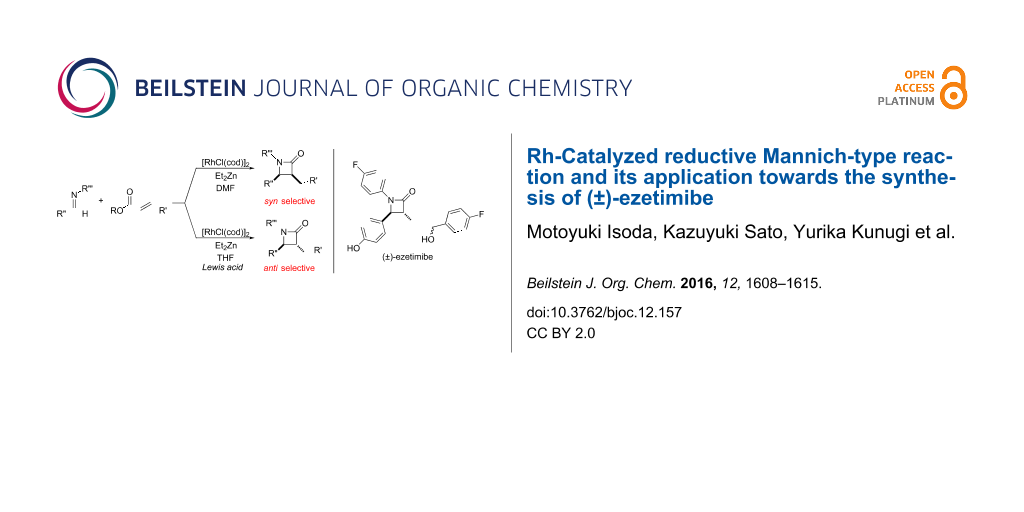
Abstract
An effective synthesis for syn-β-lactams was achieved using a Rh-catalyzed reductive Mannich-type reaction. A rhodium–hydride complex (Rh–H) derived from diethylzinc (Et2Zn) and a Rh catalyst was used for the 1,4-reduction of an α,β-unsaturated ester to give a Reformatsky-type reagent, which in turn, reacted with an imine to give the syn-β-lactam. Additionally, the reaction was applied to the synthesis of (±)-ezetimibe, a potent β-lactamic cholesterol absorption inhibitor.

Graphical Abstract
Introduction
The Mannich reaction is an important and classical C–C bond-forming reaction between an enolizable carbonyl compound and an imine to give the corresponding β-aminocarbonyl compound. For example, Shibasaki and his colleague reported the asymmetric Mannich reaction using a Lewis acid catalyst [1]. (L)-Proline is known as an excellent promoter for the Mannich reaction [2-6], and besides this, the reaction of the silyl enol ether derivatives with imines was used as an effective method [7-9]. In this situation, a wide variety of Mannich-type reactions have been reported to give β-amino esters and/or β-lactams by using metal enolates [10-17].
In contrast, most of reductive Mannich-type reactions using imines and α,β-unsaturated carbonyl compounds gave β-amino esters, but there are only a few reports for a direct synthesis of β-lactams by reductive Mannich-type reactions [14,18-21]. We recently reported a reductive Mannich-type reaction using a combination of RhCl(PPh3)3 and Et2Zn to give the corresponding syn-β-lactams from α,β-unsaturated esters and imines in good to excellent yields together with a small amount of the β-amino esters (Scheme 1) [22,23]. The reaction is very noteworthy because the formation of syn-β-lactams is particularly rare [24-27]. Additionally, various imines can be used for this reaction, and the corresponding β-lactams showed very high syn selectivity. However, some of the reactions using β-substituted α,β-unsaturated esters did not afford the products or gave the products in very low yield (Scheme 2). Herein, we report an expansion for our previous reductive Mannich-type reaction and mechanistic studies for the stereoselectivity of this reaction. Finally the method is applied for the new synthesis of ezetimibe, a potent β-lactamic cholesterol absorption inhibitor.
![[1860-5397-12-157-i1]](/bjoc/content/inline/1860-5397-12-157-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The synthesis of syn-β-lactams using a reductive Mannich-type reaction.
Scheme 1: The synthesis of syn-β-lactams using a reductive Mannich-type reaction.
![[1860-5397-12-157-i2]](/bjoc/content/inline/1860-5397-12-157-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Previous results using β-substituted α,β-unsaturated esters.
Scheme 2: Previous results using β-substituted α,β-unsaturated esters.
Results and Discussion
First of all, we tried to solve the drawbacks of the previously reported method and found that changing the catalyst to [RhCl(cod)]2 greatly improved the reaction. The results from the reaction of the [RhCl(cod)]2 catalyst with various α,β-unsaturated esters are summarized in Table 1. Most of the synthesized β-lactams were obtained with dramatically improved yields, although the β,β-disubstituted α,β-unsaturated ester did not give the product owing to the bulkiness of the β-position (Table 1, entries 1–8). It is interesting that ethyl sorbate (2g) gave the corresponding anti-β-lactam 3Ag in a moderate yield but with an anti-β-lactam stereochemistry indicating that the reaction proceeded via 1,6-reduction, then the resulting nucleophile was trapped by the imine at the α-position (Table 1, entry 9). By using [RhCl(cod)]2, the α,β-unsaturated lactone was also converted to the product with a small improvement in the yield (Table 1, entry 11). Acrylamide 2i also gave the corresponding β-aminoamide 4Ai in a low yield (Table 1, entry 12).
Table 1: Rh-catalyzed Mannich-type reaction using various α,β-unsaturated esters.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-157-i7.svg?max-width=637&scale=1.0)
|
||||
| Entry | Substrate 2 | Rh cat. (mol %) | Product | Yield (%)a |
|---|---|---|---|---|
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-157-i8.svg?max-width=637&scale=1.0)
2a |
RhCl(PPh3)3 (2) |
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-157-i9.svg?max-width=637&scale=1.0)
3Aa |
88
[syn/anti = 96:4]b |
| 2 | [RhCl(cod)]2 (1) |
78
[syn/anti = 88:12]b |
||
| 3 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-157-i10.svg?max-width=637&scale=1.0)
2b |
RhCl(PPh3)3 (4) |
![[Graphic 5]](/bjoc/content/inline/1860-5397-12-157-i11.svg?max-width=637&scale=1.0)
3Ab |
nd |
| 4 | [RhCl(cod)]2 (2) | 66c | ||
| 5 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-12-157-i12.svg?max-width=637&scale=1.0)
2d |
RhCl(PPh3)3 (2) |
![[Graphic 7]](/bjoc/content/inline/1860-5397-12-157-i13.svg?max-width=637&scale=1.0)
3Ad |
34c |
| 6 | [RhCl(cod)]2 (2) | 77c | ||
| 7 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-12-157-i14.svg?max-width=637&scale=1.0)
2e |
[RhCl(cod)]2 (2) |
![[Graphic 9]](/bjoc/content/inline/1860-5397-12-157-i15.svg?max-width=637&scale=1.0)
3Ae |
98 |
| 8 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-12-157-i16.svg?max-width=637&scale=1.0)
2f |
[RhCl(cod)]2 (2) |
![[Graphic 11]](/bjoc/content/inline/1860-5397-12-157-i17.svg?max-width=637&scale=1.0)
3Af |
nd |
| 9 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-12-157-i18.svg?max-width=637&scale=1.0)
2g |
[RhCl(cod)]2 (2) |
![[Graphic 13]](/bjoc/content/inline/1860-5397-12-157-i19.svg?max-width=637&scale=1.0)
3Ag |
59d
[E/Z = 93:7]e |
| 10 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-12-157-i20.svg?max-width=637&scale=1.0)
2h |
RhCl(PPh3)3 (2) |
![[Graphic 15]](/bjoc/content/inline/1860-5397-12-157-i21.svg?max-width=637&scale=1.0)
3Ah |
11d |
| 11 | [RhCl(cod)]2 (2) | 20d | ||
| 12 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-12-157-i22.svg?max-width=637&scale=1.0)
2i |
[RhCl(cod)]2 (2) |
![[Graphic 17]](/bjoc/content/inline/1860-5397-12-157-i23.svg?max-width=637&scale=1.0)
4Ai |
27
[syn/anti = 18:82]b |
aIsolated yield. bDiastereomeric ratio [syn/anti] after purification. cThe syn product was obtained as the sole product. dThe anti product was obtained as the sole product. eE/Z ratio by 1H NMR.
In the reaction using α,β-unsaturated lactone 2h, the β-lactam anti-3Ah that has a hydroxy group on the side chain was obtained in a low yield (Table 1, entry 11). This result was of interest because the reaction is applicable to the synthesis of ezetimibe. Ezetimibe is an inhibitor of the cholesterol transporter Niemann–Pick C1 Like 1 Protein (NPC1L1), and is used as a strong cholesterol absorption inhibitor that reduces plasma low-density lipoprotein fraction (LDL-C) [28-30]. Recent studies on the function of NPC1L1 suggest that NPC1L1 may be involved with the internal uptake of the hepatitis C virus (HCV) and absorption of vitamin K [31,32]. Although it has a simple mono β-lactamic structure and appears easy to synthesize, there are relatively few reports for the synthesis of ezetimibe because of its three asymmetric centers [33-35].
First, we examined the reaction conditions using a pilot reaction using imine 1B and 5,6-dihydro-2H-pyran-2-one (2h) (Table 2). Unfortunately, the reaction conditions as above gave a complex mixture that did not contain the desired products as shown in Table 2, entry 1. Heating the reaction mixture at 80 °C gave β-aminolactone 4Bh in a very low yield (Table 2, entry 2). When DME and THF were used as the solvents, the desired β-lactam was obtained in low yields (Table 2, entries 3 and 4, respectively). We hypothesized that the low yield was caused by the instability of 2h and the low reactivity of imine 1B under the reaction conditions. Therefore, we added a Lewis acid to assist in the formation of the iminium salt in an effort to improve the reactivity. It was clear that Et2Zn was only working as a hydride source but was ineffective as the Lewis acid. Instead when zinc chloride (ZnCl2) was added into the mixture, the yield was improved to 30% (Table 2, entry 5). BF3·Et2O was found to be the most suitable Lewis acid, and the desired β-lactam (anti-3Bh) was obtained in moderate yield (Table 2, entry 9).
Table 2: Examination of the reaction conditions using a pilot reaction.
![[Graphic 18]](/bjoc/content/inline/1860-5397-12-157-i24.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Solvent | Temp. | Additive (equiv) | Yield (%)a | |
|---|---|---|---|---|---|
| 3Bh | 4Bh | ||||
| 1 | DMF | 0 °C → rt | none | 0 | 0 |
| 2 | DMF | 80 °C | none | 0 | 8 |
| 3 | DME | 0 °C → rt | none | 17 | 0 |
| 4 | THF | 0 °C → rt | none | 20 | 0 |
| 5 | THF | rt | ZnCl2 (1.2) | 30 | 0 |
| 6 | THF | rt | ZnCl2 (10 mol %) | 19 | 0 |
| 7 | THF | rt | InBr3 (1.2) | 0 | 0 |
| 8 | THF | rt | Al(OiPr)3 (1.2) | 0 | 9 |
| 9 | THF | rt | BF3·Et2O (1.2) | 46 | 0 |
aIsolated yield.
Based on these results, we applied this reaction to the synthesis of ezetimibe (Scheme 3). The reaction of imine 1B with lactone 2j proceeded smoothly and gave the desired β-lactam product, anti-3Bj, in 58% yield. Fortunately, since 2j was stable compared to 2h under the reaction conditions, the yield of β-lactam anti-3Bj was somewhat improved. After deprotection using Pd/C and H2 the target product, (±)-ezetimibe was obtained in 80% yield and its overall yield was 46%.
![[1860-5397-12-157-i3]](/bjoc/content/inline/1860-5397-12-157-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: A new synthetic route for ezetimibe.
Scheme 3: A new synthetic route for ezetimibe.
In some of our previous publications [22,23,36], we proposed a reaction mechanism as shown in Figure 1. In the initial step, the Rh catalyst reacted with Et2Zn to give a rhodium–hydride complex 6 via the elimination of ethylene from the rhodium–ethyl complex 5. The formation of rhodium enolate 7 involved the 1,4-reduction of α,β-unsaturated ester 2 by 6 and the transmetalation with a zinc species to give the Reformatsky-type reagent Int A. This intermediate Int A reacts immediately with the imine to give the corresponding intermediate Int B. Subsequently, intramolecular cyclization of Int B gave the desired β-lactam 3. On the other hand, the possibility of alternative mechanism could not be denied which concerned with the formation of ketene from Int A to give the β-lactam, directly. However, we thought that the reaction proceeded through the above mechanism, because our previous results using similar conditions did not give the corresponding products such as β-propiolactones. It is well known that the substituents on the α- or β-position of α,β-unsaturated carbonyl compounds affect the yields and stereoselectivities [37]. A rhodium–hydride complex derived from [RhCl(cod)]2 seems to be suitable for the reactivity and selectivity in comparison with the corresponding rhodium–hydride complex from RhCl(PPh3)3 which has bulkier ligand.
![[1860-5397-12-157-1]](/bjoc/content/figures/1860-5397-12-157-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Plausible mechanism for the Rh-catalyzed reductive Mannich-type reaction.
Figure 1: Plausible mechanism for the Rh-catalyzed reductive Mannich-type reaction.
The addition of a Lewis acid contributed not only to the activation the of imine but also the anti stereoselectivity of this reaction (Scheme 4). Furthermore, we reported in the previous paper that the configuration of the Int A species was identified as that of the E-enolate by the trapping procedure of tert-butyl acrylate (2k) with TMS-Cl. In addition, the same reaction gave only the syn-β-amino ester syn-4Ak in 86% and it was speculated that the reaction was proceeding via a linear transition state (Figure 2) [23].
![[1860-5397-12-157-i4]](/bjoc/content/inline/1860-5397-12-157-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Effect of the Lewis acid addition.
Scheme 4: Effect of the Lewis acid addition.
![[1860-5397-12-157-2]](/bjoc/content/figures/1860-5397-12-157-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Reaction of 2k and 1A and the configuration of Int A.
Figure 2: Reaction of 2k and 1A and the configuration of Int A.
When the reaction proceeded via a linear transition state, the reaction would be proposed to involve six competing transition state models (model A–F) as shown in Scheme 5. All these have steric repulsion but only the model B could be expected to have the interaction between the lone pair of the imine and the enolate metal. It seems the model B would be more stable than others to give the syn product. On the other hand, the addition of Lewis acid to the reaction mixture gave the iminium salt immediately and it would obstruct the stabilized metal interaction in model B’ of Scheme 6. This is meaning that the model F’ would be the most stable model, and the formation of the anti form product might increase relatively.
![[1860-5397-12-157-i5]](/bjoc/content/inline/1860-5397-12-157-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Transition-state model without Lewis acid.
Scheme 5: Transition-state model without Lewis acid.
![[1860-5397-12-157-i6]](/bjoc/content/inline/1860-5397-12-157-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Transition-state model with Lewis acid.
Scheme 6: Transition-state model with Lewis acid.
Conclusion
In conclusion, we found improved conditions for reductive Mannich-type reaction using [RhCl(cod)]2 with Et2Zn, and the reaction proceeded smoothly to give various syn-β-lactams even if β-substituted α,β-unsaturated esters were employed as the substrates. In addition, we could also provide some perspectives on the reaction mechanism and the stereochemistry. When the reaction was carried out with imines and α,β-unsaturated lactones, similar β-lactams with anti-selectivities were obtained. Furthermore the yield and anti selectivity could be increased by adding Lewis acid, and we succeeded in achieving the effective synthesis of (±)-ezetimibe by using this reaction.
Experimental
General information
1H NMR and 13C NMR spectra were recorded on JNM-GX400 spectrometers and ECZS-400 spectrometers. 19F NMR spectra were recorded on Hitachi FT-NMR R-90H spectrometers. Chemical shifts of 1H NMR and 13C NMR are reported in ppm from tetramethylsilane (TMS) as an internal standard. Chemical shifts of 19F NMR are reported in ppm from benzotrifluoride (BTF) as an internal standard. All data are reported with the chemical shift(s), relative integration value, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), and coupling constants (Hz). Mass spectra were obtained on JEOL JMS-700T spectrometers. IR spectra were recorded on a Hitachi 270-30 infrared spectrophotometer. Melting points were measured on a Yanagimoto micro melting point apparatus MP-S3. Analytical gas–liquid chromatography (GLC) was carried out on a Hitachi G-3500 gas chromatograph (column; TC-5 0.25 mm × 15 m, carrier; He). Peak areas were calculated on a Hitachi D-2500 Chromato-Integrator.
Materials
Tetrahydrofuran (THF) was purchased from Kanto Chemical Co. Inc. (Tokyo, Japan) in the dehydrated form. N,N-Dimethylformamide (DMF) and dichloromethane (CH2Cl2) were distilled over CaH2 and phosphorus pentoxide directly before use, respectively. All imines were prepared from corresponding aldehydes and amines. Methyl acrylate was distilled directly before use. Other commercially available reagents were used without further purification. All experiments were carried out under an argon atmosphere in flame-dried glassware using standard inert techniques for introducing reagents and solvents unless otherwise noted.
General procedure for the syn selective synthesis of azetidin-2-one
In a manner closely related to a procedure in reference [23]: Imine (1) and α,β-unsaturated ester (2, 2 equiv) were added to a solution of 2 mol % of [RhCl(cod)]2 in DMF at 0 °C. Then, 1.0 M Et2Zn in hexane (3 equiv) was gradually added to the mixture at room temperature, and the mixture was stirred for 24 h. The mixture was quenched with sat. NH4Cl and extracted with AcOEt. The AcOEt layer was washed with sat. NaCl and dried over MgSO4. The solvent was removed in vacuo, and the residue was purified by column chromatography to give the corresponding syn-azetidin-2-one. The stereochemistry of the products were determined by the NOE between the CH3 and C6H5 groups on the azetidin-2-one ring and the coupling constant between each proton of C3 and C4 (syn form: J = 5.0–6.0 Hz, anti form: J = 2.0–3.0 Hz).
General procedure for the anti selective synthesis of azetidin-2-one by the addition of BF3·Et2O
To a solution of [RhCl(cod)]2 (2 mol %) in DMF at room temperature was added imine (1). Then, BF3·Et2O (1.2 equiv) was added to the mixture and stirred for 30 min at the same temperature. Subsequently, 5,6-dihydro-2H-pyran-2-one (2, 1.2 equiv) was added to the mixture and then 1.0 M Et2Zn in hexane (3 equiv) was gradually added to the mixture at room temperature, and the mixture was stirred for 24 h. The mixture was quenched with sat. NH4Cl and extracted with AcOEt. The AcOEt layer was washed with sat. NaCl and dried over MgSO4. The solvent was removed in vacuo, and the residue was purified by column chromatography to give the corresponding anti-azetidin-2-one.
Supporting Information
| Supporting Information File 1: Experimental details, characterization of the compounds, X-ray crystallographic analysis of anti-3Ai, and 1H, 13C NMR spectra. | ||
| Format: PDF | Size: 2.1 MB | Download |
References
-
Yamasaki, S.; Iida, T.; Shibasaki, M. Tetrahedron Lett. 1999, 40, 307–310. doi:10.1016/S0040-4039(98)02297-7
Return to citation in text: [1] -
Liu, M.; Sibi, M. P. Tetrahedron 2002, 58, 7991–8035. doi:10.1016/S0040-4020(02)00991-2
Return to citation in text: [1] -
Taggi, A. E.; Hafez, A. M.; Lectka, T. Acc. Chem. Res. 2003, 36, 10–19. doi:10.1021/ar020137p
Return to citation in text: [1] -
Córdova, A. Acc. Chem. Res. 2004, 37, 102–112. doi:10.1021/ar030231l
Return to citation in text: [1] -
Notz, W.; Tanaka, F.; Barbas, C. F., III. Acc. Chem. Res. 2004, 37, 580–591. doi:10.1021/ar0300468
Return to citation in text: [1] -
Córdova, A. Chem. – Eur. J. 2004, 10, 1987–1997. doi:10.1002/chem.200305646
Return to citation in text: [1] -
Jacobsen, M. F.; Ionita, L.; Skrydstrup, T. J. Org. Chem. 2004, 69, 4792–4796. doi:10.1021/jo0358170
Return to citation in text: [1] -
Hamada, T.; Manabe, K.; Kobayashi, S. J. Am. Chem. Soc. 2004, 126, 7768–7769. doi:10.1021/ja048607t
Return to citation in text: [1] -
Ihori, Y.; Yamashita, Y.; Ishitani, H.; Kobayashi, S. J. Am. Chem. Soc. 2005, 127, 15528–15535. doi:10.1021/ja053524d
Return to citation in text: [1] -
Nishiwaki, N.; Knudsen, K. R.; Gothelf, K. V.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2001, 40, 2992–2995. doi:10.1002/1521-3773(20010817)40:16<2992::AID-ANIE2992>3.0.CO;2-3
Return to citation in text: [1] -
Rao, I. N.; Prabhakaran, E. N.; Das, S. K.; Iqbal, J. J. Org. Chem. 2003, 68, 4079–4082. doi:10.1021/jo020559c
Return to citation in text: [1] -
Li, Z.; Li, C.-J. J. Am. Chem. Soc. 2005, 127, 3672–3673. doi:10.1021/ja050058j
Return to citation in text: [1] -
Suto, Y.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2007, 129, 500–501. doi:10.1021/ja068226a
Return to citation in text: [1] -
Prieto, O.; Lam, H. W. Org. Biomol. Chem. 2008, 6, 55–57. doi:10.1039/B715839D
Return to citation in text: [1] [2] -
Youn, S. W.; Song, J.-H.; Jung, D.-I. J. Org. Chem. 2008, 73, 5658–5661. doi:10.1021/jo800914c
Return to citation in text: [1] -
Jiang, J.; Xu, H.-D.; Xi, J.-B.; Ren, B.-Y.; Lv, F.-P.; Guo, X.; Jiang, L.-Q.; Zhang, Z.-Y.; Hu, W.-H. J. Am. Chem. Soc. 2011, 133, 8428–8431. doi:10.1021/ja201589k
Return to citation in text: [1] -
Zhang, H.-X.; Nie, J.; Cai, H.; Ma, J.-A. Org. Lett. 2014, 16, 2542–2545. doi:10.1021/ol500929d
Return to citation in text: [1] -
Palomo, C.; Aizpurua, J. M.; Gracenea, J. J. J. Org. Chem. 1999, 64, 1693–1698. doi:10.1021/jo981574d
Return to citation in text: [1] -
Townes, J. A.; Evans, M. A.; Queffelec, J.; Taylor, S. J.; Morken, J. P. Org. Lett. 2002, 4, 2537–2540. doi:10.1021/ol020106u
Return to citation in text: [1] -
Nishiyama, H.; Ishikawa, J.; Shiomi, T. Tetrahedron Lett. 2007, 48, 7841–7844. doi:10.1016/j.tetlet.2007.08.135
Return to citation in text: [1] -
Du, Y.; Xu, L.-W.; Shimizu, Y.; Oisaki, K.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2008, 130, 16146–16147. doi:10.1021/ja8069727
Return to citation in text: [1] -
Sato, K.; Isoda, M.; Ohata, S.; Morita, S.; Tarui, A.; Omote, M.; Kumadaki, I.; Ando, A. Adv. Synth. Catal. 2012, 354, 510–514. doi:10.1002/adsc.201100463
Return to citation in text: [1] [2] -
Isoda, M.; Sato, K.; Funakoshi, M.; Omote, M.; Tarui, A.; Omote, M.; Ando, A. J. Org. Chem. 2015, 80, 8398–8405. doi:10.1021/acs.joc.5b01233
Return to citation in text: [1] [2] [3] [4] -
Taggi, A. E.; Hafez, A. M.; Wack, H.; Young, B.; Ferraris, D.; Lectka, T. J. Am. Chem. Soc. 2002, 124, 6626–6635. doi:10.1021/ja0258226
Return to citation in text: [1] -
Chen, L.; Zhao, G.; Ding, Y. Tetrahedron Lett. 2003, 44, 2611–2614. doi:10.1016/S0040-4039(03)00187-4
Return to citation in text: [1] -
Wang, Y.; Liang, Y.; Jiao, L.; Du, D.-M.; Xu, J. J. Org. Chem. 2006, 71, 6983–6990. doi:10.1021/jo0611521
Return to citation in text: [1] -
Jiao, L.; Liang, Y.; Xu, J. J. Am. Chem. Soc. 2006, 128, 6060–6069. doi:10.1021/ja056711k
Return to citation in text: [1] -
Clader, J. W.; Burnett, D. A.; Caplen, M. A.; Domalski, M. F.; Dugar, S.; Vaccaro, W.; Sher, R.; Browne, M. E.; Zhao, H.; Burrier, R. E.; Salisbury, B.; Davis, H. R., Jr. J. Med. Chem. 1996, 39, 3684–3693. doi:10.1021/jm960405n
Return to citation in text: [1] -
Rosenblum, S. B.; Huynh, T.; Afonso, A.; Davis, H. R., Jr.; Yumibe, N.; Clader, J. W.; Burnett, D. A. J. Med. Chem. 1998, 41, 973–980. doi:10.1021/jm970701f
Return to citation in text: [1] -
Clader, J. W. J. Med. Chem. 2004, 47, 1–9. doi:10.1021/jm030283g
Return to citation in text: [1] -
Sainz, B., Jr.; Barretto, N.; Martin, D. N.; Hiraga, N.; Imamura, M.; Hussain, S.; Marsh, K. A.; Yu, X.; Chayama, K.; Alrefai, W. A.; Uprichard, S. L. Nat. Med. 2012, 18, 281–285. doi:10.1038/nm.2581
Return to citation in text: [1] -
Takada, T.; Yamanashi, Y.; Konishi, K.; Yamamoto, T.; Toyoda, Y.; Masuo, Y.; Yamamoto, H.; Suzuki, H. Sci. Transl. Med. 2015, 7, 275ra23. doi:10.1126/scitranslmed.3010329
Return to citation in text: [1] -
Kværnø, L.; Werder, M.; Hauser, H.; Carreira, E. M. J. Med. Chem. 2005, 48, 6035–6053. doi:10.1021/jm050422p
Return to citation in text: [1] -
Michalak, M.; Stodulski, M.; Stecko, S.; Mames, A.; Panfil, I.; Soluch, M.; Furman, B.; Chmielewski, M. J. Org. Chem. 2011, 76, 6931–6936. doi:10.1021/jo2010846
Return to citation in text: [1] -
Śnieżek, M.; Stecko, S.; Panfil, I.; Furman, B.; Chmielewski, M. J. Org. Chem. 2013, 78, 7048–7057. doi:10.1021/jo400807c
Return to citation in text: [1] -
Isoda, M.; Sato, K.; Tokura, Y.; Tarui, A.; Omote, M.; Ando, A. Chem. Pharm. Bull. 2014, 62, 956–961. doi:10.1248/cpb.c14-00223
Return to citation in text: [1] -
Noyori, R.; Umeda, I.; Ishigami, T. J. Org. Chem. 1972, 10, 1542–1545. doi:10.1021/jo00975a017
Return to citation in text: [1]
| 1. | Yamasaki, S.; Iida, T.; Shibasaki, M. Tetrahedron Lett. 1999, 40, 307–310. doi:10.1016/S0040-4039(98)02297-7 |
| 14. | Prieto, O.; Lam, H. W. Org. Biomol. Chem. 2008, 6, 55–57. doi:10.1039/B715839D |
| 18. | Palomo, C.; Aizpurua, J. M.; Gracenea, J. J. J. Org. Chem. 1999, 64, 1693–1698. doi:10.1021/jo981574d |
| 19. | Townes, J. A.; Evans, M. A.; Queffelec, J.; Taylor, S. J.; Morken, J. P. Org. Lett. 2002, 4, 2537–2540. doi:10.1021/ol020106u |
| 20. | Nishiyama, H.; Ishikawa, J.; Shiomi, T. Tetrahedron Lett. 2007, 48, 7841–7844. doi:10.1016/j.tetlet.2007.08.135 |
| 21. | Du, Y.; Xu, L.-W.; Shimizu, Y.; Oisaki, K.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2008, 130, 16146–16147. doi:10.1021/ja8069727 |
| 10. | Nishiwaki, N.; Knudsen, K. R.; Gothelf, K. V.; Jørgensen, K. A. Angew. Chem., Int. Ed. 2001, 40, 2992–2995. doi:10.1002/1521-3773(20010817)40:16<2992::AID-ANIE2992>3.0.CO;2-3 |
| 11. | Rao, I. N.; Prabhakaran, E. N.; Das, S. K.; Iqbal, J. J. Org. Chem. 2003, 68, 4079–4082. doi:10.1021/jo020559c |
| 12. | Li, Z.; Li, C.-J. J. Am. Chem. Soc. 2005, 127, 3672–3673. doi:10.1021/ja050058j |
| 13. | Suto, Y.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2007, 129, 500–501. doi:10.1021/ja068226a |
| 14. | Prieto, O.; Lam, H. W. Org. Biomol. Chem. 2008, 6, 55–57. doi:10.1039/B715839D |
| 15. | Youn, S. W.; Song, J.-H.; Jung, D.-I. J. Org. Chem. 2008, 73, 5658–5661. doi:10.1021/jo800914c |
| 16. | Jiang, J.; Xu, H.-D.; Xi, J.-B.; Ren, B.-Y.; Lv, F.-P.; Guo, X.; Jiang, L.-Q.; Zhang, Z.-Y.; Hu, W.-H. J. Am. Chem. Soc. 2011, 133, 8428–8431. doi:10.1021/ja201589k |
| 17. | Zhang, H.-X.; Nie, J.; Cai, H.; Ma, J.-A. Org. Lett. 2014, 16, 2542–2545. doi:10.1021/ol500929d |
| 7. | Jacobsen, M. F.; Ionita, L.; Skrydstrup, T. J. Org. Chem. 2004, 69, 4792–4796. doi:10.1021/jo0358170 |
| 8. | Hamada, T.; Manabe, K.; Kobayashi, S. J. Am. Chem. Soc. 2004, 126, 7768–7769. doi:10.1021/ja048607t |
| 9. | Ihori, Y.; Yamashita, Y.; Ishitani, H.; Kobayashi, S. J. Am. Chem. Soc. 2005, 127, 15528–15535. doi:10.1021/ja053524d |
| 23. | Isoda, M.; Sato, K.; Funakoshi, M.; Omote, M.; Tarui, A.; Omote, M.; Ando, A. J. Org. Chem. 2015, 80, 8398–8405. doi:10.1021/acs.joc.5b01233 |
| 2. | Liu, M.; Sibi, M. P. Tetrahedron 2002, 58, 7991–8035. doi:10.1016/S0040-4020(02)00991-2 |
| 3. | Taggi, A. E.; Hafez, A. M.; Lectka, T. Acc. Chem. Res. 2003, 36, 10–19. doi:10.1021/ar020137p |
| 4. | Córdova, A. Acc. Chem. Res. 2004, 37, 102–112. doi:10.1021/ar030231l |
| 5. | Notz, W.; Tanaka, F.; Barbas, C. F., III. Acc. Chem. Res. 2004, 37, 580–591. doi:10.1021/ar0300468 |
| 6. | Córdova, A. Chem. – Eur. J. 2004, 10, 1987–1997. doi:10.1002/chem.200305646 |
| 23. | Isoda, M.; Sato, K.; Funakoshi, M.; Omote, M.; Tarui, A.; Omote, M.; Ando, A. J. Org. Chem. 2015, 80, 8398–8405. doi:10.1021/acs.joc.5b01233 |
| 31. | Sainz, B., Jr.; Barretto, N.; Martin, D. N.; Hiraga, N.; Imamura, M.; Hussain, S.; Marsh, K. A.; Yu, X.; Chayama, K.; Alrefai, W. A.; Uprichard, S. L. Nat. Med. 2012, 18, 281–285. doi:10.1038/nm.2581 |
| 32. | Takada, T.; Yamanashi, Y.; Konishi, K.; Yamamoto, T.; Toyoda, Y.; Masuo, Y.; Yamamoto, H.; Suzuki, H. Sci. Transl. Med. 2015, 7, 275ra23. doi:10.1126/scitranslmed.3010329 |
| 22. | Sato, K.; Isoda, M.; Ohata, S.; Morita, S.; Tarui, A.; Omote, M.; Kumadaki, I.; Ando, A. Adv. Synth. Catal. 2012, 354, 510–514. doi:10.1002/adsc.201100463 |
| 23. | Isoda, M.; Sato, K.; Funakoshi, M.; Omote, M.; Tarui, A.; Omote, M.; Ando, A. J. Org. Chem. 2015, 80, 8398–8405. doi:10.1021/acs.joc.5b01233 |
| 36. | Isoda, M.; Sato, K.; Tokura, Y.; Tarui, A.; Omote, M.; Ando, A. Chem. Pharm. Bull. 2014, 62, 956–961. doi:10.1248/cpb.c14-00223 |
| 28. | Clader, J. W.; Burnett, D. A.; Caplen, M. A.; Domalski, M. F.; Dugar, S.; Vaccaro, W.; Sher, R.; Browne, M. E.; Zhao, H.; Burrier, R. E.; Salisbury, B.; Davis, H. R., Jr. J. Med. Chem. 1996, 39, 3684–3693. doi:10.1021/jm960405n |
| 29. | Rosenblum, S. B.; Huynh, T.; Afonso, A.; Davis, H. R., Jr.; Yumibe, N.; Clader, J. W.; Burnett, D. A. J. Med. Chem. 1998, 41, 973–980. doi:10.1021/jm970701f |
| 30. | Clader, J. W. J. Med. Chem. 2004, 47, 1–9. doi:10.1021/jm030283g |
| 37. | Noyori, R.; Umeda, I.; Ishigami, T. J. Org. Chem. 1972, 10, 1542–1545. doi:10.1021/jo00975a017 |
| 24. | Taggi, A. E.; Hafez, A. M.; Wack, H.; Young, B.; Ferraris, D.; Lectka, T. J. Am. Chem. Soc. 2002, 124, 6626–6635. doi:10.1021/ja0258226 |
| 25. | Chen, L.; Zhao, G.; Ding, Y. Tetrahedron Lett. 2003, 44, 2611–2614. doi:10.1016/S0040-4039(03)00187-4 |
| 26. | Wang, Y.; Liang, Y.; Jiao, L.; Du, D.-M.; Xu, J. J. Org. Chem. 2006, 71, 6983–6990. doi:10.1021/jo0611521 |
| 27. | Jiao, L.; Liang, Y.; Xu, J. J. Am. Chem. Soc. 2006, 128, 6060–6069. doi:10.1021/ja056711k |
| 22. | Sato, K.; Isoda, M.; Ohata, S.; Morita, S.; Tarui, A.; Omote, M.; Kumadaki, I.; Ando, A. Adv. Synth. Catal. 2012, 354, 510–514. doi:10.1002/adsc.201100463 |
| 23. | Isoda, M.; Sato, K.; Funakoshi, M.; Omote, M.; Tarui, A.; Omote, M.; Ando, A. J. Org. Chem. 2015, 80, 8398–8405. doi:10.1021/acs.joc.5b01233 |
| 33. | Kværnø, L.; Werder, M.; Hauser, H.; Carreira, E. M. J. Med. Chem. 2005, 48, 6035–6053. doi:10.1021/jm050422p |
| 34. | Michalak, M.; Stodulski, M.; Stecko, S.; Mames, A.; Panfil, I.; Soluch, M.; Furman, B.; Chmielewski, M. J. Org. Chem. 2011, 76, 6931–6936. doi:10.1021/jo2010846 |
| 35. | Śnieżek, M.; Stecko, S.; Panfil, I.; Furman, B.; Chmielewski, M. J. Org. Chem. 2013, 78, 7048–7057. doi:10.1021/jo400807c |
© 2016 Isoda et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)