Abstract
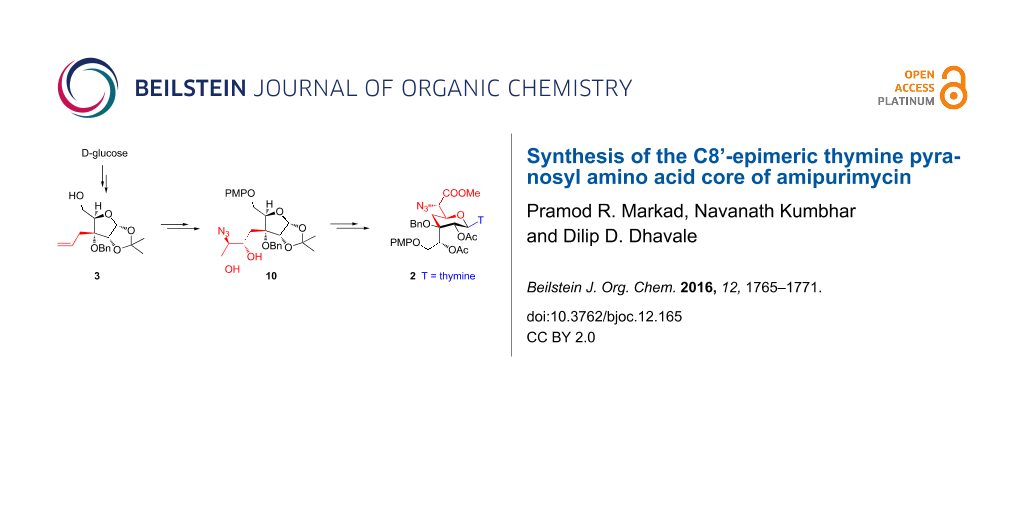
The C8’-epimeric pyranosyl amino acid core 2 of amipurimycin was synthesized from D-glucose derived alcohol 3 in 13 steps and 14% overall yield. Thus, the Sharpless asymmetric epoxidation of allyl alcohol 7 followed by trimethyl borate mediated regio-selective oxirane ring opening with azide, afforded azido diol 10. The acid-catalyzed 1,2-acetonide ring opening in 10 concomitantly led to the formation of the pyranose ring skeleton to give 2,7-dioxabicyclo[3.2.1]octane 12. Functional group manipulation in 12 gave 21 that on stereoselective β-glycosylation afforded the pyranosyl thymine nucleoside 2 – a core of amipurimycin.

Graphical Abstract
Introduction
Peptidyl nucleoside antibiotics are a class of complex molecules that encompass an extensive array of natural products [1]. The notable structural features of peptidyl nucleosides are responsible for their miscellaneous biological activities such as antitumor, antiviral, antibacterial and antifungal [2]. Peptidyl nucleosides in which the sugar part is in the furanose form are common, however, the sugar framework in the pyranose form, with a nucleobase and a peptide linker at either ends, are rare in nature. A few examples of this category are amipurimycin and miharamycin that are known as antifungal agents [2]. Amipurimycin (1) isolated from Streptomyces novoguineensis sp. nov., displays antifungal activity against pyricularia oryzae – a causative agent in rice blast disease [3,4]. Goto and co-workers have proposed the primary structure of amipurimycin (1, Figure 1) that involves (a) a unique pyranosyl amino acid ring skeleton with a hydroxy group and a branched 1,2-dihydroxyethyl side chain at C3’, (b) a glycosidic β-linked purine nucleobase, and (c) a five-membered cis-pentacin framework coupled to C6’ via the N-terminus of an amino acid [5]. The absolute configurations at the C6’, C2” and C3” of the cis-pentacin are still undefined. Thus, the partially unresolved structure, a potent antifungal activity, the unexplored mode of action and the limited synthetic study make amipurimycin (1) an attractive target for futher investigation. As of now, a total synthesis of 1 is not known. The reported methodologies are mainly focused on construction of the exigent central pyranosyl amino acid fragment with or without the C3’-branched chain and nucleobase, frequently using carbohydrate substrate [6-15]. In a non-carbohydrate approach, Garner and co-workers have exploited a cycloaddition pathway between a poly-oxygenated diene and Garner’s aldehyde for constructing the carbohydrate core of amipurimycin [9]. Recently, Datta and co-workers reported the first synthesis of a fully-functionalized thymine analogue of amipurimycin, utilizing D-serine as a starting material [6]. In this regard, our group has recently reported the synthesis of the C3’-branched carbohydrate core of amipurimycin starting from D-glucose [15]. In the continuation of this area, we now report the synthesis of the amipurimycin pyranose core comprising of (a) a hydroxy and 1,2-dihydroxyethyl side chain at C3’ having a C8’ epimeric center, (b) an C5’ amino acid pendant and (c) the thymine nucleobase (2). Our results in this regard are described herein.
![[1860-5397-12-165-1]](/bjoc/content/figures/1860-5397-12-165-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Antifungal antibiotic amipurimycin (1).
Figure 1: Antifungal antibiotic amipurimycin (1).
Result and Discussion
As shown in retrosynthetic analysis (Scheme 1), we envisioned that the substituted 2,7-dioxabicyclo[3.2.1]octane A could be glycosylated stereoselectively with the requisite nucleobase to give β-nucleoside pyranosyl skeleton 2. The bridged bicyclic system A was visualized from the azido diol B. Thus, hydrolysis of the 1,2-acetonide functionality in B will lead to in situ generation of oxocarbenium ion at C1 to which concomitant addition of a hydroxy group (present in the side chain at C3) will give the requisite pyranose ring skeleton. Intermediate B could be derived from the allyl alcohol C by using the Sharpless asymmetric epoxidation followed by regioselective epoxide ring opening with an azide nucleophile. The synthesis of allyl alcohol C from D-glucose was reported by us earlier.
![[1860-5397-12-165-i1]](/bjoc/content/inline/1860-5397-12-165-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Our synthesis started with the homoallyl alcohol 3 (with defined ‘R’ absolute configuration at the C3-quaternary center) that is obtained from D-glucose as reported earlier by us in 37% overall yield [15] (Scheme 2). Selective protection of the C5 primary hydroxy group as PMP ether using p-methoxyphenol under Mitsunobu reaction conditions afforded 4 that on benzylation of the C3 hydroxy group (NaH and benzyl bromide in DMF) gave compound 5. Upjohn dihydroxylation of 5 using K2OsO4·2H2O followed by oxidative cleavage of diol with silica-supported NaIO4 gave aldehyde that was directly reacted with ethyl 2-(triphenylphosphoranylidene)acetate to give α,β-unsaturated ester 6 (E/Z = 92:8) in 83% yield over three steps. Reduction of ester 6 (E-isomer) with DIBAL-H gave allyl alcohol 7 – a synthone for the Sharpless asymmetric epoxidation (SAE) [16,17].
![[1860-5397-12-165-i2]](/bjoc/content/inline/1860-5397-12-165-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 1,3-anhydrosugar 12 and 13.
Scheme 2: Synthesis of 1,3-anhydrosugar 12 and 13.
Thus, allyl alcohol 7 was subjected for SAE first using (+)-DET that afforded a diastereomeric mixture of epoxy alcohols 8 and 9 in the ratio of 88:12 (based on the 1H NMR analysis) in 85% yield. Similarly, use of (–)-DET in SAE afforded epoxide 8 and 9 in the ratio of 18:82 in 83% yield. With the understanding of SAE mnemonic, we assigned the absolute configuration in epoxide 8 as 7S,8S and in epoxide 9 as 7R,8R. Subsequently, major isomers of epoxy alcohols 8 and 9 were individually subjected to regioselective epoxide ring opening using trimethyl borate and NaN3 in DMF that afforded azido diol 10 and 11 as major isomers, respectively [18,19]. In the next step, individual hydrolysis of the 1,2-acetonide group in 10 and 11 using TFA–H2O (3:1) provided the corresponding 1,3-anhydrosugar 12 and 13 in good yield.
The formation of 1,3-anhydrosugar 12/13 could be explained as follows (Scheme 3). Thus, treatment of 10/11 with TFA–H2O resulted in the opening of the 1,2-acetonide functionality and generation of an oxocarbenium ion Y. Intermolecular and reversible addition of water would lead to hemiacetal Z, however, intramolecular and irreversible attack of the secondary hydroxy group to the oxocarbenium ion Y led to a stable six-membered pyranose ring compound thus shifting the equilibrium in favour of bridged bicyclic system 12/13.
![[1860-5397-12-165-i3]](/bjoc/content/inline/1860-5397-12-165-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Formation of 2,7-dioxabicyclo[3.2.1]octane 12/13.
Scheme 3: Formation of 2,7-dioxabicyclo[3.2.1]octane 12/13.
In order to validate the configurational assignments at the newly generated stereocenters, the coupling constants of protons at C4’ and C5’ in the pyranose ring of 12/13 are noticed to be decisive. In compound 12, both the C4’ methylene protons were found to be merged and appeared as a multiplet at δ 2.10–2.16 ppm. Therefore, we converted 12 into its acetyl derivative 14 that allowed us to predict the absolute configuration at the newly generated stereocenters at C5’ and C6’ (corresponding to the C7 and C8 of epoxide 8) [20]. In the 1H NMR of 14, appearance of a triplet at δ 2.30 (J = 12.7 Hz) and a doublet of doublet at δ 2.09 (J = 12.7 and 4.7 Hz), integrating for one proton each, were assigned to the methylene protons at C4’. Based on the coupling constant and nOe studies (Figure 2), the signal at δ 2.30 was assigned to the axially oriented proton at C4’. As this proton appeared as a triplet with a large coupling constant of 12.7 Hz (vicinal and geminal), the adjacent C5’ proton therefore assigned axial orientation indicating 5’S absolute configuration as anticipated from the SAE mnemonic in compound 8. As the C5’ and C6’ stereocenters in 14 are derived from the regioselective SN2 opening of epoxide 8 by NaN3, the configuration at the C6’ (carrying azido group) was therefore assigned as 6’R.
![[1860-5397-12-165-2]](/bjoc/content/figures/1860-5397-12-165-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Conformational analysis of 13 and 14.
Figure 2: Conformational analysis of 13 and 14.
In the 1H NMR spectrum of anhydrosugar 13, both the C4’ protons appeared as two doublets of doublets at δ 2.47 (J = 14.1 and 8.3 Hz) and δ 2.34 (J = 14.2 and 7.4 Hz). The vicinal coupling constants of 8.3 and 7.4 Hz between the C4’ and C5’ proton suggested distortion in the conformation of the six-membered ring of the bridged system. Since we have assigned the (5’S) configuration at C5’ in 12, the anhydrosugar 13 expected to have the opposite-5’R configuration. Considering this fact, the alkyl pendant (azidoethanol group) in 13 is assumed to be axially oriented. However, this spatial arrangement gives rise to strong steric interactions between the alkyl pendant and C8’ aryloxymethylene group (Figure 2). In order to avoid this strong repulsive interaction, compound 13 adopts relatively more stable twist-boat conformation as shown in Figure 2. The adoption of the twist-boat conformation generates dihedral angles between the H5’ and H4’a/H4’e as ≈150°/25°. This justifies the observed coupling constants in the 1H NMR between H5’ and H4’ (JH4’a, H5’a = 8.3 Hz and JH4’e, H5’a = 7.4 Hz) based on the Karplus equation [21]. This observation also confirms the assigned configurations as 5’R and 6’S in 13.
The experimental results on conformational preferences of 12 and 13 were corroborated using geometry-optimized density functional theory (DFT). The geometrically optimized preferred conformations of 12 and 13 are depicted in Figure 3, and geometrical parameters for torsion angles and intramolecular hydrogen bonding interactions are given in Table 1. As shown in Figure 3, the preferred conformation of 12 was found to be a stable distorted chair conformation (a). The significant hydrogen bonding interactions between ring oxygen O5’ with H7’ and O7’ with H5’a are accountable for preserving the stable 4C1 chair conformation of compound 12 by maintaining the values 159.77°/42.86° for the θ (H4’a–C4’–C5’–H5’a) and φ (H4’e–C4’–C5’–H5’a) torsion angles (Table 1). Similarly, for compound 13 the preferred conformation was observed to be a twist-boat conformation (Figure 3b). The intramolecular interactions and hydrogen bonding provided additional stability to maintain the twist-boat conformation of 13 (Figure 3) and to adopt the dihedral angles of 143.94°/27.32° between H5’a and H4’a/H4’e, respectively. This supports our earlier assignments made using 1H NMR studies.
![[1860-5397-12-165-3]](/bjoc/content/figures/1860-5397-12-165-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Geometrically optimized conformation of 12 and 13 respectively by DFT study.
Figure 3: Geometrically optimized conformation of 12 and 13 respectively by DFT study.
Table 1: Geometrical parameters for hydrogen bonding and torsion angles for 12 and 13.
| Comp. | Atoms involved in H-bonds | Distance in Å | Angle in degree | Torsion angle values | Energy in atomic units (au) |
|---|---|---|---|---|---|
| 12 |
O5’···H-C7’
O7’···H-C5’ O3’···H-O2’ O9’···H-C10’ |
2.480
2.550 2.190 2.290 |
98.74
99.13 112.34 151.77 |
θ = H4’a–C4’–C5’–H5’
a = 200.23° (−159.77) Φ = H4’e–C4’–C5’–H5’ a = 317.14° (−42.86) |
−1583.424 au |
| 13 |
O5’···H-C7’
O3’···H-O2’ O9’···H-C10’ |
2.426
2.124 2.372 |
99.68
114.39 122.31 |
θ = H4’a–C4–C5–H5’
a = 143.94° φ = H4’e–C4–C5–H5’ a = 27.32° |
−1583.420 au |
After confirming the absolute configurations of newly generated stereocenters, we continued our synthesis with anhydrosugar 12 as its configurations are matching with that of target compound. Thus, TEMPO-mediated selective oxidation of the primary hydroxy group in 12 to acid functionality followed by esterification using diazomethane afforded azido methyl ester 15. Acetylation of 15 with acetic anhydride in pyridine gave acetate derivative 16 in 95% yield. Having fully functionalized intermediate 16 in hand, we thought to incorporate the purine nucleobase using Vorbrüggen conditions. Thus, reaction of glycosyl donor 16 with bis(trimethylsilyl)-2-(N-acetylamino)-6-chloropurine 17, under a variety of reaction conditions, of solvents, temperature, Lewis acids as well as the use of the thymine nucleobase 18 (Scheme 4) failed to provide the desired nucleoside.
![[1860-5397-12-165-i4]](/bjoc/content/inline/1860-5397-12-165-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Knowing the fact that the glycosylation reaction is severely influenced by numerous factors including solvent, Lewis acid, and protecting groups on the nucleobase or sugar; we thought of synthesizing the peracylated anhydrosugar to alter its reactivity towards glycosylation [22]. In this regard, anhydrosugar 15 was subjected to 10% Pd/C and Et3SiH (for deprotection of the benzyl functionality and reduction of the azide to an amine) affording a crude product which was directly reacted with CbzCl to afford compound 19 (Scheme 5). Compound 19 on reaction with ceric ammonium nitrate (deprotection of PMP ether) followed by acetyl protection of the resultant triol using Ac2O in pyridine gave triacetate derivative 20. The triacetate 20 was then subjected for glycosylation reaction under similar reaction conditions as above which failed to give the desired product.
![[1860-5397-12-165-i5]](/bjoc/content/inline/1860-5397-12-165-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Glycosylation attempt by changing protections.
Scheme 5: Glycosylation attempt by changing protections.
Alternatively, compound 16 was treated with Ac2O with a catalytic amount of H2SO4 that afforded pyranosyl acetate 21 as an α:β anomeric mixture (1.5:1) in good yield (Scheme 6). Glycosylation of 21 with bis-silylated 2-(N-acetylamino)-6-chloropurine under a variety of reaction conditions was found to be unsuccessful. However, glycosylation of 21 with bis(trimethylsilyl)thymine in the presence of TMSOTf in dichloromethane led to the stereoselective formation of β-thymine analogue 2 in 42% (71% based on recovered starting material) yield [23]. In the 1H NMR spectrum of 2, the large coupling constant between H1’ and H2’ (J = 9.4 Hz) [5] indicated their relative diaxial orientation confirming the formation of β-glycosylated product 2. In this reaction, the preferred attack of the thymine nucleobase from the β-face is being assisted by the acetoxy group at C2 due to neighbouring group participation [24]. The orthogonally protected thymine analogue 2 is an important intermediate and could be utilized further for the synthesis of amipurimycin 1 and its analogues for structure–activity relationship (SAR) studies.
![[1860-5397-12-165-i6]](/bjoc/content/inline/1860-5397-12-165-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, we have utilized the skeleton of D-glucose-derived homoallyl alcohol 3 as a chiral podium for efficient synthesis of the 8’R-glycosyl amino acid core of amipurimycin. With this protocol, we have synthesized the 6’S amino acid fragment of amipurimycin. Utilization of a similar protocol with azido diol 11 could be employed for the synthesis of the corresponding 6’R fragment. Compound 2 is an important intermediate towards the synthesis of amipurimycin and its analogues for development of SAR and efforts in this regard are in progress.
Acknowledgements
We are thankful to the Department of Science and Technology, New Delhi (SERB, Project File No. EMR/2014/000873) for providing financial support and the Central Instrumentation Facility of Savitribai Phule Pune University for providing analytical support. PRM is thankful to CSIR, New Delhi for senior research fellowship. NK is thankful to Dr. D. S. Kothari fellowship (BSR/BL/14-15/0203) from the UGC, New Delhi.
References
-
Knapp, S. Chem. Rev. 1995, 95, 1859–1876. doi:10.1021/cr00038a006
Return to citation in text: [1] -
Ichikawa, S.; Matsuda, A. Expert Opin. Ther. Pat. 2007, 17, 487–498. doi:10.1517/13543776.17.5.487
Return to citation in text: [1] [2] -
Harada, S.; Kishi, T. J. Antibiot. 1977, 30, 11–16. doi:10.7164/antibiotics.30.11
Return to citation in text: [1] -
Iwasa, T.; Kishi, T.; Matsuura, K.; Wakae, O. J. Antibiot. 1977, 30, 1–10. doi:10.7164/antibiotics.30.1
Return to citation in text: [1] -
Goto, T.; Toya, Y.; Ohgi, T.; Kondo, T. Tetrahedron Lett. 1982, 23, 1271–1274. doi:10.1016/S0040-4039(00)87080-X
Return to citation in text: [1] [2] -
Stauffer, C. S.; Datta, A. J. Org. Chem. 2008, 73, 4166–4174. doi:10.1021/jo8004815
Return to citation in text: [1] [2] -
Xue, J.; Guo, Z. J. Carbohydr. Chem. 2008, 27, 51–69. doi:10.1080/07328300802030787
Return to citation in text: [1] -
Bhaket, P.; Stauffer, C. S.; Datta, A. J. Org. Chem. 2004, 69, 8594–8601. doi:10.1021/jo048586l
Return to citation in text: [1] -
Garner, P.; Yoo, J. U.; Sarabu, R.; Kennedy, V. O.; Youngs, W. J. Tetrahedron 1998, 54, 9303–9316. doi:10.1016/S0040-4020(98)00585-7
Return to citation in text: [1] [2] -
Czernecki, S.; Franco, S.; Valery, J.-M. J. Org. Chem. 1997, 62, 4845–4847. doi:10.1021/jo962266n
Return to citation in text: [1] -
Czernecki, S.; Valery, J.-M.; Wilkens, R. Bull. Chem. Soc. Jpn. 1996, 69, 1347–1351. doi:10.1246/bcsj.69.1347
Return to citation in text: [1] -
Rauter, A. P.; Fernandes, A. C.; Czernecki, S.; Valery, J.-M. J. Org. Chem. 1996, 61, 3594–3598. doi:10.1021/jo952220e
Return to citation in text: [1] -
Casiraghi, G.; Colombo, L.; Rassu, G.; Spanu, P. J. Org. Chem. 1991, 56, 6523–6527. doi:10.1021/jo00023a015
Return to citation in text: [1] -
Hara, K.; Fujimoto, H.; Sato, K.-I.; Hashimoto, H.; Yoshimura, J. Carbohydr. Res. 1987, 159, 65–79. doi:10.1016/S0008-6215(00)90006-5
Return to citation in text: [1] -
Mane, R. S.; Ghosh, S.; Chopade, B. A.; Reiser, O.; Dhavale, D. D. J. Org. Chem. 2011, 76, 2892–2895. doi:10.1021/jo102193q
Return to citation in text: [1] [2] [3] -
Sharpless, K. B. Catalytic Asymmetric Epoxidation of Allylic alcohols.. In Catalytic asymmetric synthesis; Ojima, I., Ed.; VCH Publishers: New York, 1993; pp 103 ff.
Return to citation in text: [1] -
Xia, Q.-H.; Ge, H.-Q.; Ye, C.-P.; Liu, Z.-M.; Su, K.-X. Chem. Rev. 2005, 105, 1603–1662. doi:10.1021/cr0406458
Return to citation in text: [1] -
Sasaki, M.; Tanino, K.; Hirai, A.; Miyashita, M. Org. Lett. 2003, 5, 1789–1791. doi:10.1021/ol034455f
Return to citation in text: [1] -
Formation of other regio-isomer was not observed, this was confirmed by reaction of 10 with NaIO4 and analyzing reaction mixture with 1H NMR for aldehyde signal.
Return to citation in text: [1] -
Compound 3 to 10 follows standard carbohydrate assignments (numbering). However for convenience, compound 12, 13, 14 and 16–21 follows assignments relative to amipurimycin.
Return to citation in text: [1] -
Jackman, L. M.; Cotton, F. A., Eds. Dynamic nuclear magnetic resonance spectroscopy; Academic Press INC: London, 1975.
Return to citation in text: [1] -
Vorbrüggen, H.; Ruh-Pohlenz, C. Org. React. 2000, 55. doi:10.1002/0471264180.or055.01
Return to citation in text: [1] -
In the glycosylation reaction, use of acetonitrile as solvent instead of DCE has resulted in the formation of 16 and not the glycosylated product.
Return to citation in text: [1] -
Vorbrüggen, H.; Höfle, G. Chem. Ber. 1981, 114, 1256–1268. doi:10.1002/cber.19811140405
Return to citation in text: [1]
| 5. | Goto, T.; Toya, Y.; Ohgi, T.; Kondo, T. Tetrahedron Lett. 1982, 23, 1271–1274. doi:10.1016/S0040-4039(00)87080-X |
| 24. | Vorbrüggen, H.; Höfle, G. Chem. Ber. 1981, 114, 1256–1268. doi:10.1002/cber.19811140405 |
| 5. | Goto, T.; Toya, Y.; Ohgi, T.; Kondo, T. Tetrahedron Lett. 1982, 23, 1271–1274. doi:10.1016/S0040-4039(00)87080-X |
| 22. | Vorbrüggen, H.; Ruh-Pohlenz, C. Org. React. 2000, 55. doi:10.1002/0471264180.or055.01 |
| 3. | Harada, S.; Kishi, T. J. Antibiot. 1977, 30, 11–16. doi:10.7164/antibiotics.30.11 |
| 4. | Iwasa, T.; Kishi, T.; Matsuura, K.; Wakae, O. J. Antibiot. 1977, 30, 1–10. doi:10.7164/antibiotics.30.1 |
| 23. | In the glycosylation reaction, use of acetonitrile as solvent instead of DCE has resulted in the formation of 16 and not the glycosylated product. |
| 2. | Ichikawa, S.; Matsuda, A. Expert Opin. Ther. Pat. 2007, 17, 487–498. doi:10.1517/13543776.17.5.487 |
| 20. | Compound 3 to 10 follows standard carbohydrate assignments (numbering). However for convenience, compound 12, 13, 14 and 16–21 follows assignments relative to amipurimycin. |
| 2. | Ichikawa, S.; Matsuda, A. Expert Opin. Ther. Pat. 2007, 17, 487–498. doi:10.1517/13543776.17.5.487 |
| 21. | Jackman, L. M.; Cotton, F. A., Eds. Dynamic nuclear magnetic resonance spectroscopy; Academic Press INC: London, 1975. |
| 15. | Mane, R. S.; Ghosh, S.; Chopade, B. A.; Reiser, O.; Dhavale, D. D. J. Org. Chem. 2011, 76, 2892–2895. doi:10.1021/jo102193q |
| 16. | Sharpless, K. B. Catalytic Asymmetric Epoxidation of Allylic alcohols.. In Catalytic asymmetric synthesis; Ojima, I., Ed.; VCH Publishers: New York, 1993; pp 103 ff. |
| 17. | Xia, Q.-H.; Ge, H.-Q.; Ye, C.-P.; Liu, Z.-M.; Su, K.-X. Chem. Rev. 2005, 105, 1603–1662. doi:10.1021/cr0406458 |
| 6. | Stauffer, C. S.; Datta, A. J. Org. Chem. 2008, 73, 4166–4174. doi:10.1021/jo8004815 |
| 18. | Sasaki, M.; Tanino, K.; Hirai, A.; Miyashita, M. Org. Lett. 2003, 5, 1789–1791. doi:10.1021/ol034455f |
| 19. | Formation of other regio-isomer was not observed, this was confirmed by reaction of 10 with NaIO4 and analyzing reaction mixture with 1H NMR for aldehyde signal. |
| 9. | Garner, P.; Yoo, J. U.; Sarabu, R.; Kennedy, V. O.; Youngs, W. J. Tetrahedron 1998, 54, 9303–9316. doi:10.1016/S0040-4020(98)00585-7 |
| 6. | Stauffer, C. S.; Datta, A. J. Org. Chem. 2008, 73, 4166–4174. doi:10.1021/jo8004815 |
| 7. | Xue, J.; Guo, Z. J. Carbohydr. Chem. 2008, 27, 51–69. doi:10.1080/07328300802030787 |
| 8. | Bhaket, P.; Stauffer, C. S.; Datta, A. J. Org. Chem. 2004, 69, 8594–8601. doi:10.1021/jo048586l |
| 9. | Garner, P.; Yoo, J. U.; Sarabu, R.; Kennedy, V. O.; Youngs, W. J. Tetrahedron 1998, 54, 9303–9316. doi:10.1016/S0040-4020(98)00585-7 |
| 10. | Czernecki, S.; Franco, S.; Valery, J.-M. J. Org. Chem. 1997, 62, 4845–4847. doi:10.1021/jo962266n |
| 11. | Czernecki, S.; Valery, J.-M.; Wilkens, R. Bull. Chem. Soc. Jpn. 1996, 69, 1347–1351. doi:10.1246/bcsj.69.1347 |
| 12. | Rauter, A. P.; Fernandes, A. C.; Czernecki, S.; Valery, J.-M. J. Org. Chem. 1996, 61, 3594–3598. doi:10.1021/jo952220e |
| 13. | Casiraghi, G.; Colombo, L.; Rassu, G.; Spanu, P. J. Org. Chem. 1991, 56, 6523–6527. doi:10.1021/jo00023a015 |
| 14. | Hara, K.; Fujimoto, H.; Sato, K.-I.; Hashimoto, H.; Yoshimura, J. Carbohydr. Res. 1987, 159, 65–79. doi:10.1016/S0008-6215(00)90006-5 |
| 15. | Mane, R. S.; Ghosh, S.; Chopade, B. A.; Reiser, O.; Dhavale, D. D. J. Org. Chem. 2011, 76, 2892–2895. doi:10.1021/jo102193q |
| 15. | Mane, R. S.; Ghosh, S.; Chopade, B. A.; Reiser, O.; Dhavale, D. D. J. Org. Chem. 2011, 76, 2892–2895. doi:10.1021/jo102193q |
© 2016 Markad et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)