Abstract
Benzo[1,2-b:4,5-b′]dithiophene (BDT) is an excellent building block for constructing π-conjugated molecules for the use in organic solar cells. In this paper, four 4,8-bis(5-alkyl-2-thienyl)benzo[1,2-b:4,5-b′]dithiophene (TBDT)-containing A–π–D–π–A-type small molecules (COOP-nHT-TBDT, n = 1, 2, 3, 4), having 2-cyano-3-octyloxy-3-oxo-1-propenyl (COOP) as terminal group and regioregular oligo(3-hexylthiophene) (nHT) as the π-conjugated bridge unit were synthesized. The optical and electrochemical properties of these compounds were systematically investigated. All these four compounds displayed broad absorption bands over 350–600 nm. The optical band gap becomes narrower (from 1.94 to 1.82 eV) and the HOMO energy levels increased (from −5.68 to −5.34 eV) with the increase of the length of the π-conjugated bridge. Organic solar cells using the synthesized compounds as the electron donor and PC61BM as the electron acceptor were fabricated and tested. Results showed that compounds with longer oligothiophene π-bridges have better power conversion efficiency and higher device stability. The device based on the quaterthiophene-bridged compound 4 gave a highest power conversion efficiency of 5.62% with a VOC of 0.93 V, JSC of 9.60 mA·cm−2, and a FF of 0.63.
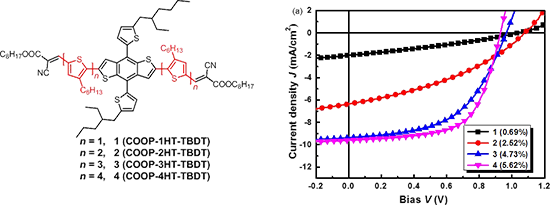
Graphical Abstract
Introduction
Solution-processed organic solar cells (OSCs) are considered to be one of the most promising renewable energy technologies because of the advantages of low cost, lightweight, flexibility, and great potentials in large-scale production [1,2]. In the past few years, OSCs based on polymers have achieved power conversion efficiencies (PCEs) of over 11% [3,4]. Meanwhile, OSCs based on conjugated small molecules attracted also enormous attentions due to their ease of synthesis, defined chemical structure, low batch-to-batch variation, and good reproducibility in photovoltaic performance [5-7]. To date, PCEs of more than 9% for small molecule OSCs (SMOSCs) have been reported [8-12].
Among various electron-donating moieties, benzo[1,2-b:4,5-b′]dithiophene (BDT) has been widely used as the central building block for constructing high-performance A–π–D–π–A-type organic semiconductors for organic solar cells, where A represents the terminal electron acceptor unit, D represents the core electron donor unit, and π represents the conjugated π-bridge [13,14], and a maximum PCE of 9.95% was reported for a terthiophene-bridged small molecule with a BDT core [8]. Currently, there are three main structure modifications of A–π–D–π–A-type molecules with a BDT core. One is the substitution on the 4,8-positions of the BDT core with aromatic units, including alkyl/alkoxyl/alkylthiol-substituted phenyl groups [15], thienyl group [16-18], and thienothiophene [17,19]. Structure modifications of the BDT core with aromatic units extend the π-conjugation of BDT unit to a two-dimensional structure, which increases intermolecular interactions, and consequently improves the device performance. The other one is to attach different electron acceptor units at the terminal of the molecules, including: dicyanovinyl [20,21], cyanoacetate [20-23], rhodanine [8,14,17,19,23], 1,3-indandione [16], and diketopyrolpyrol (DPP) moieties [24,25]. Changing the electron-withdrawing strength of the terminal electron acceptor unit, on the other hand, will change the intramolecular charge transfer state and tune the light absorption ability, which will consequently change the photovoltaic performance of the materials as well.
The third possible structure modification of BDT derivatives is to tune the conjugation length of the π-bridges. In this respect, oligothiophenes, including monotiophene [16,20], bithiophene [16], terthiophene [8,14,15,17-19,21-23,26], quaterthiophene and quinquethiophene [27], and cyclopentadithiophene [28] have been utilized as the π-bridge in constructing conjugated molecules with a BDT core. Among these 3,3''-dihexyl-2,2':5':2''-terthiophene (3T) is the most widely used π-bridge. It is worth to mention that 3-alkylthiophen can be coupled in different ways at the 2- and 5-positions, yielding oligo(3-alkylthiophene)s with different isomeric structures. In order to minimize the synthesis efforts, structurally symmetric oligothiophene units are mostly used for constructing A–π–D–π–A-type molecules with a BDT core. Interestingly, although various terthiophene-based derivatives with a BDT core have been reported, there is only one paper that reported the synthesis and characterization of BDT derivatives based on oligothiophene π-bridges with more than three thiophene units [27], where symmetric quater- and quinquethiophenes were used as the π-conjugation bridge. Surprisingly, the quaterthiophene-bridged compound showed the worst photovoltaic performance when blending with a fullerene derivative as the photoactive layer. This was ascribed to the influence of the orientation of the alkyl side chains. Although regioregular oligo(3-alkylthiophene)s are better building blocks for studying the effect of the π-conjugation length, only regioregular terthiophene (rr-3T) was reported to be used as the π-bridge unit in BDT derivatives [21,22]. BDT derivatives based on regioregular bi- or quaterthiophene have not been reported, and there is no systematically investigation on the effect of the π-conjugation length yet.
To better understand the effect of the conjugation length on the molecular structure and properties of the conjugated molecules with BDT core, we report here a series of A–π–D–π–A-type conjugated molecules with a regioregular oligo(3-hexylthiophene) chain as the π-bridge unit. The optical and electrochemical properties of these compounds were systematically investigated. Organic solar cells based these conjugated small molecules as the electron donor were fabricated and tested. In addition, long-term stability of these solar cells was also studied, and a general structure–property–performance relationship of these type of molecules is evaluated, which could serve as a useful guideline for further molecular design and synthesis for organic solar cells.
Results and Discussion
Synthesis and structure characterization of COOP-nHT-TBDT
The synthetic routes to compounds 1–4 (COOP-nHT-TBDT, n = 1–4; COOP = 2-cyano-3-octyloxy-3-oxo-1-propenyl, nHT = oligo(3-hexylthiophene), TBDT = 4,8-bis(5-alkyl-2-thienyl)benzo[1,2-b:4,5-b′]dithiophene) is shown in Scheme 1. The bithiophene building block, 3,4'-dihexyl-5'-iodo-2,2'-bithiophene-5-carbaldehyde (11) was synthesized by an ipso-substitution of 10, which was synthesized by a Suzuki coupling of 9 with 6, with ICl. The regioregular terthiophene and quaterthiophene building blocks were synthesized according to a similar synthetic route starting in high yields. The aldehyde precursors with BDT core CHO-nHT-TBDTs 17–20 were synthesized through a Pd-catalyzed Stille coupling reaction of 9, 11, 13 and 15 with 16, respectively (reaction iv), and the final compounds, COOP-nHT-TBDTs 1–4, were obtained by Knoevenagel condensations of CHO-nHT-TBDTs 17–20 with octylcyanoacetate (reaction v). Since all these compounds have multiple alkyl chains, they are soluble in common organic solvents, so that the final compounds can be processed well in solution. Complete characterization of both the intermediate and the final compounds was performed by 1H NMR, 13C NMR and mass spectrometry (see details in Supporting Information File 1).
![[1860-5397-12-169-i1]](/bjoc/content/inline/1860-5397-12-169-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic route to compounds 1–4 with BDT core. Reagents and conditions: i) [Pd2(dba)3]·CHCl3, HP(t-Bu)3BF4, K2CO3, THF; ii) 1. n-BuLi/THF, 2. 2-isopropyloxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane; iii) ICl/THF; iv) Pd(PPh3)4, DMF, 80 °C; v) octyl cyanoacetate, piperidine, CHCl3, reflux.
Scheme 1: Synthetic route to compounds 1–4 with BDT core. Reagents and conditions: i) [Pd2(dba)3]·CHCl3, HP(t-...
Optical properties
Figure 1 presents the UV–vis absorption spectra of COOP-nHT-TBDTs 1–4 in solution and in thin solid films. The spectroscopic data were collected and listed in Table 1. In dilute chloroform solution, all these compounds show intensive absorption bands from 350 to 600 nm with a gradual increase of the molar extinction coefficient. Interestingly, the three molecules 2, 3, and 4 display one broad absorption band peaking at 494, 491, and 485 nm, respectively, while two absorption bands peaking at 440 and 498 nm were found for 1. Elongation of the π-conjugated bridge leads to a slight hypochromic shift of the absorption band, presumably ascribed to a disorder of the complex structures of the π-conjugation chain, or due to the steric hindrance effects of the alkyl side chains for the bigger molecules [29,30]. The absorption onset wavelength increases slightly with the increase of the π-bridge chain length, suggesting an extended π-conjugation system for the compounds with longer oligothiophene chains.
![[1860-5397-12-169-1]](/bjoc/content/figures/1860-5397-12-169-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: UV−vis absorption spectra of COOP-nHT-TBDTs 1–4 (a) in chloroform solution (5.0 × 10−5 mol·L-1) and (b) in solid films.
Figure 1: UV−vis absorption spectra of COOP-nHT-TBDTs 1–4 (a) in chloroform solution (5.0 × 10−5 mol·L-1) and...
Table 1: Optical and electrochemical properties of 1–4 in comparison to other compounds in the literature.
| compound |
λmaxsol
[nm]a |
εmaxsol
[mol−1·L·cm−1]b |
λmaxfilm
[nm] |
Egopt
[eV]c |
E0ox
[V]d |
E0red
[V]d,e |
EHOMO
[eV]f |
ELUMO
[eV]f |
Egcv
[eV]g |
ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 440 (498)h | 64,500 | 580 (547)h | 1.94 | 0.66 | −1.53 | −5.68 (−5.38)i | −3.65 (−3.35)i | 2.03 | [20] |
| 2 | 494 | 68,800 | 558 (602)h | 1.86 | 0.50 | −1.62 | −5.52 (−5.22)i | −3.56 (−3.26)i | 1.96 | this work |
| 3 | 491 | 80,700 | 576 (625)h | 1.82 | 0.42 | −1.63 | −5.43 (−5.13)i | −3.55 (−3.25)i | 1.88 | this work |
| 4 | 485 | 93,900 | 570 (614)h | 1.82 | 0.32 | −1.64 | −5.35 (−5.05)i | −3.53 (−3.23)i | 1.82 | this work |
| DCAO3TBDT | 494 | 72,000 | 560 | 1.84 | — | — | −5.04i | −3.24i | 1.80 | [23] |
| TBDTCNR | 488 | — | 578 | 1.75 | — | — | −5.40i | −3.63i | 1.77 | [21] |
aIn CHCl3 (5.0 × 10−5 mol·L−1); bextinction coefficient was obtained by linearly fitting the absorbance as a function of the concentration; coptical band gap, calculated from the absorption onset wavelength (λonset) in solid films according to the equation Egopt (eV) = 1240/λonset (nm); dmeasured in CHCl3 solution (1.0 × 10−3 mol·L−1); eirreversible wave: E0red was estimated as the potential where ipc = 0.855 × ipcmax; fcalculated from the cyclic voltammograms, EHOMO = −[Eoxonset + 5.1] (eV), ELUMO = −[Eredonset + 5.1] (eV); gelectrochemical band gap Egcv= EHOMO − ELUMO = −[Eoxonset − Eredonset] (eV); hshoulder peak; icalculated from the cyclic voltammograms, EHOMO = −[Eoxonset + 4.8] (eV), ELUMO = −[Eredonset + 4.8] (eV).
In the solid state, absorption spectra of these compounds (see Figure 1b) are remarkably broadened and red-shifted relative to those in solutions, which is attributable to strong π–π stacking interaction between the molecular backbones in the solid films [31,32]. It is noticeable that compound 1 in the film shows two shoulder peaks in the long wavelength range, while compounds 2, 3, and 4 have only one shoulder peak. The optical band gaps calculated from the onsets of absorption edge of the four molecules are 1.94, 1.86, 1.82 and 1.82 eV, respectively, in agreement with the BDT derivatives reported in the literatures [21,23].
Electrochemical properties
Cyclic voltammetry (CV) was applied to investigate the energy levels of 1–4. The cyclic voltammograms of these four compounds are presented in Figure 2 and the electrochemical data are listed in Table 1. As can be seen from Figure 2, compounds 1 and 2 exhibited two reversible oxidation processes in the positive range, while 3 and 4 showed multiple oxidation processes, suggesting more oxidation processes of the π-conjugation bridge units for the larger molecules. The first oxidation potentials (E0ox vs Fc+/Fc) for COOP-nHT-TBDTs were measured to be 0.66, 0.50, 0.42, and 0.32 V, for n = 1, 2, 3, and 4, respectively, indicating a decreasing trend with the increase of the π-bridge chain length. Meanwhile, one irreversible reduction process was found for all these four compounds in the negative potential range. Except for 1, which showed a reduction potential (E0red) of −1.53 eV, the other three compounds showed almost identical E0red of −1.60 V, indicating that the reduction process is mainly due to the reduction of the terminal COOP group. The onset oxidation potentials (Eoxonset) and onset reduction potentials (Eredonset) determined from the CV results are also listed in Table 1. The frontier molecular orbital energy levels (HOMO/LUMO) and also the energy band gaps of these compounds were calculated according to the method reported in our previous paper [20], where the ferrocene/ferrocenium couple (Fc+/Fc) was used as the standard, and the vacuum energy level of Fc+/Fc was taken as −5.1 eV [33]. As can be seen from this table, the HOMO energy levels of COOP-nHT-TBDTs increased slightly from −5.68 to −5.34 V with increasing π-conjugation bridge length. Except for 1, which has a LUMO level of −3.65 eV, the other three compounds possess similar LUMO level at −3.55 eV, attributed to the same electron-withdrawing terminal units. The LUMO energy levels of COOP-nHT-TBDTs are more than 0.3 eV higher than that of PC61BM [34], which provides a sufficient driving force for electron transfer from COOP-nHT-TBDTs to PC61BM (Figure 2b). On the other hand, the low-lying HOMO energy level of COOP-nHT-TBDTs would be beneficial for achieving a high open circuit voltage (VOC), since VOC of organic solar cells is directly related to the difference of the HOMO energy of the donor material and the LUMO energy of the acceptor.
![[1860-5397-12-169-2]](/bjoc/content/figures/1860-5397-12-169-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: (a) Cyclic voltammograms of 1–4 measured in CH2Cl2 solution (1.0 × 10−3 mol·L−1) with 0.1 mol·L−1 Bu4NPF6 at a scan rate of 100 mV·s−1; (b) HOMO and LUMO energy levels of these compounds.
Figure 2: (a) Cyclic voltammograms of 1–4 measured in CH2Cl2 solution (1.0 × 10−3 mol·L−1) with 0.1 mol·L−1 Bu...
Photovoltaic performance
Bulk heterojunction (BHJ) solar cells with a device structure of ITO/PEDOT:PSS (30 nm)/photoactive layer/LiF (1.5 nm)/Al (100 nm) were fabricated and tested, where the blended solid film of the synthesized small molecules as donor and PC61BM as acceptor was used as the photoactive layer. The photovoltaic performances of 1 has been reported in our previous paper [20], and the PV performance data of the best cell are listed in Table 2 for comparison. The photovoltaic performance of cells based on compounds 2, 3, and 4 were carefully optimized by varying the D/A blend ratio. Figure S4 and S5 (Supporting Information File 1) depict the current density–voltage (J–V) curves and the external quantum efficiency (EQE) spectra of cells based on 2, 3, and 4, and the photovoltaic performance data are listed in Table 2. As can be seen from Table 2, the optimal D/A blend ratio for 2:PC61BM based cells was found to be 1:0.6, which showed a maximum PCE of 2.52% (and an average PCE of 2.35%) with a high VOC of 1.07 V, JSC of 6.36 mA·cm−2, and FF of 37% for the best cell. The optimal D/A blend ratio was found to be 1:0.4 for cells based on 3 and 4 (Table 2, entry 6 and 10), which is different to that of cells based on 1 and 2. High PCEs of 4.73% and 5.62% were achieved for cells with 3 and 4, respectively, which are much higher than that of the devices based on smaller molecules. Obviously, with the extension of conjugation π-bridges, the PV performance of the COOP-nHT-TBDTs enhanced gradually. Since VOC decreases slightly with the increase of the conjugation length of the π-bridge, such a device performance enhancement was mainly ascribed to the increase of JSC and FF (Table 2). EQE spectra comparison clearly confirmed the higher photon-to-electron conversion efficiency for the bigger molecules (Figure 3b). In addition, the photo responses of devices based on 3 and 4 cover a wavelength range from 380 to 700 nm, which is wider than that of devices based on 1 and 2 (Figure 3b, insert), agreeing with the absorption spectra of the corresponding thin solid films (Figure 1b). Solar cells based on a 4:PC71BM photoactive layer were also fabricated and tested. However, the PC71BM-based devices showed a slightly decreased performance compared to the PC61BM-based devices (Figure S5, Supporting Information File 1). Using additives or post-thermal annealing did not improve device performance. We speculate that impurities in PC71BM or the non-ideal interface between PEDOT:PSS and the photoactive layer could be the reason for the lower device performance. However, further experiments are still need to fully understand the detailed reasons. Nevertheless, the PCE of 5.62% for the 4:PC61BM cells is among the best performance for cells based on COOP-capped BDT derivatives [21-23,35-37].
Table 2: Photovoltaic properties of COOP-nHT-TBDT:PC61BM-based devices.
| entry | donor | D/A ratio [w/w] | VOC [V] | JSC [mA·cm−2]a | FF [%] | PCE [%] | average PCE [%] (± std. dev.)b |
|---|---|---|---|---|---|---|---|
| 1 | 1c | 1:0.6 | 1.04 | 2.28 | 29 | 0.69 | 0.61 (± 0.072) |
| 2 | 2 | 1:0.4 | 1.04 | 5.30 | 36 | 2.01 | 1.89 (± 0.24) |
| 3 | 1:0.6 | 1.07 | 6.36 | 37 | 2.52 | 2.35 (± 0.10) | |
| 4 | 1:0.8 | 1.06 | 5.20 | 36 | 1.98 | 1.78 (± 0.17) | |
| 5 | 3 | 1:0.2 | 0.97 | 5.99 | 47 | 2.73 | 2.64 (± 0.08) |
| 6 | 1:0.4 | 0.97 | 9.38 | 52 | 4.73 | 4.58 (± 0.16) | |
| 7 | 1:0.6 | 0.97 | 8.94 | 47 | 4.07 | 3.79 (± 0.18) | |
| 8 | 1:0.8 | 0.95 | 6.35 | 37 | 2.23 | 1.97 (± 0.21) | |
| 9 | 4 | 1:0.2 | 0.93 | 6.07 | 59 | 3.33 | 3.14 (± 0.15) |
| 10 | 1:0.4 | 0.93 | 9.60 | 63 | 5.62 | 5.27 (± 0.21) | |
| 11 | 1:0.6 | 0.91 | 7.73 | 44 | 3.07 | 2.75 (± 0.27) | |
| 12 | 1:0.8 | 0.91 | 5.72 | 40 | 2.08 | 1.93 (± 0.21) | |
adetermined by convoluting the spectral response with the AM 1.5G spectrum (100 mW·cm−2); bstandard deviation was calculated over eight individual devices; cdata from [20].
![[1860-5397-12-169-3]](/bjoc/content/figures/1860-5397-12-169-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: (a) J–V curves of the best COOP-nHT-TBDT:PC61BM solar cells; (b) EQE spectra of the corresponding cells, Inset: normalized EQE spectra of four best devices, showing the difference in spectrum response wavelength range.
Figure 3: (a) J–V curves of the best COOP-nHT-TBDT:PC61BM solar cells; (b) EQE spectra of the corresponding c...
Surface morphology of the blended films
The surface morphology of the COOP-nHT-TBDT:PC61BM films was scrutinized with atomic force microscopy (AFM). Figure 4 depicts the topological images of the blended films prepared under the optimized conditions. The surface roughness for the COOP-nHT-TBDT:PC61BM blended films was measured to be 1.23, 2.14, 0.96 and 2.84 nm for films based on 1, 2, 3, and 4, respectively, demonstrating a reasonable surface smoothness for these films. Obviously, crystalline domains can be seen in these films, among which, the 2:PC61BM and 4:PC61BM blended films showed larger crystalline domains compared to the films based on 3 and 4. Such a nanomorphology difference could be ascribed to the chemical structure difference of the π-conjugation bridges, demonstrating a possible odd–even effect. Nevertheless, the large crystalline domains of the 4-based film lead to a better charge carrier mobility (vide infra), which is beneficial for device performance.
![[1860-5397-12-169-4]](/bjoc/content/figures/1860-5397-12-169-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: AFM height images of COOP-nHT-TBDT:PC61BM blended films: (a) 1, 1:0.6 (w/w); (b) 2, 1:0.6 (w/w); (c) 3, 1:0.4 (w/w); (d) 4, 1:0.4 (w/w).
Figure 4: AFM height images of COOP-nHT-TBDT:PC61BM blended films: (a) 1, 1:0.6 (w/w); (b) 2, 1:0.6 (w/w); (c...
Charge-carrier mobility of the blended films
To further understand the influence of the chemical structure on the device performance, the hole mobility of these compounds in blended films was measured using the space-charge-limited current method (SCLC). The device structure studied here was ITO/PEDOT:PSS/COOP-nHT-TBDT:PC61BM/MoO3/Al, and the thin film deposition method is similar to that for solar cell fabrication. The analysis method was described in detail in our previous paper [34]. The hole transport mobilities of COOP-nHT-TBDT were measured to be 2.01 × 10−6, 1.81 × 10−6, 4.60 × 10−4 and 8.22 × 10−4 cm2·V−1·s−1 for 1, 2, 3, and 4, respectively. Obviously, the largest molecule 4 displays the highest hole mobility, which could be owing to the formation of large crystalline domains in 4:PC61BM blended film, as shown in Figure 5. The high hole mobility for the larger molecules could be one of the reasons for the higher power conversion efficiency for devices based on COOP-nHT-TBDT:PC61BM (Table 2).
![[1860-5397-12-169-5]](/bjoc/content/figures/1860-5397-12-169-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: J–V curves of COOP-nHT-TBDT:PC61BM-based hole-only devices.
Figure 5: J–V curves of COOP-nHT-TBDT:PC61BM-based hole-only devices.
Long-term stability
Finally, the long-term stability of these COOP-nHT-TBDT-based solar cells was tested. Devices for stability test were fabricated according to the optimized conditions described above, and these devices showed an initial performance similar to the best PCE as listed in Table 2 for each compound. Figure 6 presents the evolution of VOC, JSC, FF and PCE of un-encapsulated COOP-nHT-TBDT:PC61BM cells tested in N2 atmosphere under continuous illumination. To fully simulate the degradation behaviour of solar cells under working conditions, an external load to match the maximum power output point (mpp) was attached to each device, which was described in our previous report [38]. Similar to the previous report, the VOC of the devices decreased only slightly during light illumination. However, the JSC decreased to 42%, 12%, 10% and 4% of their initial value for cells based on 1, 2, 3 and 4, respectively (Figure 6b). In addition, the FF of these four devices decreased very slowly (Figure 6c) during aging. Overall, the 4-based devices showed the highest device stability with only 10% decay of its initial device performance, whereas the 1 showed the worst device stability, in which a decrease of 48% was measured. Surprisingly, 2 showed also higher device stability when compared to 3, which was mainly due to a more stable FF. More nanomophology stability was supposed to be main reason for the higher stability of the larger molecules, since larger molecules have a higher energy barrier for diffusion. However, more experiments are still needed to further understand the stability improvement of the larger molecules.
![[1860-5397-12-169-6]](/bjoc/content/figures/1860-5397-12-169-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: (a) VOC, (b) JSC, (c) FF, and (d) PCE decay of the COOP-nHT-TBDT:PC61BM solar cells. Note all these data are normalized to their initial value.
Figure 6: (a) VOC, (b) JSC, (c) FF, and (d) PCE decay of the COOP-nHT-TBDT:PC61BM solar cells. Note all these...
Conclusion
Four small A–π–D–π–A molecules with BDT core (COOP-nHT-TBDT, n = 1, 2, 3, 4) with regioregular oligo(3-hexylthiophene) π-bridges were synthesized and characterized. The length of the π-conjugation bridges has a significant impact on the optical, electrochemical properties and, in consequence, the device performance. With the elongation of the conjugated chain, broader absorption bands and narrower optical band gaps were observed for this type of compound, which would be beneficial for increasing JSC in solar cell applications. A high VOC of 0.9–1.0 V was achieved for the COOP-nHT-TBDT:PC61BM cells, owing to the low-lying HOMO levels of these compounds. The lengthening of the conjugated π-bridges improves the performance of the COOP-nHT-TBDT:PC61BM cells, and a maximum PCE of 5.62% with a VOC of 0.93 V, JSC of 9.60 mA·cm−2, and FF of 0.63 was achieved for the 4:PC61BM-based device. In addition, improved device stability was also found for the larger molecules, which could be ascribed to the higher stability of the nanomorphology.
Supporting Information
Supporting Information features experimental details about the synthesis of COOP-nHT-TBDT, the determination of molecular molar extinction coefficient of COOP-nHT-TBDT, the J–V curves and EQE spectra of organic solar cells based on COOP-nHT-TBDT at different blend ratios, a J–V comparison of devices based on 4:PC61BM and 4:PC71BM, UV–vis absorption spectra of COOP-nHT-TBDT:PC61BM blended films, as well as the NMR and MALDI–TOF MS spectra of COOP-nHT-TBDT.
| Supporting Information File 1: Additional experimental data. | ||
| Format: PDF | Size: 2.2 MB | Download |
Acknowledgements
The work is financially supported by the National Natural Science Foundation of China (21274163), Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA09020201), and the ''Hundred Talents Project'' (Initialization Support) of the Chinese Academy of Sciences.
References
-
Lu, L.; Zheng, T.; Wu, Q.; Schneider, A. M.; Zhao, D.; Yu, L. Chem. Rev. 2015, 115, 12666–12731. doi:10.1021/acs.chemrev.5b00098
Return to citation in text: [1] -
Nielsen, T. D.; Cruickshank, C.; Foged, S.; Thorsen, J.; Krebs, F. C. Sol. Energy Mater. Sol. Cells 2010, 94, 1553–1571. doi:10.1016/j.solmat.2010.04.074
Return to citation in text: [1] -
Yusoff, A. R. b. M.; Kim, D.; Kim, H. P.; Shneider, F. K.; da Silva, W. J.; Jang, J. Energy Environ. Sci. 2015, 8, 303–316. doi:10.1039/C4EE03048F
Return to citation in text: [1] -
Zhao, J.; Li, Y.; Yang, G.; Jiang, K.; Lin, H.; Ade, H.; Ma, W.; Yan, H. Nat. Energy 2016, 1, 15027. doi:10.1038/nenergy.2015.27
Return to citation in text: [1] -
Roncali, J.; Leriche, P.; Blanchard, P. Adv. Mater. 2014, 26, 3821–3838. doi:10.1002/adma.201305999
Return to citation in text: [1] -
Ni, W.; Wan, X.; Li, M.; Wang, Y.; Chen, Y. Chem. Commun. 2015, 51, 4936–4950. doi:10.1039/C4CC09758K
Return to citation in text: [1] -
Long, G.; Wan, X.; Chen, Y. Acc. Chem. Res. 2013, 46, 2645–2655. doi:10.1021/ar400088c
Return to citation in text: [1] -
Kan, B.; Zhang, Q.; Li, M.; Wan, X.; Ni, W.; Long, G.; Wang, Y.; Yang, X.; Feng, H.; Chen, Y. J. Am. Chem. Soc. 2014, 136, 15529–15532. doi:10.1021/ja509703k
Return to citation in text: [1] [2] [3] [4] -
Zhang, Q.; Kan, B.; Liu, F.; Long, G.; Wan, X.; Chen, X.; Zuo, Y.; Ni, W.; Zhang, H.; Li, M.; Hu, Z.; Huang, F.; Cao, Y.; Liang, Z.; Zhang, M.; Russell, T. P.; Chen, Y. Nat. Photonics 2015, 9, 35–41. doi:10.1038/nphoton.2014.269
Return to citation in text: [1] -
Kan, B.; Li, M.; Zhang, Q.; Liu, F.; Wan, X.; Wang, Y.; Ni, W.; Long, G.; Yang, X.; Feng, H.; Zuo, Y.; Zhang, M.; Huang, F.; Cao, Y.; Russell, T. P.; Chen, Y. J. Am. Chem. Soc. 2015, 137, 3886–3893. doi:10.1021/jacs.5b00305
Return to citation in text: [1] -
Sun, K.; Xiao, Z.; Lu, S.; Zajaczkowski, W.; Pisula, W.; Hanssen, E.; White, J. M.; Williamson, R. M.; Subbiah, J.; Ouyang, J.; Holmes, A. B.; Wong, W. W. H.; Jones, D. J. Nat. Commun. 2015, 6, 6013. doi:10.1038/ncomms7013
Return to citation in text: [1] -
Liu, Y.; Chen, C.-C.; Hong, Z.; Gao, J.; Yang, Y.; Zhou, H.; Dou, L.; Li, G.; Yang, Y. Sci. Rep. 2013, 3, 3356. doi:10.1038/srep03356
Return to citation in text: [1] -
Li, M.; Ni, W.; Wan, X.; Zhang, Q.; Kan, B.; Chen, Y. J. Mater. Chem. A 2015, 3, 4765–4776. doi:10.1039/C4TA06452F
Return to citation in text: [1] -
Cui, C.; Guo, X.; Min, J.; Guo, B.; Cheng, X.; Zhang, M.; Brabec, C. J.; Li, Y. Adv. Mater. 2015, 27, 7469–7475. doi:10.1002/adma.201503815
Return to citation in text: [1] [2] [3] -
Qiu, B.; Yuan, J.; Xiao, X.; He, D.; Qiu, L.; Zou, Y.; Zhang, Z.-g.; Li, Y. ACS Appl. Mater. Interfaces 2015, 7, 25237–25246. doi:10.1021/acsami.5b07066
Return to citation in text: [1] [2] -
Shen, S.; Jiang, P.; He, C.; Zhang, J.; Shen, P.; Zhang, Y.; Yi, Y.; Zhang, Z.; Li, Z.; Li, Y. Chem. Mater. 2013, 25, 2274–2281. doi:10.1021/cm400782q
Return to citation in text: [1] [2] [3] [4] -
Zhou, J.; Zuo, Y.; Wan, X.; Long, G.; Zhang, Q.; Ni, W.; Liu, Y.; Li, Z.; He, G.; Li, C.; Kan, B.; Li, M.; Chen, Y. J. Am. Chem. Soc. 2013, 135, 8484–8487. doi:10.1021/ja403318y
Return to citation in text: [1] [2] [3] [4] -
Du, Z.; Chen, W.; Qiu, M.; Chen, Y.; Wang, N.; Wang, T.; Sun, M.; Yu, D.; Yang, R. Phys. Chem. Chem. Phys. 2015, 17, 17391–17398. doi:10.1039/C5CP02632F
Return to citation in text: [1] [2] -
Kan, B.; Zhang, Q.; Liu, F.; Wan, X.; Wang, Y.; Ni, W.; Yang, X.; Zhang, M.; Zhang, H.; Russell, T. P.; Chen, Y. Chem. Mater. 2015, 27, 8414–8423. doi:10.1021/acs.chemmater.5b03889
Return to citation in text: [1] [2] [3] -
Yin, N.; Wang, L.; Ma, Y.; Lin, Y.; Wu, J.; Luo, Q.; Yang, H.-B.; Ma, C.-Q.; Zhao, X. Dyes Pigm. 2015, 120, 299–306. doi:10.1016/j.dyepig.2015.04.030
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Patra, D.; Huang, T.-Y.; Chiang, C.-C.; Maturana, R. O. V.; Pao, C.-W.; Ho, K.-C.; Wei, K.-H.; Chu, C.-W. ACS Appl. Mater. Interfaces 2013, 5, 9494–9500. doi:10.1021/am4021928
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Liu, Y.; Wan, X.; Wang, F.; Zhou, J.; Long, G.; Tian, J.; Chen, Y. Adv. Mater. 2011, 23, 5387–5391. doi:10.1002/adma.201102790
Return to citation in text: [1] [2] [3] [4] -
Zhou, J.; Wan, X.; Liu, Y.; Zuo, Y.; Li, Z.; He, G.; Long, G.; Ni, W.; Li, C.; Su, X.; Chen, Y. J. Am. Chem. Soc. 2012, 134, 16345–16351. doi:10.1021/ja306865z
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Lin, Y.; Ma, L.; Li, Y.; Liu, Y.; Zhu, D.; Zhan, X. Adv. Energy Mater. 2013, 3, 1166–1170. doi:10.1002/aenm.201300181
Return to citation in text: [1] -
Huang, J.; Zhan, C.; Zhang, X.; Zhao, Y.; Lu, Z.; Jia, H.; Jiang, B.; Ye, J.; Zhang, S.; Tang, A.; Liu, Y.; Pei, Q.; Yao, J. ACS Appl. Mater. Interfaces 2013, 5, 2033–2039. doi:10.1021/am302896u
Return to citation in text: [1] -
Lin, Y.; Ma, L.; Li, Y.; Liu, Y.; Zhu, D.; Zhan, X. Adv. Energy Mater. 2014, 4, 1300626. doi:10.1002/aenm.201300626
Return to citation in text: [1] -
Tang, A.; Zhan, C.; Yao, J. Chem. Mater. 2015, 27, 4719–4730. doi:10.1021/acs.chemmater.5b01350
Return to citation in text: [1] [2] -
Kumar, C. V.; Cabau, L.; Viterisi, A.; Biswas, S.; Sharma, G. D.; Palomares, E. J. Phys. Chem. C 2015, 119, 20871–20879. doi:10.1021/acs.jpcc.5b07130
Return to citation in text: [1] -
Patra, D.; Chiang, C.-C.; Chen, W.-A.; Wei, K.-H.; Wu, M.-C.; Chu, C.-W. J. Mater. Chem. A 2013, 1, 7767–7774. doi:10.1039/c3ta11544e
Return to citation in text: [1] -
Chu, H.-C.; Sahu, D.; Hsu, Y.-C.; Padhy, H.; Patra, D.; Lin, J.-T.; Bhattacharya, D.; Lu, K.-L.; Wei, K.-H.; Lin, H.-C. Dyes Pigm. 2012, 93, 1488–1497. doi:10.1016/j.dyepig.2011.09.012
Return to citation in text: [1] -
Li, Z.; He, G.; Wan, X.; Liu, Y.; Zhou, J.; Long, G.; Zuo, Y.; Zhang, M.; Chen, Y. Adv. Energy Mater. 2012, 2, 74–77. doi:10.1002/aenm.201100572
Return to citation in text: [1] -
Long, G.; Wan, X.; Kan, B.; Liu, Y.; He, G.; Li, Z.; Zhang, Y.; Zhang, Y.; Zhang, Q.; Zhang, M.; Chen, Y. Adv. Energy Mater. 2013, 3, 639–646. doi:10.1002/aenm.201300046
Return to citation in text: [1] -
Cardona, C. M.; Li, W.; Kaifer, A. E.; Stockdale, D.; Bazan, G. C. Adv. Mater. 2011, 23, 2367–2371. doi:10.1002/adma.201004554
Return to citation in text: [1] -
Wu, J.; Ma, Y.; Wu, N.; Lin, Y.; Lin, J.; Wang, L.; Ma, C.-Q. Org. Electron. 2015, 23, 28–38. doi:10.1016/j.orgel.2015.04.003
Return to citation in text: [1] [2] -
Lim, N.; Cho, N.; Paek, S.; Kim, C.; Lee, J. K.; Ko, J. Chem. Mater. 2014, 26, 2283–2288. doi:10.1021/cm5004092
Return to citation in text: [1] -
Deng, D.; Zhang, Y.; Yuan, L.; He, C.; Lu, K.; Wei, Z. Adv. Energy Mater. 2014, 4, 1400538. doi:10.1002/aenm.201400538
Return to citation in text: [1] -
Du, Z.; Chen, W.; Chen, Y.; Qiao, S.; Bao, X.; Wen, S.; Sun, M.; Han, L.; Yang, R. J. Mater. Chem. A 2014, 2, 15904–15911. doi:10.1039/C4TA03314K
Return to citation in text: [1] -
Wu, N.; Luo, Q.; Bao, Z.; Lin, J.; Li, Y.-Q.; Ma, C.-Q. Sol. Energy Mater. Sol. Cells 2015, 141, 248–259. doi:10.1016/j.solmat.2015.05.039
Return to citation in text: [1]
| 20. | Yin, N.; Wang, L.; Ma, Y.; Lin, Y.; Wu, J.; Luo, Q.; Yang, H.-B.; Ma, C.-Q.; Zhao, X. Dyes Pigm. 2015, 120, 299–306. doi:10.1016/j.dyepig.2015.04.030 |
| 33. | Cardona, C. M.; Li, W.; Kaifer, A. E.; Stockdale, D.; Bazan, G. C. Adv. Mater. 2011, 23, 2367–2371. doi:10.1002/adma.201004554 |
| 34. | Wu, J.; Ma, Y.; Wu, N.; Lin, Y.; Lin, J.; Wang, L.; Ma, C.-Q. Org. Electron. 2015, 23, 28–38. doi:10.1016/j.orgel.2015.04.003 |
| 1. | Lu, L.; Zheng, T.; Wu, Q.; Schneider, A. M.; Zhao, D.; Yu, L. Chem. Rev. 2015, 115, 12666–12731. doi:10.1021/acs.chemrev.5b00098 |
| 2. | Nielsen, T. D.; Cruickshank, C.; Foged, S.; Thorsen, J.; Krebs, F. C. Sol. Energy Mater. Sol. Cells 2010, 94, 1553–1571. doi:10.1016/j.solmat.2010.04.074 |
| 13. | Li, M.; Ni, W.; Wan, X.; Zhang, Q.; Kan, B.; Chen, Y. J. Mater. Chem. A 2015, 3, 4765–4776. doi:10.1039/C4TA06452F |
| 14. | Cui, C.; Guo, X.; Min, J.; Guo, B.; Cheng, X.; Zhang, M.; Brabec, C. J.; Li, Y. Adv. Mater. 2015, 27, 7469–7475. doi:10.1002/adma.201503815 |
| 16. | Shen, S.; Jiang, P.; He, C.; Zhang, J.; Shen, P.; Zhang, Y.; Yi, Y.; Zhang, Z.; Li, Z.; Li, Y. Chem. Mater. 2013, 25, 2274–2281. doi:10.1021/cm400782q |
| 20. | Yin, N.; Wang, L.; Ma, Y.; Lin, Y.; Wu, J.; Luo, Q.; Yang, H.-B.; Ma, C.-Q.; Zhao, X. Dyes Pigm. 2015, 120, 299–306. doi:10.1016/j.dyepig.2015.04.030 |
| 8. | Kan, B.; Zhang, Q.; Li, M.; Wan, X.; Ni, W.; Long, G.; Wang, Y.; Yang, X.; Feng, H.; Chen, Y. J. Am. Chem. Soc. 2014, 136, 15529–15532. doi:10.1021/ja509703k |
| 9. | Zhang, Q.; Kan, B.; Liu, F.; Long, G.; Wan, X.; Chen, X.; Zuo, Y.; Ni, W.; Zhang, H.; Li, M.; Hu, Z.; Huang, F.; Cao, Y.; Liang, Z.; Zhang, M.; Russell, T. P.; Chen, Y. Nat. Photonics 2015, 9, 35–41. doi:10.1038/nphoton.2014.269 |
| 10. | Kan, B.; Li, M.; Zhang, Q.; Liu, F.; Wan, X.; Wang, Y.; Ni, W.; Long, G.; Yang, X.; Feng, H.; Zuo, Y.; Zhang, M.; Huang, F.; Cao, Y.; Russell, T. P.; Chen, Y. J. Am. Chem. Soc. 2015, 137, 3886–3893. doi:10.1021/jacs.5b00305 |
| 11. | Sun, K.; Xiao, Z.; Lu, S.; Zajaczkowski, W.; Pisula, W.; Hanssen, E.; White, J. M.; Williamson, R. M.; Subbiah, J.; Ouyang, J.; Holmes, A. B.; Wong, W. W. H.; Jones, D. J. Nat. Commun. 2015, 6, 6013. doi:10.1038/ncomms7013 |
| 12. | Liu, Y.; Chen, C.-C.; Hong, Z.; Gao, J.; Yang, Y.; Zhou, H.; Dou, L.; Li, G.; Yang, Y. Sci. Rep. 2013, 3, 3356. doi:10.1038/srep03356 |
| 16. | Shen, S.; Jiang, P.; He, C.; Zhang, J.; Shen, P.; Zhang, Y.; Yi, Y.; Zhang, Z.; Li, Z.; Li, Y. Chem. Mater. 2013, 25, 2274–2281. doi:10.1021/cm400782q |
| 5. | Roncali, J.; Leriche, P.; Blanchard, P. Adv. Mater. 2014, 26, 3821–3838. doi:10.1002/adma.201305999 |
| 6. | Ni, W.; Wan, X.; Li, M.; Wang, Y.; Chen, Y. Chem. Commun. 2015, 51, 4936–4950. doi:10.1039/C4CC09758K |
| 7. | Long, G.; Wan, X.; Chen, Y. Acc. Chem. Res. 2013, 46, 2645–2655. doi:10.1021/ar400088c |
| 16. | Shen, S.; Jiang, P.; He, C.; Zhang, J.; Shen, P.; Zhang, Y.; Yi, Y.; Zhang, Z.; Li, Z.; Li, Y. Chem. Mater. 2013, 25, 2274–2281. doi:10.1021/cm400782q |
| 38. | Wu, N.; Luo, Q.; Bao, Z.; Lin, J.; Li, Y.-Q.; Ma, C.-Q. Sol. Energy Mater. Sol. Cells 2015, 141, 248–259. doi:10.1016/j.solmat.2015.05.039 |
| 3. | Yusoff, A. R. b. M.; Kim, D.; Kim, H. P.; Shneider, F. K.; da Silva, W. J.; Jang, J. Energy Environ. Sci. 2015, 8, 303–316. doi:10.1039/C4EE03048F |
| 4. | Zhao, J.; Li, Y.; Yang, G.; Jiang, K.; Lin, H.; Ade, H.; Ma, W.; Yan, H. Nat. Energy 2016, 1, 15027. doi:10.1038/nenergy.2015.27 |
| 24. | Lin, Y.; Ma, L.; Li, Y.; Liu, Y.; Zhu, D.; Zhan, X. Adv. Energy Mater. 2013, 3, 1166–1170. doi:10.1002/aenm.201300181 |
| 25. | Huang, J.; Zhan, C.; Zhang, X.; Zhao, Y.; Lu, Z.; Jia, H.; Jiang, B.; Ye, J.; Zhang, S.; Tang, A.; Liu, Y.; Pei, Q.; Yao, J. ACS Appl. Mater. Interfaces 2013, 5, 2033–2039. doi:10.1021/am302896u |
| 17. | Zhou, J.; Zuo, Y.; Wan, X.; Long, G.; Zhang, Q.; Ni, W.; Liu, Y.; Li, Z.; He, G.; Li, C.; Kan, B.; Li, M.; Chen, Y. J. Am. Chem. Soc. 2013, 135, 8484–8487. doi:10.1021/ja403318y |
| 19. | Kan, B.; Zhang, Q.; Liu, F.; Wan, X.; Wang, Y.; Ni, W.; Yang, X.; Zhang, M.; Zhang, H.; Russell, T. P.; Chen, Y. Chem. Mater. 2015, 27, 8414–8423. doi:10.1021/acs.chemmater.5b03889 |
| 20. | Yin, N.; Wang, L.; Ma, Y.; Lin, Y.; Wu, J.; Luo, Q.; Yang, H.-B.; Ma, C.-Q.; Zhao, X. Dyes Pigm. 2015, 120, 299–306. doi:10.1016/j.dyepig.2015.04.030 |
| 21. | Patra, D.; Huang, T.-Y.; Chiang, C.-C.; Maturana, R. O. V.; Pao, C.-W.; Ho, K.-C.; Wei, K.-H.; Chu, C.-W. ACS Appl. Mater. Interfaces 2013, 5, 9494–9500. doi:10.1021/am4021928 |
| 22. | Liu, Y.; Wan, X.; Wang, F.; Zhou, J.; Long, G.; Tian, J.; Chen, Y. Adv. Mater. 2011, 23, 5387–5391. doi:10.1002/adma.201102790 |
| 23. | Zhou, J.; Wan, X.; Liu, Y.; Zuo, Y.; Li, Z.; He, G.; Long, G.; Ni, W.; Li, C.; Su, X.; Chen, Y. J. Am. Chem. Soc. 2012, 134, 16345–16351. doi:10.1021/ja306865z |
| 20. | Yin, N.; Wang, L.; Ma, Y.; Lin, Y.; Wu, J.; Luo, Q.; Yang, H.-B.; Ma, C.-Q.; Zhao, X. Dyes Pigm. 2015, 120, 299–306. doi:10.1016/j.dyepig.2015.04.030 |
| 16. | Shen, S.; Jiang, P.; He, C.; Zhang, J.; Shen, P.; Zhang, Y.; Yi, Y.; Zhang, Z.; Li, Z.; Li, Y. Chem. Mater. 2013, 25, 2274–2281. doi:10.1021/cm400782q |
| 17. | Zhou, J.; Zuo, Y.; Wan, X.; Long, G.; Zhang, Q.; Ni, W.; Liu, Y.; Li, Z.; He, G.; Li, C.; Kan, B.; Li, M.; Chen, Y. J. Am. Chem. Soc. 2013, 135, 8484–8487. doi:10.1021/ja403318y |
| 18. | Du, Z.; Chen, W.; Qiu, M.; Chen, Y.; Wang, N.; Wang, T.; Sun, M.; Yu, D.; Yang, R. Phys. Chem. Chem. Phys. 2015, 17, 17391–17398. doi:10.1039/C5CP02632F |
| 8. | Kan, B.; Zhang, Q.; Li, M.; Wan, X.; Ni, W.; Long, G.; Wang, Y.; Yang, X.; Feng, H.; Chen, Y. J. Am. Chem. Soc. 2014, 136, 15529–15532. doi:10.1021/ja509703k |
| 14. | Cui, C.; Guo, X.; Min, J.; Guo, B.; Cheng, X.; Zhang, M.; Brabec, C. J.; Li, Y. Adv. Mater. 2015, 27, 7469–7475. doi:10.1002/adma.201503815 |
| 17. | Zhou, J.; Zuo, Y.; Wan, X.; Long, G.; Zhang, Q.; Ni, W.; Liu, Y.; Li, Z.; He, G.; Li, C.; Kan, B.; Li, M.; Chen, Y. J. Am. Chem. Soc. 2013, 135, 8484–8487. doi:10.1021/ja403318y |
| 19. | Kan, B.; Zhang, Q.; Liu, F.; Wan, X.; Wang, Y.; Ni, W.; Yang, X.; Zhang, M.; Zhang, H.; Russell, T. P.; Chen, Y. Chem. Mater. 2015, 27, 8414–8423. doi:10.1021/acs.chemmater.5b03889 |
| 23. | Zhou, J.; Wan, X.; Liu, Y.; Zuo, Y.; Li, Z.; He, G.; Long, G.; Ni, W.; Li, C.; Su, X.; Chen, Y. J. Am. Chem. Soc. 2012, 134, 16345–16351. doi:10.1021/ja306865z |
| 34. | Wu, J.; Ma, Y.; Wu, N.; Lin, Y.; Lin, J.; Wang, L.; Ma, C.-Q. Org. Electron. 2015, 23, 28–38. doi:10.1016/j.orgel.2015.04.003 |
| 15. | Qiu, B.; Yuan, J.; Xiao, X.; He, D.; Qiu, L.; Zou, Y.; Zhang, Z.-g.; Li, Y. ACS Appl. Mater. Interfaces 2015, 7, 25237–25246. doi:10.1021/acsami.5b07066 |
| 20. | Yin, N.; Wang, L.; Ma, Y.; Lin, Y.; Wu, J.; Luo, Q.; Yang, H.-B.; Ma, C.-Q.; Zhao, X. Dyes Pigm. 2015, 120, 299–306. doi:10.1016/j.dyepig.2015.04.030 |
| 8. | Kan, B.; Zhang, Q.; Li, M.; Wan, X.; Ni, W.; Long, G.; Wang, Y.; Yang, X.; Feng, H.; Chen, Y. J. Am. Chem. Soc. 2014, 136, 15529–15532. doi:10.1021/ja509703k |
| 20. | Yin, N.; Wang, L.; Ma, Y.; Lin, Y.; Wu, J.; Luo, Q.; Yang, H.-B.; Ma, C.-Q.; Zhao, X. Dyes Pigm. 2015, 120, 299–306. doi:10.1016/j.dyepig.2015.04.030 |
| 21. | Patra, D.; Huang, T.-Y.; Chiang, C.-C.; Maturana, R. O. V.; Pao, C.-W.; Ho, K.-C.; Wei, K.-H.; Chu, C.-W. ACS Appl. Mater. Interfaces 2013, 5, 9494–9500. doi:10.1021/am4021928 |
| 21. | Patra, D.; Huang, T.-Y.; Chiang, C.-C.; Maturana, R. O. V.; Pao, C.-W.; Ho, K.-C.; Wei, K.-H.; Chu, C.-W. ACS Appl. Mater. Interfaces 2013, 5, 9494–9500. doi:10.1021/am4021928 |
| 22. | Liu, Y.; Wan, X.; Wang, F.; Zhou, J.; Long, G.; Tian, J.; Chen, Y. Adv. Mater. 2011, 23, 5387–5391. doi:10.1002/adma.201102790 |
| 23. | Zhou, J.; Wan, X.; Liu, Y.; Zuo, Y.; Li, Z.; He, G.; Long, G.; Ni, W.; Li, C.; Su, X.; Chen, Y. J. Am. Chem. Soc. 2012, 134, 16345–16351. doi:10.1021/ja306865z |
| 35. | Lim, N.; Cho, N.; Paek, S.; Kim, C.; Lee, J. K.; Ko, J. Chem. Mater. 2014, 26, 2283–2288. doi:10.1021/cm5004092 |
| 36. | Deng, D.; Zhang, Y.; Yuan, L.; He, C.; Lu, K.; Wei, Z. Adv. Energy Mater. 2014, 4, 1400538. doi:10.1002/aenm.201400538 |
| 37. | Du, Z.; Chen, W.; Chen, Y.; Qiao, S.; Bao, X.; Wen, S.; Sun, M.; Han, L.; Yang, R. J. Mater. Chem. A 2014, 2, 15904–15911. doi:10.1039/C4TA03314K |
| 28. | Kumar, C. V.; Cabau, L.; Viterisi, A.; Biswas, S.; Sharma, G. D.; Palomares, E. J. Phys. Chem. C 2015, 119, 20871–20879. doi:10.1021/acs.jpcc.5b07130 |
| 8. | Kan, B.; Zhang, Q.; Li, M.; Wan, X.; Ni, W.; Long, G.; Wang, Y.; Yang, X.; Feng, H.; Chen, Y. J. Am. Chem. Soc. 2014, 136, 15529–15532. doi:10.1021/ja509703k |
| 14. | Cui, C.; Guo, X.; Min, J.; Guo, B.; Cheng, X.; Zhang, M.; Brabec, C. J.; Li, Y. Adv. Mater. 2015, 27, 7469–7475. doi:10.1002/adma.201503815 |
| 15. | Qiu, B.; Yuan, J.; Xiao, X.; He, D.; Qiu, L.; Zou, Y.; Zhang, Z.-g.; Li, Y. ACS Appl. Mater. Interfaces 2015, 7, 25237–25246. doi:10.1021/acsami.5b07066 |
| 17. | Zhou, J.; Zuo, Y.; Wan, X.; Long, G.; Zhang, Q.; Ni, W.; Liu, Y.; Li, Z.; He, G.; Li, C.; Kan, B.; Li, M.; Chen, Y. J. Am. Chem. Soc. 2013, 135, 8484–8487. doi:10.1021/ja403318y |
| 18. | Du, Z.; Chen, W.; Qiu, M.; Chen, Y.; Wang, N.; Wang, T.; Sun, M.; Yu, D.; Yang, R. Phys. Chem. Chem. Phys. 2015, 17, 17391–17398. doi:10.1039/C5CP02632F |
| 19. | Kan, B.; Zhang, Q.; Liu, F.; Wan, X.; Wang, Y.; Ni, W.; Yang, X.; Zhang, M.; Zhang, H.; Russell, T. P.; Chen, Y. Chem. Mater. 2015, 27, 8414–8423. doi:10.1021/acs.chemmater.5b03889 |
| 21. | Patra, D.; Huang, T.-Y.; Chiang, C.-C.; Maturana, R. O. V.; Pao, C.-W.; Ho, K.-C.; Wei, K.-H.; Chu, C.-W. ACS Appl. Mater. Interfaces 2013, 5, 9494–9500. doi:10.1021/am4021928 |
| 22. | Liu, Y.; Wan, X.; Wang, F.; Zhou, J.; Long, G.; Tian, J.; Chen, Y. Adv. Mater. 2011, 23, 5387–5391. doi:10.1002/adma.201102790 |
| 23. | Zhou, J.; Wan, X.; Liu, Y.; Zuo, Y.; Li, Z.; He, G.; Long, G.; Ni, W.; Li, C.; Su, X.; Chen, Y. J. Am. Chem. Soc. 2012, 134, 16345–16351. doi:10.1021/ja306865z |
| 26. | Lin, Y.; Ma, L.; Li, Y.; Liu, Y.; Zhu, D.; Zhan, X. Adv. Energy Mater. 2014, 4, 1300626. doi:10.1002/aenm.201300626 |
| 27. | Tang, A.; Zhan, C.; Yao, J. Chem. Mater. 2015, 27, 4719–4730. doi:10.1021/acs.chemmater.5b01350 |
| 31. | Li, Z.; He, G.; Wan, X.; Liu, Y.; Zhou, J.; Long, G.; Zuo, Y.; Zhang, M.; Chen, Y. Adv. Energy Mater. 2012, 2, 74–77. doi:10.1002/aenm.201100572 |
| 32. | Long, G.; Wan, X.; Kan, B.; Liu, Y.; He, G.; Li, Z.; Zhang, Y.; Zhang, Y.; Zhang, Q.; Zhang, M.; Chen, Y. Adv. Energy Mater. 2013, 3, 639–646. doi:10.1002/aenm.201300046 |
| 21. | Patra, D.; Huang, T.-Y.; Chiang, C.-C.; Maturana, R. O. V.; Pao, C.-W.; Ho, K.-C.; Wei, K.-H.; Chu, C.-W. ACS Appl. Mater. Interfaces 2013, 5, 9494–9500. doi:10.1021/am4021928 |
| 23. | Zhou, J.; Wan, X.; Liu, Y.; Zuo, Y.; Li, Z.; He, G.; Long, G.; Ni, W.; Li, C.; Su, X.; Chen, Y. J. Am. Chem. Soc. 2012, 134, 16345–16351. doi:10.1021/ja306865z |
| 23. | Zhou, J.; Wan, X.; Liu, Y.; Zuo, Y.; Li, Z.; He, G.; Long, G.; Ni, W.; Li, C.; Su, X.; Chen, Y. J. Am. Chem. Soc. 2012, 134, 16345–16351. doi:10.1021/ja306865z |
| 21. | Patra, D.; Huang, T.-Y.; Chiang, C.-C.; Maturana, R. O. V.; Pao, C.-W.; Ho, K.-C.; Wei, K.-H.; Chu, C.-W. ACS Appl. Mater. Interfaces 2013, 5, 9494–9500. doi:10.1021/am4021928 |
| 29. | Patra, D.; Chiang, C.-C.; Chen, W.-A.; Wei, K.-H.; Wu, M.-C.; Chu, C.-W. J. Mater. Chem. A 2013, 1, 7767–7774. doi:10.1039/c3ta11544e |
| 30. | Chu, H.-C.; Sahu, D.; Hsu, Y.-C.; Padhy, H.; Patra, D.; Lin, J.-T.; Bhattacharya, D.; Lu, K.-L.; Wei, K.-H.; Lin, H.-C. Dyes Pigm. 2012, 93, 1488–1497. doi:10.1016/j.dyepig.2011.09.012 |
| 20. | Yin, N.; Wang, L.; Ma, Y.; Lin, Y.; Wu, J.; Luo, Q.; Yang, H.-B.; Ma, C.-Q.; Zhao, X. Dyes Pigm. 2015, 120, 299–306. doi:10.1016/j.dyepig.2015.04.030 |
| 27. | Tang, A.; Zhan, C.; Yao, J. Chem. Mater. 2015, 27, 4719–4730. doi:10.1021/acs.chemmater.5b01350 |
| 21. | Patra, D.; Huang, T.-Y.; Chiang, C.-C.; Maturana, R. O. V.; Pao, C.-W.; Ho, K.-C.; Wei, K.-H.; Chu, C.-W. ACS Appl. Mater. Interfaces 2013, 5, 9494–9500. doi:10.1021/am4021928 |
| 22. | Liu, Y.; Wan, X.; Wang, F.; Zhou, J.; Long, G.; Tian, J.; Chen, Y. Adv. Mater. 2011, 23, 5387–5391. doi:10.1002/adma.201102790 |
© 2016 Yin et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)