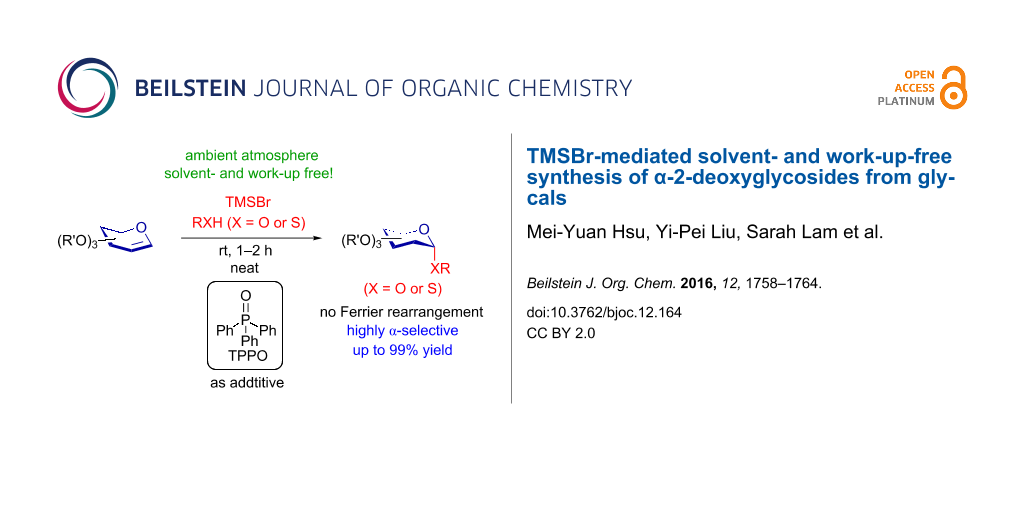
Abstract
The thio-additions of glycals were efficiently promoted by a stoichiometric amount of trimethylsilyl bromide (TMSBr) to produce S-2-deoxyglycosides under solvent-free conditions in good to excellent yields. In addition, with triphenylphosphine oxide as an additive, the TMSBr-mediated direct glycosylations of glycals with a large range of alcohols were highly α-selective.

Graphical Abstract
Introduction
Deoxyglycosides are essential moieties of numerous bioactive natural products, and are prevalent subunits in antitumor and antibiotic agents [1-3]. Furthermore, 2-deoxy- and 2,6-dideoxyglycosides are crucial components for the pharmacology and bioactivity of many biologically active compounds [4], and were recently observed to inhibit cancer growth [5]. Because of the relevance of 2-deoxyglycosides, great efforts have been made in researching the assembly of oligosaccharides containing these sugars [6,7]. However, the absence of a neighbouring group at C2 causes poor stereoselectivity and high susceptibility to hydrolysis, which are the main obstacles to constructing glycosidic linkages stereoselectively [8]. Some approaches, such as the AgPF6-DTBMS [9] and preactivation approach [10], can directly yield stereoselective glycosylations. Indirect methods that utilize auxiliary groups at C2, including halogen atoms [11-18], thio [19-21], and seleno groups [22-25], 1,2-migratory glycosylations that involve sulfur [26-33], oxygen [34], or nitrogen [35-37] atoms as directing groups and long-range directing functionalities at C6 [10,38-42] have also been developed to improve the stereoselectivity. However, additional required steps involving the introduction and removal of directing groups are reducing the efficiency.
Thioglycosides are some of the most commonly used donors for glycosylation reactions because of their high stability and reactivity [43]. Numerous stereoselective synthetic methods that use 2-deoxythioglycosides have been reported [9,10,44-49]. We recently developed a glycosyl chloride-mediated synthesis of highly α-selective 2-deoxyglucosides by using 2-deoxythioglucosides [50]. In the literature, to synthesize 2-deoxythioglycosides, a highly toxic tin hydride reagent was used to produce S-2-deoxysugars from glycosyl bromide through an anomeric glycosyl radical and acetate rearrangement, followed by subsequent thioglycosylation to afford 2-deoxythioglycosides as anomeric mixtures [10]. Glycals have been considered as alternative precursors for producing 2-deoxythioglycosides as well as oligosaccharides. Several methods based on the use of glycals in the presence of Lewis acids for S- or O-2-deoxyglycoside preparations have been developed [51-63]. However, based on the hard and soft (Lewis) acids and bases (HSAB) theory, hard acids would coordinate to the harder O3 in glycals in preference to the softer alkene to initiate an undesired Ferrier rearrangement, leading to the formation of a considerable amount of 2,3-unsatuated glycosides. This constitutes the major competitive reaction pathway in acid-catalysed 2-deoxyglycosylation of glycals [52,57,59]. Besides, unfavourable conditions involving the use of expensive or toxic metal complexes, high temperatures, and long reaction times are usually required in most of the aforementioned methods.
Furthermore, organic solvents in laboratories are associated with numerous health hazards [64], and most of them are consumed during chemical reactions, work-up and purification procedures. Especially, dichloromethane, one of the most general solvents for glycosylation reactions, is acknowledged as an acute inhalation hazard and carcinogen [65,66]. To date, only a few studies of glycosylation under neat conditions have been published. In these methods either the need of heating [67-69] or the use of ball milling [70-72] was demanded. Moreover, the selectivity was manipulated by the neighbouring group effect on C2 [67,69-71], which is absent in 2-deoxyglycosides. Mild, work-up- and solvent-free reaction conditions for highly stereoselective 2-deoxyglycosylation is therefore desirable. Here, we present a solvent- and work-up-free approach to prepare S- and α-selective O-2-deoxyglycosides from glycals.
Results and Discussion
In our preliminary study, 77% yield of 2-deoxythiolglucoside 2 was produced exclusively when glucal 1 in the presence of p-thiocresol was promoted by a stoichiometric amount of TMSBr under neat conditions at room temperature under ambient atmosphere (Table 1, α:β = 2:1, entry 1). Without work-up and washing, S-2-deoxyglycoside 2 could be directly isolated and purified by flash column chromatography. We further extended the scope of the reaction to other glycals by using TMSBr as the promoter under neat conditions (Table 1, entries 2–6). For per-O-benzylated glucal (3) and per-O-acetylated rhamnal (5), the corresponding thiol-2-deoxyglycosides 7 (61%, α:β = 2:1) and 10 (76%, α:β = 2:1) were produced in good yields with moderate stereoselectivity (Table 1, entry 2 and 5). Interestingly, as shown in Table 1, entries 1 and 2, the inductive effect of the substituents played critical roles in their reactivity. Electron-withdrawing groups, but not electron-donating groups, in glycals were likely to enhance the reactivity. The effect of the donor conformation on the stereoselectivity of the glycosylation was probed by galactal 4 and fucal 6. S-2-Deoxygalactoside 8 (87%, α:β = 3:1, Table 1, entry 3) and S-2-deoxyfucoside 11 (89%, α:β = 3:1, Table 1, entry 6) were produced in high yields with slightly superior selectivity. As shown in Table 1, entry 4, a prolonged reaction time led to further reaction of 8 to give 22% of a dithiol acetal side product 9.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-164-i3.svg?max-width=637&scale=1.0)
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-164-i4.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-164-i5.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-164-i6.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-12-164-i7.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-12-164-i8.svg?max-width=637&scale=1.0)
![[Graphic 7]](/bjoc/content/inline/1860-5397-12-164-i9.svg?max-width=637&scale=1.0)
![[Graphic 8]](/bjoc/content/inline/1860-5397-12-164-i10.svg?max-width=637&scale=1.0)
![[Graphic 9]](/bjoc/content/inline/1860-5397-12-164-i11.svg?max-width=637&scale=1.0)
![[Graphic 10]](/bjoc/content/inline/1860-5397-12-164-i12.svg?max-width=637&scale=1.0)
![[Graphic 11]](/bjoc/content/inline/1860-5397-12-164-i13.svg?max-width=637&scale=1.0)
![[Graphic 12]](/bjoc/content/inline/1860-5397-12-164-i14.svg?max-width=637&scale=1.0)
![[Graphic 13]](/bjoc/content/inline/1860-5397-12-164-i15.svg?max-width=637&scale=1.0)
Inspired by the results obtained in the synthesis of S-2-deoxyglycosides, we explored the use of numerous alcohols as acceptors in order to directly synthesize O-2-deoxyglycosides from glycals. In Table 2 the reaction of glucal 1 and benzyl alcohol (12) under similar reaction conditions was tested and O-benzyl 2-deoxyglucoside 25 was produced in 59% yield in the presence of TMSBr (α:β = 3:1, Table 2, entry 1). To further improve the α-selectivity and the yield, various additives were screened (Table 2, entries 2–11) [73-77]. Several participating solvents, dimethylformamide (DMF) (64%, Table 2, entry 2) [76], acetonitrile (ACN) [74] (44%, Table 2, entry 3), tetrahydrofuran (THF) (56%, Table 2, entry 4), and dioxane [75] (50%, Table 2, entry 5) were tested as glycosylation modulators and similar yields of 25 were obtained, but their α-selectivities dramatically improved to α:β = 10:1. In addition, 25 was afforded in 67% with excellent α-selectivity (α:β = 10:1) with the addition of dimethyl sulfide (DMS) (Table 2, entry 6). However, the basic additive 2,4,6-tri-tert-butylpyridine (TTBP) produced 25 with poor selectivity (52%, α:β = 2:1, Table 2, entry 7). Furthermore, various phosphine and phosphine oxide reagents were added in O-2-deoxyglycosylation reactions (Table 2, entries 8–11); surprisingly, the desired product 25 exhibited a high yield with excellent α-selectivity (78%, α:β = 10:1, Table 2, entry 11) with TPPO [77].
Table 2: Additives in TMSBr-mediated 2-deoxyglycosylation of glucal 1.
![[Graphic 14]](/bjoc/content/inline/1860-5397-12-164-i16.svg?max-width=637&scale=1.0)
|
|||
| Entry | Additive | Yield | Ratio (α:β) |
|---|---|---|---|
| 1 | None | 59% | 3:1 |
| 2 | DMF | 64% | 10:1 |
| 3 | ACN | 44% | 10:1 |
| 4 | THF | 56% | 10:1 |
| 5 | dioxane | 50% | 10:1 |
| 6 | DMS | 67% | 10:1 |
| 7 | 2,4,6-tri-tert-butylpyridine (TTBP) | 52% | 2:1 |
| 8 | triphenylphosphine (TPP) | 73% | 7:1 |
| 9a | diphenyl phosphate (DPP) | 25% | 5:1 |
| 10 | trimethyl phosphine oxide (TMPO) | 50% | 2:1 |
| 11 | triphenyl phosphine oxide (TPPO) | 78% | 10:1 |
a1 was recovered in 46%.
Encouraged by these results, we attempted to extend the scope of the glycosylation of 3,4,6-O-acetyl- and O-benzylglucal (1 and 3) with other acceptors (Table 3). Under the optimized conditions, glucal 1 reacted with numerous primary, secondary, and tertiary alcohols, including methanol (13), allyl alcohol (14), isopropanol (15), tert-butanol (16), 5-azidopentanol (17), cyclohexanol (18) and 2-adamantanol (19), to give O-2-deoxyglucosides in high yields (74–90%) and α-selectivities (α:β = 7–10:1, Table 3, entries 2–8). Regarding the glycosylation with amino acid derivatives, L-serine 20 and threonine derivative 21, increased ratio of β-glucosides were formed in their corresponding products 33 (71%, 5:1, Table 3, entry 9) and 34 (79%, 4:1, Table 3, entry 10). For the use of monosaccharides as acceptors, primary monosaccharides 22 and 23 gave disaccharides 35 (80%, Table 3, entry 11) and 36 (78%, Table 3, entry 12) respectively in high yields with moderate α-selectivity (α:β = 4:1). Surprisingly, in the glycosylation with secondary monosaccharide acceptor 24, α-disaccharide 37 (56%, Table 3, entry 13) was isolated as the sole product. For per-O-benzylated glucal 3, a higher yield of 38 (97%, Table 3, entry 14) was produced with good selectivity (α:β = 5:1) in the presence of TPPO when compared to the additive-free conditions (79%, Table 3, entry 15). For aliphatic alcohols (13–19), glycosylation products (39–45) were always obtained in excellent yields (75–95%) and moderate selectivities (α:β = 3–5:1, Table 3, entries 16–22). The reaction with amino acid residues 20 and 21 (Table 3, entries 23 and 24) produced aminosugars 46 (68%, α:β = 5:1) and 47 (74%, α:β = 4:1) in good yields with moderate α-selectivity. Disaccharides 48 (90%, α:β = 4:1, Table 3, entry 25) and 49 (80%, α:β = 4:1, Table 3, entry 26) were formed in high yields with moderate selectivity similar to the examples of the products of primary monosaccharide acceptors 22 and 23. Finally, the secondary monosaccharide acceptor 24 (Table 3, entry 27) also underwent complete α-selective glycosylation, producing α-disaccharide 50 (67%) as the only product. According to our study, the glycosylation of per-O-acetylated glucal 1 with aliphatic alcohols 12–19 showed better α-selectivities as compared to the per-O-benzylated glucal 3. However, with amino acid derivatives 20 and 21 and monosaccharides 22–24 as acceptors, similar α-selectivities were attained with both glucals 1 and 3.
Table 3: TMSBr-mediated 2-deoxyglycosylation of glucals 1 and 3.
![[Graphic 15]](/bjoc/content/inline/1860-5397-12-164-i17.svg?max-width=637&scale=1.0)
|
||||
| Entry | Donor | Acceptor | Product | Yield (α:β) |
|---|---|---|---|---|
| 1 | 1 | benzyl alcohol (12) | 25 | 78% (10:1) |
| 2 | 1 | methanol (13) | 26 | 82% (10:1) |
| 3 | 1 | 3-propenol (14) | 27 | 78% (7:1) |
| 4 | 1 | isopropanol (15) | 28 | 74% (9:1) |
| 5 | 1 | tert-butanol (16) | 29 | 76% (8:1) |
| 6 | 1 | 5-azidopentanol (17) | 30 | 90% (10:1) |
| 7 | 1 | cyclohexanol (18) | 31 | 89% (10:1) |
| 8 | 1 | 2-adamantanol (19) | 32 | 86% (10:1) |
| 9a | 1 | 20 | 33 | 71% (5:1) |
| 10a | 1 | 21 | 34 | 79% (4:1) |
| 11 | 1 | 22 | 35 | 80% (4:1) |
| 12a | 1 | 23 | 36 | 78% (4:1) |
| 13a | 1 | 24 | 37 | 56% α only |
| 14 | 3 | 12 | 38 | 97% (5:1) |
| 15b | 3 | 12 | 38 | 79% (6:1) |
| 16 | 3 | 13 | 39 | 95% (4:1) |
| 17 | 3 | 14 | 40 | 91% (5:1) |
| 18 | 3 | 15 | 41 | 93% (5:1) |
| 19 | 3 | 16 | 42 | 85% (3:1) |
| 20 | 3 | 17 | 43 | 89% (5:1) |
| 21 | 3 | 18 | 44 | 81% (5:1) |
| 22 | 3 | 19 | 45 | 75% (5:1) |
| 23a | 3 | 20 | 46 | 68% (5:1) |
| 24a | 3 | 21 | 47 | 74% (4:1) |
| 25 | 3 | 22 | 48 | 90% (4:1) |
| 26a | 3 | 23 | 49 | 80% (4:1) |
| 27a | 3 | 24 | 50 | 67% α only |
aA minimum amount of CH2Cl2 was added for solubility. bWithout the addition of TPPO.
The results using acetylated galactal 4 were summarized in Table 4. The reactions using several aliphatic alcohols (12, 14–18) as acceptors yielded the desired compounds (51, 53–57) in excellent yields (90–99%) with high α-selectivities (α:β = 7–9:1, Table 4, entries 1, 3–7). Glycosylation with MeOH (13); however, produced 52 in an excellent yield but lower selectivity (95%, α:β = 4:1, Table 4, entry 2). The bulky acceptor 2-adamantanol (19) produced compound 58 in a decreased yield but with excellent selectivity because of its low solubility (43%, α:β = 13:1, Table 4, entry 8). L-Serine and threonine derivatives 20 and 21 reacted with galactal 4 to give the glycosylated amino acids 59 and 60 (Table 4, entries 9 and 10) in excellent selectivities (59, α only; 60, α:β = 9:1) but in different yields (59, 50%; 60, 97%). In Table 4, entry 11, disaccharide 61 was acquired in the presence of 22 with a moderate yield and selectivity (50%, α:β = 4:1). When primary monosaccharide 23 was used as the acceptor, disaccharide 62 was provided in an excellent yield and selectivity (94%, α:β = 10:1, Table 4, entry 12). Additionally, a 60% yield of the α-only product 63 was observed exclusively when the secondary hydroxyl glucoside 24 was used (Table 4, entry 13). Notably, the disubstituted side product was not observed in this reaction.
Table 4: TMSBr-mediated 2-deoxyglycosylation of O-acetyl galactal 4.
![[Graphic 16]](/bjoc/content/inline/1860-5397-12-164-i18.svg?max-width=637&scale=1.0)
|
|||
| Entry | Acceptor | Product | Yield (α/β) |
|---|---|---|---|
| 1 | 12 | 51 | quant. (9:1) |
| 2 | 13 | 52 | 95% (4:1) |
| 3 | 14 | 53 | 90% (8:1) |
| 4 | 15 | 54 | quant. (7:1) |
| 5 | 16 | 55 | quant. (8:1) |
| 6 | 17 | 56 | quant. (9:1) |
| 7 | 18 | 57 | 99% (9:1) |
| 8a | 19 | 58 | 43% (13:1) |
| 9a | 20 | 59 | 50 % α only |
| 10a | 21 | 60 | 97% (9:1) |
| 11 | 22 | 61 | 50% (4:1) |
| 12a | 23 | 62 | 94% (10:1) |
| 13a | 24 | 63 | 60% α only |
aA minimum amount of CH2Cl2 was added for solubility.
On the basis of these results, we demonstrated the applicability of the methodology in oligosaccharide synthesis by synthesising trisaccharide 66 in two sequential steps (Scheme 1). Monosaccharide acceptor 64 underwent the TMSBr-mediated nucleophilic addition to glucal 1 to produce exclusively disaccharide 65 (97%, α:β = 7:1) in an excellent yield with high α-selectivity. Remarkably, the 1-thiol group remained intact after the formation of disaccharide 65. Subsequently, 65 was coupled with the primary hydroxy saccharide acceptor 23 through a chloride-mediated preactivation glycosylation to afford 66 in 71% yield with moderate selectivity (α:β = 1:2) [16].
![[1860-5397-12-164-i1]](/bjoc/content/inline/1860-5397-12-164-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Iterative synthesis of trisaccharide 66.
Scheme 1: Iterative synthesis of trisaccharide 66.
Two possible mechanisms are proposed for the α-selectivity observed here (Scheme 2). It is well-accepted that the acid-catalysed nucleophilic addition of an alcohol to a glycal is likely to proceed through the formation of an oxocarbenium ion via the protonation at C2 [6,63]. In the presence of TPPO, the oxocarbenium cation is stabilized by the ion–dipole interaction with TPPO oriented preferably at the pseudoequatorial position [78] and the ensuing SN2-like displacement by the alcohol contributes to the improvement of the α-selectivity (Scheme 2, route A). Alternatively, it is possible that a 2-deoxyglycosyl bromide is first generated mainly in the more stable α-form [61]. The glycosyl bromide intermediate then undergoes double SN2-like substitution by TPPO and the alcohol to give the α-glycoside as the major product [77] (Scheme 2, route B).
![[1860-5397-12-164-i2]](/bjoc/content/inline/1860-5397-12-164-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed mechanisms for TMSBr-mediated synthesis of 2-deoxyglycosides in the presence of TPPO.
Scheme 2: Proposed mechanisms for TMSBr-mediated synthesis of 2-deoxyglycosides in the presence of TPPO.
Conclusion
A simple, efficient, and environmentally friendly method for preparing S- and O-2-deoxyglycosides was established. S-2-Deoxyglycosides were obtained with moderate α-selectivity when glycals and thiocresol were treated with a stoichiometric amount of TMSBr in neat conditions. Extension of this approach to hydroxy acceptors provided an efficient method to construct the glycosyl bonds between the 2-deoxysugars and the acceptors in good to excellent yields with high α-selectivity in the presence of TPPO, which served as an additive that improved both glycosylation yield and α-selectivity. Without the use of excess solvents, toxic reagents, special equipment, and high temperature, reactions were complete in a few hours at room temperature under ambient atmosphere. Ferrier rearranged products and other side products were not observed. As these reactions were clean, tedious work-up and extraction processes could be obviated prior to purification by flash column chromatography. The utility of this glycosylation method was highlighted by an iterative synthesis of trisaccharide 66.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures, compound characterization data, and copies of NMR spectra. | ||
| Format: PDF | Size: 9.3 MB | Download |
References
-
Langenhan, J. M.; Peters, N. R.; Guzei, I. A.; Hoffmann, M.; Thorson, J. S. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 12305–12310. doi:10.1073/pnas.0503270102
Return to citation in text: [1] -
Daniel, P. T.; Koert, U.; Schuppan, J. Angew. Chem., Int. Ed. 2006, 45, 872–893. doi:10.1002/anie.200502698
Angew. Chem. 2006, 118, 886–908. doi:10.1002/ange.200502698
Return to citation in text: [1] -
Iyer, A. K. V.; Zhou, M.; Azad, N.; Elbaz, H.; Wang, L.; Rogalsky, D. K.; Rojanasakul, Y.; O’Doherty, G. A.; Langenhan, J. M. ACS Med. Chem. Lett. 2010, 1, 326–330. doi:10.1021/ml1000933
Return to citation in text: [1] -
Nicolaou, K. C.; Mitchel, H. J. Angew. Chem., Int. Ed. 2001, 40, 1576–1624. doi:10.1002/1521-3773(20010504)40:9<1576::AID-ANIE15760>3.0.CO;2-G
Return to citation in text: [1] -
Xi, H.; Kurtoglu, M.; Lampidis, T. J. IUBMB Life 2014, 66, 110–121. doi:10.1002/iub.1251
Return to citation in text: [1] -
Hou, D.; Lowary, T. L. Carbohydr. Res. 2009, 344, 1911–1940. doi:10.1016/j.carres.2009.07.013
Return to citation in text: [1] [2] -
Marzabadi, C. H.; Franck, R. W. Tetrahedron 2000, 56, 8385–8417. doi:10.1016/S0040-4020(00)00691-8
Return to citation in text: [1] -
Overend, W. G.; Rees, C. W.; Sequeira, J. S. J. Chem. Soc. 1962, 3429–3440. doi:10.1039/jr9620003429
Return to citation in text: [1] -
Lear, M. J.; Yoshimura, F.; Hirama, M. Angew. Chem., Int. Ed. 2001, 40, 946–949. doi:10.1002/1521-3773(20010302)40:5<946::AID-ANIE946>3.0.CO;2-G
Return to citation in text: [1] [2] -
Lu, Y.-S.; Li, Q.; Zhang, L.-H.; Ye, X.-S. Org. Lett. 2008, 10, 3445–3448. doi:10.1021/ol801190c
Return to citation in text: [1] [2] [3] [4] -
Roush, W. R.; Briner, K.; Sebesta, D. P. Synlett 1993, 264–266. doi:10.1055/s-1993-22425
Return to citation in text: [1] -
Roush, W. R.; Bennett, C. E. J. Am. Chem. Soc. 1999, 121, 3541–3542. doi:10.1021/ja984365j
Return to citation in text: [1] -
Roush, W. R.; Gung, B. W.; Bennett, C. E. Org. Lett. 1999, 1, 891–893. doi:10.1021/ol9908070
Return to citation in text: [1] -
Tatsuta, K.; Fujimoto, K.; Kinoshita, M.; Umezawa, S. Carbohydr. Res. 1977, 54, 85–104. doi:10.1016/S0008-6215(00)80558-3
Return to citation in text: [1] -
Thiem, J.; Gerken, M. J. Org. Chem. 1985, 50, 954–958. doi:10.1021/jo00207a009
Return to citation in text: [1] -
Thiem, J.; Schöttmer, B. Angew. Chem., Int. Ed. Engl. 1987, 26, 555–557. doi:10.1002/anie.198705551
Angew. Chem. 1987, 99, 591–592. doi:10.1002/ange.19870990626
Return to citation in text: [1] [2] -
Bucher, C.; Gilmour, R. Angew. Chem., Int. Ed. 2010, 49, 8724–8728. doi:10.1002/anie.201004467
Angew. Chem. 2010, 122, 8906–8910. doi:10.1002/ange.201004467
Return to citation in text: [1] -
Durantie, E.; Bucher, C.; Gilmour, R. Chem. – Eur. J. 2012, 18, 8208–8215. doi:10.1002/chem.201200468
Return to citation in text: [1] -
Ito, Y.; Ogawa, T. Tetrahedron Lett. 1987, 28, 2723–2726. doi:10.1016/S0040-4039(00)96191-4
Return to citation in text: [1] -
Grewal, G.; Kaila, N.; Franck, R. W. J. Org. Chem. 1992, 57, 2084–2092. doi:10.1021/jo00033a033
Return to citation in text: [1] -
Ramesh, S.; Franck, R. W. J. Chem. Soc., Chem. Commun. 1989, 960–962. doi:10.1039/C39890000960
Return to citation in text: [1] -
Barrett, A. G. M.; Miller, T. A. Tetrahedron Lett. 1988, 29, 1873–1874. doi:10.1016/S0040-4039(00)82065-1
Return to citation in text: [1] -
Perez, M.; Beau, J.-M. Tetrahedron Lett. 1989, 30, 75–78. doi:10.1016/S0040-4039(01)80327-0
Return to citation in text: [1] -
Sebesta, D. P.; Roush, W. R. J. Org. Chem. 1992, 57, 4799–4802. doi:10.1021/jo00044a010
Return to citation in text: [1] -
Nicolaou, K. C.; Pastor, J.; Barluenga, S.; Winssinger, N. Chem. Commun. 1998, 1947–1948. doi:10.1039/a804795b
Return to citation in text: [1] -
Auzanneau, F.-I.; Bundle, D. R. Carbohydr. Res. 1991, 212, 13–24. doi:10.1016/0008-6215(91)84041-C
Return to citation in text: [1] -
Zuurmond, H. M.; van der Klein, P. A. M.; van der Marel, G. A.; van Boom, J. H. Tetrahedron 1993, 49, 6501–6514. doi:10.1016/S0040-4020(01)80165-4
Return to citation in text: [1] -
Yang, Z.; Yu, B. Carbohydr. Res. 2001, 333, 105–114. doi:10.1016/S0008-6215(01)00124-0
Return to citation in text: [1] -
Yu, B.; Yang, Z. Org. Lett. 2001, 3, 377–379. doi:10.1021/ol006894+
Return to citation in text: [1] -
Sajtos, F.; Lázár, L.; Borbás, A.; Bajza, I.; Lipták, A. Tetrahedron Lett. 2005, 46, 5191–5194. doi:10.1016/j.tetlet.2005.05.112
Return to citation in text: [1] -
Lázár, L.; Bajza, I.; Jakab, Z.; Lipták, A. Synlett 2005, 2242–2244. doi:10.1055/s-2005-872244
Return to citation in text: [1] -
Hou, D.; Lowary, T. L. Org. Lett. 2007, 9, 4487–4490. doi:10.1021/ol7019108
Return to citation in text: [1] -
Hou, D.; Lowary, T. L. J. Org. Chem. 2009, 74, 2278–2289. doi:10.1021/jo900131a
Return to citation in text: [1] -
Capozzi, G.; Dios, A.; Franck, R. W.; Geer, A.; Marzabadi, C.; Menichetti, S. C.; Tamarez, M. Angew. Chem. 1996, 108, 805–807. doi:10.1002/ange.19961080710
Angew. Chem. Int. Ed. Engl. 1996, 35, 777–779. doi:10.1002/anie.199607771
Return to citation in text: [1] -
Gurjar, M. K.; Ghosh, P. K. Indian J. Chem. 1988, 27B, 1063–1064.
Return to citation in text: [1] -
Trumtel, M.; Veyrières, A.; Sinay, P. Tetrahedron Lett. 1989, 30, 2529–2532. doi:10.1016/S0040-4039(01)80442-1
Return to citation in text: [1] -
Castro-Palomino, J. C.; Schmidt, R. R. Synlett 1998, 501–503. doi:10.1055/s-1998-1689
Return to citation in text: [1] -
Pongdee, R.; Wu, B.; Sulikowski, G. A. Org. Lett. 2001, 3, 3523–3525. doi:10.1021/ol016593f
Return to citation in text: [1] -
Crich, D.; Vinogradova, O. J. Org. Chem. 2006, 71, 8473–8480. doi:10.1021/jo061417b
Return to citation in text: [1] -
Kim, K. S.; Park, J.; Lee, Y. J.; Seo, Y. S. Angew. Chem., Int. Ed. 2003, 42, 459–462. doi:10.1002/anie.200390139
Angew. Chem. 2003, 115, 475–478. doi:10.1002/ange.200390107
Return to citation in text: [1] -
Park, J.; Boltje, T. J.; Boons, G.-J. Org. Lett. 2008, 10, 4367–4370. doi:10.1021/ol801833n
Return to citation in text: [1] -
Lu, Y.-S.; Li, Q.; Wang, Y.; Ye, X.-S. Synlett 2010, 1519–1524. doi:10.1055/s-0029-1219943
Return to citation in text: [1] -
Codée, J. D. C.; Litjens, R. E. J. N.; van den Bos, L. J.; Overkleeft, H. S.; van der Marel, G. A. Chem. Soc. Rev. 2005, 34, 769–782. doi:10.1039/b417138c
Return to citation in text: [1] -
Zhang, G.; Fang, L.; Zhu, L.; Aimiuwu, J. E.; Shen, J.; Cheng, H.; Muller, M. T.; Lee, G. E.; Sun, D.; Wang, P. G. J. Med. Chem. 2005, 48, 5269–5278. doi:10.1021/jm050144u
Return to citation in text: [1] -
Zhang, G.; Fang, L.; Zhu, L.; Zhong, Y.; Wang, P. G.; Sun, D. J. Med. Chem. 2006, 49, 1792–1799. doi:10.1021/jm050916m
Return to citation in text: [1] -
Fan, E.; Shi, W.; Lowary, T. L. J. Org. Chem. 2007, 72, 2917–2928. doi:10.1021/jo062542q
Return to citation in text: [1] -
Paul, S.; Jayaraman, N. Carbohydr. Res. 2007, 342, 1305–1314. doi:10.1016/j.carres.2007.02.030
Return to citation in text: [1] -
Braccini, I.; Derouet, C.; Esnault, J.; de Penhoat, C. H.; Mallet, J.-M.; Michon, V.; Sinay, P. Carbohydr. Res. 1993, 246, 23–41. doi:10.1016/0008-6215(93)84021-W
Return to citation in text: [1] -
Jaunzems, J.; Sourkouni-Argirusi, G.; Jesberger, M.; Kirschning, A. Tetrahedron Lett. 2003, 44, 637–639. doi:10.1016/S0040-4039(02)02708-9
Return to citation in text: [1] -
Verma, V. P.; Wang, C.-C. Chem. – Eur. J. 2013, 19, 846–851. doi:10.1002/chem.201203418
Return to citation in text: [1] -
Beau, J.-M.; Sinay, P. Tetrahedron Lett. 1985, 26, 6185–6188. doi:10.1016/S0040-4039(00)95048-2
Return to citation in text: [1] -
Bolitt, V.; Mioskowski, C.; Lee, S.-G.; Falck, J. R. J. Org. Chem. 1990, 55, 5812–5813. doi:10.1021/jo00310a006
Return to citation in text: [1] [2] -
Mereyala, H. B.; Ravi, D. Tetrahedron Lett. 1991, 32, 7317–7320. doi:10.1016/0040-4039(91)80508-4
Return to citation in text: [1] -
Yadav, J. S.; Reddy, B. V. S.; Reddy, K. B.; Satyanarayana, M. Tetrahedron Lett. 2002, 43, 7009–7012. doi:10.1016/S0040-4039(02)01584-8
Return to citation in text: [1] -
Paul, S.; Jayaraman, N. Carbohydr. Res. 2004, 339, 2197–2204. doi:10.1016/j.carres.2004.07.010
Return to citation in text: [1] -
Sherry, B. D.; Loy, R. N.; Toste, F. D. J. Am. Chem. Soc. 2004, 126, 4510–4511. doi:10.1021/ja031895t
Return to citation in text: [1] -
Palmier, S.; Vauzeilles, B.; Beau, J.-M. Org. Biomol. Chem. 2003, 1, 1097–1098. doi:10.1039/b301805a
Return to citation in text: [1] [2] -
Yadav, J. S.; Reddy, B. V. S.; Bhasker, E. V.; Raghavendra, S.; Narsaiah, A. V. Tetrahedron Lett. 2007, 48, 677–680. doi:10.1016/j.tetlet.2006.11.103
Return to citation in text: [1] -
Lin, H.-C.; Pan, J.-F.; Chen, Y.-B.; Lin, Z.-P.; Lin, C.-H. Tetrahedron 2011, 67, 6362–6368. doi:10.1016/j.tet.2011.05.124
Return to citation in text: [1] [2] -
Balmond, E. I.; Coe, D. M.; Galan, M. C.; McGarrigle, E. M. Angew. Chem., Int. Ed. 2012, 51, 9152–9155. doi:10.1002/anie.201204505
Return to citation in text: [1] -
Cui, X.-K.; Zhong, M.; Meng, X.-B.; Li, Z.-J. Carbohydr. Res. 2012, 358, 19–22. doi:10.1016/j.carres.2012.06.004
Return to citation in text: [1] [2] -
Balmond, E. I.; Benito-Alifonso, D.; Coe, D. M.; Alder, R. W.; McGarrigle, E. M.; Galan, M. C. Angew. Chem., Int. Ed. 2014, 53, 8190–8194. doi:10.1002/anie.201403543
Return to citation in text: [1] -
Kimura, T.; Takahashi, D.; Toshima, K. J. Org. Chem. 2015, 80, 9552–9562. doi:10.1021/acs.joc.5b01542
Return to citation in text: [1] [2] -
Dick, F. D. Occup. Environ. Med. 2006, 63, 221–226. doi:10.1136/oem.2005.022400
Return to citation in text: [1] -
Rioux, J. P.; Myers, R. A. M. J. Emerg. Med. 1988, 6, 227–238. doi:10.1016/0736-4679(88)90330-7
Return to citation in text: [1] -
Lefevre, P. A.; Ashby, J. Carcinogenesis 1989, 10, 1067–1072. doi:10.1093/carcin/10.6.1067
Return to citation in text: [1] -
Limousin, C.; Cléophax, J.; Petit, A.; Loupy, A.; Lukacs, G. J. Carbohydr. Chem. 1997, 16, 327–342. doi:10.1080/07328309708006533
Return to citation in text: [1] [2] -
de Oliveira, R. N.; de Freitas Filho, J. R.; Srivastava, R. M. Tetrahedron Lett. 2002, 43, 2141–2143. doi:10.1016/S0040-4039(02)00156-9
Return to citation in text: [1] -
Lautrette, S.; Granet, R.; Krausz, P. Chem. Commun. 2004, 586–587. doi:10.1039/b315699k
Return to citation in text: [1] [2] -
Patil, P. R.; Kartha, K. P. R. J. Carbohydr. Chem. 2008, 27, 411–419. doi:10.1080/07328300802402259
Return to citation in text: [1] [2] -
Tyagi, M.; Khurana, D.; Kartha, K. P. R. Carbohydr. Res. 2013, 379, 55–59. doi:10.1016/j.carres.2013.06.018
Return to citation in text: [1] [2] -
Kumar, V.; Yadav, N.; Kartha, K. P. R. Carbohydr. Res. 2014, 397, 18–26. doi:10.1016/j.carres.2014.08.002
Return to citation in text: [1] -
Crich, D.; Smith, M.; Yao, Q.; Picione, J. Synthesis 2001, 323–326. doi:10.1055/s-2001-10798
Return to citation in text: [1] -
Lemieux, R. U.; Ratclffe, R. M. Can. J. Chem. 1979, 57, 1244–1251. doi:10.1139/v79-203
Return to citation in text: [1] [2] -
Satoh, H.; Hansen, H. S.; Manabe, S.; van Gunsteren, W. F.; Hünenberger, P. H. J. Chem. Theory Comput. 2010, 6, 1783–1797. doi:10.1021/ct1001347
Return to citation in text: [1] [2] -
Chen, J.-H.; Ruei, J.-H.; Mong, K.-K. T. Eur. J. Org. Chem. 2014, 1827–1831. doi:10.1002/ejoc.201400006
Return to citation in text: [1] [2] -
Oka, N.; Kajino, R.; Takeuchi, K.; Nagakawa, H.; Ando, K. J. Org. Chem. 2014, 79, 7656–7664. doi:10.1021/jo500632h
Return to citation in text: [1] [2] [3] -
Bogusiak, J.; Szeja, W. Synlett 1997, 661–662. doi:10.1055/s-1997-3245
Return to citation in text: [1]
| 74. | Lemieux, R. U.; Ratclffe, R. M. Can. J. Chem. 1979, 57, 1244–1251. doi:10.1139/v79-203 |
| 75. | Satoh, H.; Hansen, H. S.; Manabe, S.; van Gunsteren, W. F.; Hünenberger, P. H. J. Chem. Theory Comput. 2010, 6, 1783–1797. doi:10.1021/ct1001347 |
| 77. | Oka, N.; Kajino, R.; Takeuchi, K.; Nagakawa, H.; Ando, K. J. Org. Chem. 2014, 79, 7656–7664. doi:10.1021/jo500632h |
| 1. | Langenhan, J. M.; Peters, N. R.; Guzei, I. A.; Hoffmann, M.; Thorson, J. S. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 12305–12310. doi:10.1073/pnas.0503270102 |
| 2. |
Daniel, P. T.; Koert, U.; Schuppan, J. Angew. Chem., Int. Ed. 2006, 45, 872–893. doi:10.1002/anie.200502698
Angew. Chem. 2006, 118, 886–908. doi:10.1002/ange.200502698 |
| 3. | Iyer, A. K. V.; Zhou, M.; Azad, N.; Elbaz, H.; Wang, L.; Rogalsky, D. K.; Rojanasakul, Y.; O’Doherty, G. A.; Langenhan, J. M. ACS Med. Chem. Lett. 2010, 1, 326–330. doi:10.1021/ml1000933 |
| 8. | Overend, W. G.; Rees, C. W.; Sequeira, J. S. J. Chem. Soc. 1962, 3429–3440. doi:10.1039/jr9620003429 |
| 43. | Codée, J. D. C.; Litjens, R. E. J. N.; van den Bos, L. J.; Overkleeft, H. S.; van der Marel, G. A. Chem. Soc. Rev. 2005, 34, 769–782. doi:10.1039/b417138c |
| 6. | Hou, D.; Lowary, T. L. Carbohydr. Res. 2009, 344, 1911–1940. doi:10.1016/j.carres.2009.07.013 |
| 7. | Marzabadi, C. H.; Franck, R. W. Tetrahedron 2000, 56, 8385–8417. doi:10.1016/S0040-4020(00)00691-8 |
| 9. | Lear, M. J.; Yoshimura, F.; Hirama, M. Angew. Chem., Int. Ed. 2001, 40, 946–949. doi:10.1002/1521-3773(20010302)40:5<946::AID-ANIE946>3.0.CO;2-G |
| 10. | Lu, Y.-S.; Li, Q.; Zhang, L.-H.; Ye, X.-S. Org. Lett. 2008, 10, 3445–3448. doi:10.1021/ol801190c |
| 44. | Zhang, G.; Fang, L.; Zhu, L.; Aimiuwu, J. E.; Shen, J.; Cheng, H.; Muller, M. T.; Lee, G. E.; Sun, D.; Wang, P. G. J. Med. Chem. 2005, 48, 5269–5278. doi:10.1021/jm050144u |
| 45. | Zhang, G.; Fang, L.; Zhu, L.; Zhong, Y.; Wang, P. G.; Sun, D. J. Med. Chem. 2006, 49, 1792–1799. doi:10.1021/jm050916m |
| 46. | Fan, E.; Shi, W.; Lowary, T. L. J. Org. Chem. 2007, 72, 2917–2928. doi:10.1021/jo062542q |
| 47. | Paul, S.; Jayaraman, N. Carbohydr. Res. 2007, 342, 1305–1314. doi:10.1016/j.carres.2007.02.030 |
| 48. | Braccini, I.; Derouet, C.; Esnault, J.; de Penhoat, C. H.; Mallet, J.-M.; Michon, V.; Sinay, P. Carbohydr. Res. 1993, 246, 23–41. doi:10.1016/0008-6215(93)84021-W |
| 49. | Jaunzems, J.; Sourkouni-Argirusi, G.; Jesberger, M.; Kirschning, A. Tetrahedron Lett. 2003, 44, 637–639. doi:10.1016/S0040-4039(02)02708-9 |
| 5. | Xi, H.; Kurtoglu, M.; Lampidis, T. J. IUBMB Life 2014, 66, 110–121. doi:10.1002/iub.1251 |
| 35. | Gurjar, M. K.; Ghosh, P. K. Indian J. Chem. 1988, 27B, 1063–1064. |
| 36. | Trumtel, M.; Veyrières, A.; Sinay, P. Tetrahedron Lett. 1989, 30, 2529–2532. doi:10.1016/S0040-4039(01)80442-1 |
| 37. | Castro-Palomino, J. C.; Schmidt, R. R. Synlett 1998, 501–503. doi:10.1055/s-1998-1689 |
| 77. | Oka, N.; Kajino, R.; Takeuchi, K.; Nagakawa, H.; Ando, K. J. Org. Chem. 2014, 79, 7656–7664. doi:10.1021/jo500632h |
| 4. | Nicolaou, K. C.; Mitchel, H. J. Angew. Chem., Int. Ed. 2001, 40, 1576–1624. doi:10.1002/1521-3773(20010504)40:9<1576::AID-ANIE15760>3.0.CO;2-G |
| 10. | Lu, Y.-S.; Li, Q.; Zhang, L.-H.; Ye, X.-S. Org. Lett. 2008, 10, 3445–3448. doi:10.1021/ol801190c |
| 38. | Pongdee, R.; Wu, B.; Sulikowski, G. A. Org. Lett. 2001, 3, 3523–3525. doi:10.1021/ol016593f |
| 39. | Crich, D.; Vinogradova, O. J. Org. Chem. 2006, 71, 8473–8480. doi:10.1021/jo061417b |
| 40. |
Kim, K. S.; Park, J.; Lee, Y. J.; Seo, Y. S. Angew. Chem., Int. Ed. 2003, 42, 459–462. doi:10.1002/anie.200390139
Angew. Chem. 2003, 115, 475–478. doi:10.1002/ange.200390107 |
| 41. | Park, J.; Boltje, T. J.; Boons, G.-J. Org. Lett. 2008, 10, 4367–4370. doi:10.1021/ol801833n |
| 42. | Lu, Y.-S.; Li, Q.; Wang, Y.; Ye, X.-S. Synlett 2010, 1519–1524. doi:10.1055/s-0029-1219943 |
| 19. | Ito, Y.; Ogawa, T. Tetrahedron Lett. 1987, 28, 2723–2726. doi:10.1016/S0040-4039(00)96191-4 |
| 20. | Grewal, G.; Kaila, N.; Franck, R. W. J. Org. Chem. 1992, 57, 2084–2092. doi:10.1021/jo00033a033 |
| 21. | Ramesh, S.; Franck, R. W. J. Chem. Soc., Chem. Commun. 1989, 960–962. doi:10.1039/C39890000960 |
| 26. | Auzanneau, F.-I.; Bundle, D. R. Carbohydr. Res. 1991, 212, 13–24. doi:10.1016/0008-6215(91)84041-C |
| 27. | Zuurmond, H. M.; van der Klein, P. A. M.; van der Marel, G. A.; van Boom, J. H. Tetrahedron 1993, 49, 6501–6514. doi:10.1016/S0040-4020(01)80165-4 |
| 28. | Yang, Z.; Yu, B. Carbohydr. Res. 2001, 333, 105–114. doi:10.1016/S0008-6215(01)00124-0 |
| 29. | Yu, B.; Yang, Z. Org. Lett. 2001, 3, 377–379. doi:10.1021/ol006894+ |
| 30. | Sajtos, F.; Lázár, L.; Borbás, A.; Bajza, I.; Lipták, A. Tetrahedron Lett. 2005, 46, 5191–5194. doi:10.1016/j.tetlet.2005.05.112 |
| 31. | Lázár, L.; Bajza, I.; Jakab, Z.; Lipták, A. Synlett 2005, 2242–2244. doi:10.1055/s-2005-872244 |
| 32. | Hou, D.; Lowary, T. L. Org. Lett. 2007, 9, 4487–4490. doi:10.1021/ol7019108 |
| 33. | Hou, D.; Lowary, T. L. J. Org. Chem. 2009, 74, 2278–2289. doi:10.1021/jo900131a |
| 11. | Roush, W. R.; Briner, K.; Sebesta, D. P. Synlett 1993, 264–266. doi:10.1055/s-1993-22425 |
| 12. | Roush, W. R.; Bennett, C. E. J. Am. Chem. Soc. 1999, 121, 3541–3542. doi:10.1021/ja984365j |
| 13. | Roush, W. R.; Gung, B. W.; Bennett, C. E. Org. Lett. 1999, 1, 891–893. doi:10.1021/ol9908070 |
| 14. | Tatsuta, K.; Fujimoto, K.; Kinoshita, M.; Umezawa, S. Carbohydr. Res. 1977, 54, 85–104. doi:10.1016/S0008-6215(00)80558-3 |
| 15. | Thiem, J.; Gerken, M. J. Org. Chem. 1985, 50, 954–958. doi:10.1021/jo00207a009 |
| 16. |
Thiem, J.; Schöttmer, B. Angew. Chem., Int. Ed. Engl. 1987, 26, 555–557. doi:10.1002/anie.198705551
Angew. Chem. 1987, 99, 591–592. doi:10.1002/ange.19870990626 |
| 17. |
Bucher, C.; Gilmour, R. Angew. Chem., Int. Ed. 2010, 49, 8724–8728. doi:10.1002/anie.201004467
Angew. Chem. 2010, 122, 8906–8910. doi:10.1002/ange.201004467 |
| 18. | Durantie, E.; Bucher, C.; Gilmour, R. Chem. – Eur. J. 2012, 18, 8208–8215. doi:10.1002/chem.201200468 |
| 34. |
Capozzi, G.; Dios, A.; Franck, R. W.; Geer, A.; Marzabadi, C.; Menichetti, S. C.; Tamarez, M. Angew. Chem. 1996, 108, 805–807. doi:10.1002/ange.19961080710
Angew. Chem. Int. Ed. Engl. 1996, 35, 777–779. doi:10.1002/anie.199607771 |
| 61. | Cui, X.-K.; Zhong, M.; Meng, X.-B.; Li, Z.-J. Carbohydr. Res. 2012, 358, 19–22. doi:10.1016/j.carres.2012.06.004 |
| 10. | Lu, Y.-S.; Li, Q.; Zhang, L.-H.; Ye, X.-S. Org. Lett. 2008, 10, 3445–3448. doi:10.1021/ol801190c |
| 16. |
Thiem, J.; Schöttmer, B. Angew. Chem., Int. Ed. Engl. 1987, 26, 555–557. doi:10.1002/anie.198705551
Angew. Chem. 1987, 99, 591–592. doi:10.1002/ange.19870990626 |
| 9. | Lear, M. J.; Yoshimura, F.; Hirama, M. Angew. Chem., Int. Ed. 2001, 40, 946–949. doi:10.1002/1521-3773(20010302)40:5<946::AID-ANIE946>3.0.CO;2-G |
| 22. | Barrett, A. G. M.; Miller, T. A. Tetrahedron Lett. 1988, 29, 1873–1874. doi:10.1016/S0040-4039(00)82065-1 |
| 23. | Perez, M.; Beau, J.-M. Tetrahedron Lett. 1989, 30, 75–78. doi:10.1016/S0040-4039(01)80327-0 |
| 24. | Sebesta, D. P.; Roush, W. R. J. Org. Chem. 1992, 57, 4799–4802. doi:10.1021/jo00044a010 |
| 25. | Nicolaou, K. C.; Pastor, J.; Barluenga, S.; Winssinger, N. Chem. Commun. 1998, 1947–1948. doi:10.1039/a804795b |
| 6. | Hou, D.; Lowary, T. L. Carbohydr. Res. 2009, 344, 1911–1940. doi:10.1016/j.carres.2009.07.013 |
| 63. | Kimura, T.; Takahashi, D.; Toshima, K. J. Org. Chem. 2015, 80, 9552–9562. doi:10.1021/acs.joc.5b01542 |
| 51. | Beau, J.-M.; Sinay, P. Tetrahedron Lett. 1985, 26, 6185–6188. doi:10.1016/S0040-4039(00)95048-2 |
| 52. | Bolitt, V.; Mioskowski, C.; Lee, S.-G.; Falck, J. R. J. Org. Chem. 1990, 55, 5812–5813. doi:10.1021/jo00310a006 |
| 53. | Mereyala, H. B.; Ravi, D. Tetrahedron Lett. 1991, 32, 7317–7320. doi:10.1016/0040-4039(91)80508-4 |
| 54. | Yadav, J. S.; Reddy, B. V. S.; Reddy, K. B.; Satyanarayana, M. Tetrahedron Lett. 2002, 43, 7009–7012. doi:10.1016/S0040-4039(02)01584-8 |
| 55. | Paul, S.; Jayaraman, N. Carbohydr. Res. 2004, 339, 2197–2204. doi:10.1016/j.carres.2004.07.010 |
| 56. | Sherry, B. D.; Loy, R. N.; Toste, F. D. J. Am. Chem. Soc. 2004, 126, 4510–4511. doi:10.1021/ja031895t |
| 57. | Palmier, S.; Vauzeilles, B.; Beau, J.-M. Org. Biomol. Chem. 2003, 1, 1097–1098. doi:10.1039/b301805a |
| 58. | Yadav, J. S.; Reddy, B. V. S.; Bhasker, E. V.; Raghavendra, S.; Narsaiah, A. V. Tetrahedron Lett. 2007, 48, 677–680. doi:10.1016/j.tetlet.2006.11.103 |
| 59. | Lin, H.-C.; Pan, J.-F.; Chen, Y.-B.; Lin, Z.-P.; Lin, C.-H. Tetrahedron 2011, 67, 6362–6368. doi:10.1016/j.tet.2011.05.124 |
| 60. | Balmond, E. I.; Coe, D. M.; Galan, M. C.; McGarrigle, E. M. Angew. Chem., Int. Ed. 2012, 51, 9152–9155. doi:10.1002/anie.201204505 |
| 61. | Cui, X.-K.; Zhong, M.; Meng, X.-B.; Li, Z.-J. Carbohydr. Res. 2012, 358, 19–22. doi:10.1016/j.carres.2012.06.004 |
| 62. | Balmond, E. I.; Benito-Alifonso, D.; Coe, D. M.; Alder, R. W.; McGarrigle, E. M.; Galan, M. C. Angew. Chem., Int. Ed. 2014, 53, 8190–8194. doi:10.1002/anie.201403543 |
| 63. | Kimura, T.; Takahashi, D.; Toshima, K. J. Org. Chem. 2015, 80, 9552–9562. doi:10.1021/acs.joc.5b01542 |
| 50. | Verma, V. P.; Wang, C.-C. Chem. – Eur. J. 2013, 19, 846–851. doi:10.1002/chem.201203418 |
| 10. | Lu, Y.-S.; Li, Q.; Zhang, L.-H.; Ye, X.-S. Org. Lett. 2008, 10, 3445–3448. doi:10.1021/ol801190c |
| 73. | Crich, D.; Smith, M.; Yao, Q.; Picione, J. Synthesis 2001, 323–326. doi:10.1055/s-2001-10798 |
| 74. | Lemieux, R. U.; Ratclffe, R. M. Can. J. Chem. 1979, 57, 1244–1251. doi:10.1139/v79-203 |
| 75. | Satoh, H.; Hansen, H. S.; Manabe, S.; van Gunsteren, W. F.; Hünenberger, P. H. J. Chem. Theory Comput. 2010, 6, 1783–1797. doi:10.1021/ct1001347 |
| 76. | Chen, J.-H.; Ruei, J.-H.; Mong, K.-K. T. Eur. J. Org. Chem. 2014, 1827–1831. doi:10.1002/ejoc.201400006 |
| 77. | Oka, N.; Kajino, R.; Takeuchi, K.; Nagakawa, H.; Ando, K. J. Org. Chem. 2014, 79, 7656–7664. doi:10.1021/jo500632h |
| 76. | Chen, J.-H.; Ruei, J.-H.; Mong, K.-K. T. Eur. J. Org. Chem. 2014, 1827–1831. doi:10.1002/ejoc.201400006 |
| 70. | Patil, P. R.; Kartha, K. P. R. J. Carbohydr. Chem. 2008, 27, 411–419. doi:10.1080/07328300802402259 |
| 71. | Tyagi, M.; Khurana, D.; Kartha, K. P. R. Carbohydr. Res. 2013, 379, 55–59. doi:10.1016/j.carres.2013.06.018 |
| 72. | Kumar, V.; Yadav, N.; Kartha, K. P. R. Carbohydr. Res. 2014, 397, 18–26. doi:10.1016/j.carres.2014.08.002 |
| 67. | Limousin, C.; Cléophax, J.; Petit, A.; Loupy, A.; Lukacs, G. J. Carbohydr. Chem. 1997, 16, 327–342. doi:10.1080/07328309708006533 |
| 69. | Lautrette, S.; Granet, R.; Krausz, P. Chem. Commun. 2004, 586–587. doi:10.1039/b315699k |
| 70. | Patil, P. R.; Kartha, K. P. R. J. Carbohydr. Chem. 2008, 27, 411–419. doi:10.1080/07328300802402259 |
| 71. | Tyagi, M.; Khurana, D.; Kartha, K. P. R. Carbohydr. Res. 2013, 379, 55–59. doi:10.1016/j.carres.2013.06.018 |
| 65. | Rioux, J. P.; Myers, R. A. M. J. Emerg. Med. 1988, 6, 227–238. doi:10.1016/0736-4679(88)90330-7 |
| 66. | Lefevre, P. A.; Ashby, J. Carcinogenesis 1989, 10, 1067–1072. doi:10.1093/carcin/10.6.1067 |
| 67. | Limousin, C.; Cléophax, J.; Petit, A.; Loupy, A.; Lukacs, G. J. Carbohydr. Chem. 1997, 16, 327–342. doi:10.1080/07328309708006533 |
| 68. | de Oliveira, R. N.; de Freitas Filho, J. R.; Srivastava, R. M. Tetrahedron Lett. 2002, 43, 2141–2143. doi:10.1016/S0040-4039(02)00156-9 |
| 69. | Lautrette, S.; Granet, R.; Krausz, P. Chem. Commun. 2004, 586–587. doi:10.1039/b315699k |
| 52. | Bolitt, V.; Mioskowski, C.; Lee, S.-G.; Falck, J. R. J. Org. Chem. 1990, 55, 5812–5813. doi:10.1021/jo00310a006 |
| 57. | Palmier, S.; Vauzeilles, B.; Beau, J.-M. Org. Biomol. Chem. 2003, 1, 1097–1098. doi:10.1039/b301805a |
| 59. | Lin, H.-C.; Pan, J.-F.; Chen, Y.-B.; Lin, Z.-P.; Lin, C.-H. Tetrahedron 2011, 67, 6362–6368. doi:10.1016/j.tet.2011.05.124 |
| 64. | Dick, F. D. Occup. Environ. Med. 2006, 63, 221–226. doi:10.1136/oem.2005.022400 |
© 2016 Hsu et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)