 3o.
The stereochemistry of the silicon-substituted chiral center in compound ent-3k was found to adopt “(S)” configuration which was unambiguously established by single crystal X-ray diffraction analysis (Figure 1) [45].
![[1860-5397-17-177-1]](/bjoc/content/figures/1860-5397-17-177-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Single crystal X-ray structure of ent-3k (CCDC 2097263).
Figure 1: Single crystal X-ray structure of ent-3k (CCDC 2097263).
To prove the scalability of this synthetic method, we examined the synthesis of 3c and ent-3d in a 1 mmol scale (Scheme 5). The products 3c and ent-3d were isolated even with better yields while the enantiomeric excess was unperturbed.
![[1860-5397-17-177-i5]](/bjoc/content/inline/1860-5397-17-177-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Preparative scale synthesis of 3c and ent-3d.
Scheme 5: Preparative scale synthesis of 3c and ent-3d.
Conclusion
In summary, we have outlined bifunctional squaramide-catalyzed 1,4-conjugate addition reaction of nitromethane to β-silyl α,β-unsaturated carbonyl compounds to access a series of chiral β-silyl nitroalkanes in high yields and good to excellent enantioselectivities at room temperature. The notable features of this reaction are access to both the (R) and (S) enantiomers of the products, solvent-free synthesis, mild reaction conditions, low catalyst loading, and use of only a small excess of nitromethane (2.5 equivalents with respect to limiting reagent).
Supporting Information
| Supporting Information File 1: Experimental data and copies of spectra. | ||
| Format: PDF | Size: 3.3 MB | Download |
References
-
Fleming, I. Sci. Synth. 2002, 4, 927–946. doi:10.1055/sos-sd-004-01012
Return to citation in text: [1] -
Fleming, I.; Barbero, A.; Walter, D. Chem. Rev. 1997, 97, 2063–2192. doi:10.1021/cr941074u
Return to citation in text: [1] [2] [3] -
Tamao, K.; Ishida, N.; Kumada, M. J. Org. Chem. 1983, 48, 2120–2122. doi:10.1021/jo00160a046
Return to citation in text: [1] -
Fleming, I.; Henning, R.; Plaut, H. J. Chem. Soc., Chem. Commun. 1984, 29–31. doi:10.1039/c39840000029
Return to citation in text: [1] -
Radner, F.; Wistrand, L.-G. Tetrahedron Lett. 1995, 36, 5093–5094. doi:10.1016/0040-4039(95)00948-c
Return to citation in text: [1] -
Yao, W.; Li, R.; Jiang, H.; Han, D. J. Org. Chem. 2018, 83, 2250–2255. doi:10.1021/acs.joc.7b03139
Return to citation in text: [1] -
Langkopf, E.; Schinzer, D. Chem. Rev. 1995, 95, 1375–1408. doi:10.1021/cr00037a011
Return to citation in text: [1] -
Denmark, S. E.; Liu, J. H.-C. Angew. Chem., Int. Ed. 2010, 49, 2978–2986. doi:10.1002/anie.200905657
Return to citation in text: [1] -
Fleming, I.; Lawrence, N. J. Tetrahedron Lett. 1990, 31, 3645–3648. doi:10.1016/s0040-4039(00)94466-6
Return to citation in text: [1] -
Fleming, I.; Ghosh, S. K. J. Chem. Soc., Chem. Commun. 1994, 2287–2288. doi:10.1039/c39940002287
Return to citation in text: [1] -
Fleming, I.; Lee, D. Tetrahedron Lett. 1996, 37, 6929–6930. doi:10.1016/0040-4039(96)01519-5
Return to citation in text: [1] -
Lee, J.; Panek, J. S. Org. Lett. 2011, 13, 502–505. doi:10.1021/ol102848w
Return to citation in text: [1] -
Wu, J.; Panek, J. S. Angew. Chem., Int. Ed. 2010, 49, 6165–6168. doi:10.1002/anie.201002220
Return to citation in text: [1] -
Pace, V.; Rae, J. P.; Procter, D. J. Org. Lett. 2014, 16, 476–479. doi:10.1021/ol4033623
Return to citation in text: [1] -
Lee, K.-s.; Hoveyda, A. H. J. Am. Chem. Soc. 2010, 132, 2898–2900. doi:10.1021/ja910989n
Return to citation in text: [1] -
Mita, T.; Sugawara, M.; Saito, K.; Sato, Y. Org. Lett. 2014, 16, 3028–3031. doi:10.1021/ol501143c
Return to citation in text: [1] -
Rong, J.; Collados, J. F.; Ortiz, P.; Jumde, R. P.; Otten, E.; Harutyunyan, S. R. Nat. Commun. 2016, 7, 13780. doi:10.1038/ncomms13780
Return to citation in text: [1] -
Sato, Y.; Takagi, C.; Shintani, R.; Nozaki, K. Angew. Chem., Int. Ed. 2017, 56, 9211–9216. doi:10.1002/anie.201705500
Return to citation in text: [1] -
Duong, H. Q.; Sieburth, S. M. J. Org. Chem. 2018, 83, 5398–5409. doi:10.1021/acs.joc.8b00116
Return to citation in text: [1] -
Su, B.; Lee, T.; Hartwig, J. F. J. Am. Chem. Soc. 2018, 140, 18032–18038. doi:10.1021/jacs.8b10428
Return to citation in text: [1] -
Huang, M.-Y.; Yang, J.-M.; Zhao, Y.-T.; Zhu, S.-F. ACS Catal. 2019, 9, 5353–5357. doi:10.1021/acscatal.9b01187
Return to citation in text: [1] -
Zhu, J.; Chen, S.; He, C. J. Am. Chem. Soc. 2021, 143, 5301–5307. doi:10.1021/jacs.1c01106
Return to citation in text: [1] -
Zhang, L.; Oestreich, M. ACS Catal. 2021, 11, 3516–3522. doi:10.1021/acscatal.1c00436
Return to citation in text: [1] -
Kranidiotis‐Hisatomi, N.; Yi, H.; Oestreich, M. Angew. Chem., Int. Ed. 2021, 60, 13652–13655. doi:10.1002/anie.202102233
Return to citation in text: [1] -
Hayama, T.; Tomoda, S.; Takeuchi, Y.; Nomura, Y. Tetrahedron Lett. 1983, 24, 2795–2796. doi:10.1016/s0040-4039(00)88025-9
Return to citation in text: [1] -
Cunico, R. F. J. Org. Chem. 1990, 55, 4474–4478. doi:10.1021/jo00301a053
Return to citation in text: [1] -
Jiang, C.-R.; Zhao, C.-L.; Guo, H.-F.; He, W. Chem. Commun. 2016, 52, 7862–7865. doi:10.1039/c6cc03840a
Return to citation in text: [1] -
Kitanosono, T.; Zhu, L.; Liu, C.; Xu, P.; Kobayashi, S. J. Am. Chem. Soc. 2015, 137, 15422–15425. doi:10.1021/jacs.5b11418
Return to citation in text: [1] -
Chowdhury, R.; Dubey, A. K.; Ghosh, S. K. Eur. J. Org. Chem. 2020, 2962–2972. doi:10.1002/ejoc.202000306
Return to citation in text: [1] [2] -
Shintani, R.; Okamoto, K.; Hayashi, T. Org. Lett. 2005, 7, 4757–4759. doi:10.1021/ol051978+
Return to citation in text: [1] -
Balskus, E. P.; Jacobsen, E. N. J. Am. Chem. Soc. 2006, 128, 6810–6812. doi:10.1021/ja061970a
Return to citation in text: [1] -
Kacprzynski, M. A.; Kazane, S. A.; May, T. L.; Hoveyda, A. H. Org. Lett. 2007, 9, 3187–3190. doi:10.1021/ol071331k
Return to citation in text: [1] -
Zhao, K.; Loh, T.-P. Chem. – Eur. J. 2014, 20, 16764–16772. doi:10.1002/chem.201403849
Return to citation in text: [1] -
Zhang, Y.; Huang, J.; Guo, Y.; Li, L.; Fu, Z.; Huang, W. Angew. Chem., Int. Ed. 2018, 57, 4594–4598. doi:10.1002/anie.201800483
Return to citation in text: [1] [2] -
Zhang, Y.; Huang, X.; Guo, J.; Wei, C.; Gong, M.; Fu, Z. Org. Lett. 2020, 22, 9545–9550. doi:10.1021/acs.orglett.0c03589
Return to citation in text: [1] [2] -
Zhang, Y.; Guo, J.; Han, J.; Zhou, X.; Cao, W.; Fu, Z. Org. Biomol. Chem. 2021, 19, 6412–6416. doi:10.1039/d1ob00981h
Return to citation in text: [1] [2] [3] -
Chowdhury, R.; Dubey, A. K.; Ghosh, S. K. J. Org. Chem. 2019, 84, 2404–2414. doi:10.1021/acs.joc.8b02412
Return to citation in text: [1] -
Chowdhury, R.; Dubey, A. K.; Ghosh, S. K. Asian J. Org. Chem. 2021, 10, 1173–1183. doi:10.1002/ajoc.202100120
Return to citation in text: [1] -
Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Org. Lett. 2005, 7, 1967–1969. doi:10.1021/ol050431s
Return to citation in text: [1] -
Wang, J.; Li, H.; Zu, L.; Jiang, W.; Xie, H.; Duan, W.; Wang, W. J. Am. Chem. Soc. 2006, 128, 12652–12653. doi:10.1021/ja065187u
Return to citation in text: [1] -
Yang, W.; Du, D.-M. Org. Lett. 2010, 12, 5450–5453. doi:10.1021/ol102294g
Return to citation in text: [1] -
Zhang, G.; Zhu, C.; Liu, D.; Pan, J.; Zhang, J.; Hu, D.; Song, B. Tetrahedron 2017, 73, 129–136. doi:10.1016/j.tet.2016.11.063
Return to citation in text: [1] -
Cholewiak, A.; Adamczyk, K.; Kopyt, M.; Kasztelan, A.; Kwiatkowski, P. Org. Biomol. Chem. 2018, 16, 4365–4371. doi:10.1039/c8ob00561c
Return to citation in text: [1] -
The ratio of 3o/4 = 90:10 (determined by 1H NMR analysis of the crude reaction mixture).
Return to citation in text: [1] -
The crystallographic data (CCDC 2097263) for ent-3k, can be obtained free of charge from the Cambridge crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Return to citation in text: [1]
| 45. | The crystallographic data (CCDC 2097263) for ent-3k, can be obtained free of charge from the Cambridge crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. |
| 1. | Fleming, I. Sci. Synth. 2002, 4, 927–946. doi:10.1055/sos-sd-004-01012 |
| 2. | Fleming, I.; Barbero, A.; Walter, D. Chem. Rev. 1997, 97, 2063–2192. doi:10.1021/cr941074u |
| 2. | Fleming, I.; Barbero, A.; Walter, D. Chem. Rev. 1997, 97, 2063–2192. doi:10.1021/cr941074u |
| 7. | Langkopf, E.; Schinzer, D. Chem. Rev. 1995, 95, 1375–1408. doi:10.1021/cr00037a011 |
| 8. | Denmark, S. E.; Liu, J. H.-C. Angew. Chem., Int. Ed. 2010, 49, 2978–2986. doi:10.1002/anie.200905657 |
| 9. | Fleming, I.; Lawrence, N. J. Tetrahedron Lett. 1990, 31, 3645–3648. doi:10.1016/s0040-4039(00)94466-6 |
| 10. | Fleming, I.; Ghosh, S. K. J. Chem. Soc., Chem. Commun. 1994, 2287–2288. doi:10.1039/c39940002287 |
| 11. | Fleming, I.; Lee, D. Tetrahedron Lett. 1996, 37, 6929–6930. doi:10.1016/0040-4039(96)01519-5 |
| 12. | Lee, J.; Panek, J. S. Org. Lett. 2011, 13, 502–505. doi:10.1021/ol102848w |
| 13. | Wu, J.; Panek, J. S. Angew. Chem., Int. Ed. 2010, 49, 6165–6168. doi:10.1002/anie.201002220 |
| 14. | Pace, V.; Rae, J. P.; Procter, D. J. Org. Lett. 2014, 16, 476–479. doi:10.1021/ol4033623 |
| 39. | Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Org. Lett. 2005, 7, 1967–1969. doi:10.1021/ol050431s |
| 40. | Wang, J.; Li, H.; Zu, L.; Jiang, W.; Xie, H.; Duan, W.; Wang, W. J. Am. Chem. Soc. 2006, 128, 12652–12653. doi:10.1021/ja065187u |
| 41. | Yang, W.; Du, D.-M. Org. Lett. 2010, 12, 5450–5453. doi:10.1021/ol102294g |
| 42. | Zhang, G.; Zhu, C.; Liu, D.; Pan, J.; Zhang, J.; Hu, D.; Song, B. Tetrahedron 2017, 73, 129–136. doi:10.1016/j.tet.2016.11.063 |
| 43. | Cholewiak, A.; Adamczyk, K.; Kopyt, M.; Kasztelan, A.; Kwiatkowski, P. Org. Biomol. Chem. 2018, 16, 4365–4371. doi:10.1039/c8ob00561c |
| 5. | Radner, F.; Wistrand, L.-G. Tetrahedron Lett. 1995, 36, 5093–5094. doi:10.1016/0040-4039(95)00948-c |
| 6. | Yao, W.; Li, R.; Jiang, H.; Han, D. J. Org. Chem. 2018, 83, 2250–2255. doi:10.1021/acs.joc.7b03139 |
| 44. | The ratio of 3o/4 = 90:10 (determined by 1H NMR analysis of the crude reaction mixture). |
| 3. | Tamao, K.; Ishida, N.; Kumada, M. J. Org. Chem. 1983, 48, 2120–2122. doi:10.1021/jo00160a046 |
| 4. | Fleming, I.; Henning, R.; Plaut, H. J. Chem. Soc., Chem. Commun. 1984, 29–31. doi:10.1039/c39840000029 |
| 36. | Zhang, Y.; Guo, J.; Han, J.; Zhou, X.; Cao, W.; Fu, Z. Org. Biomol. Chem. 2021, 19, 6412–6416. doi:10.1039/d1ob00981h |
| 2. | Fleming, I.; Barbero, A.; Walter, D. Chem. Rev. 1997, 97, 2063–2192. doi:10.1021/cr941074u |
| 29. | Chowdhury, R.; Dubey, A. K.; Ghosh, S. K. Eur. J. Org. Chem. 2020, 2962–2972. doi:10.1002/ejoc.202000306 |
| 37. | Chowdhury, R.; Dubey, A. K.; Ghosh, S. K. J. Org. Chem. 2019, 84, 2404–2414. doi:10.1021/acs.joc.8b02412 |
| 38. | Chowdhury, R.; Dubey, A. K.; Ghosh, S. K. Asian J. Org. Chem. 2021, 10, 1173–1183. doi:10.1002/ajoc.202100120 |
| 29. | Chowdhury, R.; Dubey, A. K.; Ghosh, S. K. Eur. J. Org. Chem. 2020, 2962–2972. doi:10.1002/ejoc.202000306 |
| 34. | Zhang, Y.; Huang, J.; Guo, Y.; Li, L.; Fu, Z.; Huang, W. Angew. Chem., Int. Ed. 2018, 57, 4594–4598. doi:10.1002/anie.201800483 |
| 35. | Zhang, Y.; Huang, X.; Guo, J.; Wei, C.; Gong, M.; Fu, Z. Org. Lett. 2020, 22, 9545–9550. doi:10.1021/acs.orglett.0c03589 |
| 36. | Zhang, Y.; Guo, J.; Han, J.; Zhou, X.; Cao, W.; Fu, Z. Org. Biomol. Chem. 2021, 19, 6412–6416. doi:10.1039/d1ob00981h |
| 28. | Kitanosono, T.; Zhu, L.; Liu, C.; Xu, P.; Kobayashi, S. J. Am. Chem. Soc. 2015, 137, 15422–15425. doi:10.1021/jacs.5b11418 |
| 34. | Zhang, Y.; Huang, J.; Guo, Y.; Li, L.; Fu, Z.; Huang, W. Angew. Chem., Int. Ed. 2018, 57, 4594–4598. doi:10.1002/anie.201800483 |
| 35. | Zhang, Y.; Huang, X.; Guo, J.; Wei, C.; Gong, M.; Fu, Z. Org. Lett. 2020, 22, 9545–9550. doi:10.1021/acs.orglett.0c03589 |
| 36. | Zhang, Y.; Guo, J.; Han, J.; Zhou, X.; Cao, W.; Fu, Z. Org. Biomol. Chem. 2021, 19, 6412–6416. doi:10.1039/d1ob00981h |
| 25. | Hayama, T.; Tomoda, S.; Takeuchi, Y.; Nomura, Y. Tetrahedron Lett. 1983, 24, 2795–2796. doi:10.1016/s0040-4039(00)88025-9 |
| 26. | Cunico, R. F. J. Org. Chem. 1990, 55, 4474–4478. doi:10.1021/jo00301a053 |
| 27. | Jiang, C.-R.; Zhao, C.-L.; Guo, H.-F.; He, W. Chem. Commun. 2016, 52, 7862–7865. doi:10.1039/c6cc03840a |
| 15. | Lee, K.-s.; Hoveyda, A. H. J. Am. Chem. Soc. 2010, 132, 2898–2900. doi:10.1021/ja910989n |
| 16. | Mita, T.; Sugawara, M.; Saito, K.; Sato, Y. Org. Lett. 2014, 16, 3028–3031. doi:10.1021/ol501143c |
| 17. | Rong, J.; Collados, J. F.; Ortiz, P.; Jumde, R. P.; Otten, E.; Harutyunyan, S. R. Nat. Commun. 2016, 7, 13780. doi:10.1038/ncomms13780 |
| 18. | Sato, Y.; Takagi, C.; Shintani, R.; Nozaki, K. Angew. Chem., Int. Ed. 2017, 56, 9211–9216. doi:10.1002/anie.201705500 |
| 19. | Duong, H. Q.; Sieburth, S. M. J. Org. Chem. 2018, 83, 5398–5409. doi:10.1021/acs.joc.8b00116 |
| 20. | Su, B.; Lee, T.; Hartwig, J. F. J. Am. Chem. Soc. 2018, 140, 18032–18038. doi:10.1021/jacs.8b10428 |
| 21. | Huang, M.-Y.; Yang, J.-M.; Zhao, Y.-T.; Zhu, S.-F. ACS Catal. 2019, 9, 5353–5357. doi:10.1021/acscatal.9b01187 |
| 22. | Zhu, J.; Chen, S.; He, C. J. Am. Chem. Soc. 2021, 143, 5301–5307. doi:10.1021/jacs.1c01106 |
| 23. | Zhang, L.; Oestreich, M. ACS Catal. 2021, 11, 3516–3522. doi:10.1021/acscatal.1c00436 |
| 24. | Kranidiotis‐Hisatomi, N.; Yi, H.; Oestreich, M. Angew. Chem., Int. Ed. 2021, 60, 13652–13655. doi:10.1002/anie.202102233 |
| 30. | Shintani, R.; Okamoto, K.; Hayashi, T. Org. Lett. 2005, 7, 4757–4759. doi:10.1021/ol051978+ |
| 31. | Balskus, E. P.; Jacobsen, E. N. J. Am. Chem. Soc. 2006, 128, 6810–6812. doi:10.1021/ja061970a |
| 32. | Kacprzynski, M. A.; Kazane, S. A.; May, T. L.; Hoveyda, A. H. Org. Lett. 2007, 9, 3187–3190. doi:10.1021/ol071331k |
| 33. | Zhao, K.; Loh, T.-P. Chem. – Eur. J. 2014, 20, 16764–16772. doi:10.1002/chem.201403849 |
© 2021 Dubey and Chowdhury; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)