Abstract
Two hydroxy-functionalized hyper-cross-linked ultra-microporous compounds have been synthesized by Friedel–Crafts alkylation reaction and characterised with different spectroscopic techniques. Both compounds exhibit an efficient carbon dioxide uptake over other gases like N2, H2 and O2 at room temperature. A high isosteric heat of adsorption (Qst) has been obtained for both materials because of strong interactions between polar –OH groups and CO2 molecules.
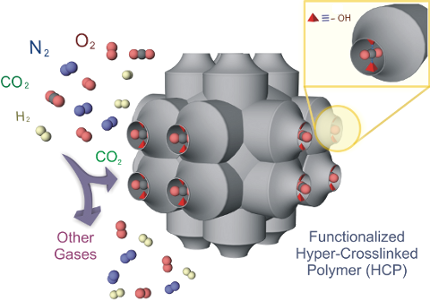
Graphical Abstract
Introduction
The increase in the earth’s average temperature, also termed as global warming, is mainly due to the effects of greenhouse gases. The impacts of global warming includes rising sea level, more likelihood of extreme events (like floods, hurricanes etc.), widespread vanishing of animal population, loss of plankton due to warming seas. There are many heat-trapping greenhouse gases present in the atmosphere (from methane to water vapour), but CO2 puts us at the greatest risk if it continues to accumulate in the atmosphere. This is due to the fact that CO2 remains in the atmosphere in a time scale of hundred years in contrast to other greenhouse gases which leave the atmosphere with relatively smaller time scale [1]. The CO2 long life in the atmosphere provides the clearest possible rationale for carbon dioxide capture and storage. Previously, different types of amine solvents were employed to study the CO2 capture, but the need of high energy to regenerate the amine solutions after CO2 capture, hinders their applications further [2]. In the domain of porous materials, zeolites, metal-organic frameworks (MOFs), cage molecules, etc. have been introduced for selective uptake of CO2 [3-5]. In terms of surface area, tuneable porosity and feasible host–guest interaction, MOFs have scored over other above mentioned porous materials [6]. But the less hydrolytic stability of metal-organic frameworks limits their real time application [7,8]. So the search for new materials having high surface area and feasible interaction with carbon dioxide like MOFs and with high chemical stability have become one of top priority for researchers.
Microporous organic polymers (MOP) are a relatively new class of porous materials, constructed from light elements like H, C, B, N, O etc. having a large surface area, small pore size and low skeletal density [9-12]. This type of materials has already been used for various purposes of applications such as gas storage, gas separation, catalysis, sensing, clean energy, etc. [13-18]. Relatively weaker coordination bonds in MOFs have been replaced with stronger covalent bonds in this type of porous compounds. This results in a high chemical stability of the microporous organic polymers, which is an essential condition for the real-time application of any compound. The last decade has witnessed advancements in synthesizing various types of microporous organic materials including covalent organic frameworks (COFs), conjugate microporous polymers (CMPs), porous polymeric networks (PPNs), porous aromatic frameworks (PAFs), covalent triazine framework (CTFs), etc. [19-24]. Hyper-cross-linked microporous organic polymers (HCPs) are a subclass of this type of porous materials. Recently, hyper-cross-linked MOPs are emerged as a new subclass, synthesized by hyper-cross linking of basic small organic building blocks by Friedel–Crafts reaction in the presence of the Lewis acid FeCl3 (as catalyst) and formaldehyde dimethyl acetal (FDA) as the cross linker [25-27]. Here, aromatic small organic compounds are used to polymerise via C–C cross coupling to produce the targeted porous and physicochemical stable organic hyper-cross-linked polymeric materials. One huge advantage of this material is the low-cost synthesis, the cost-effective formaldehyde dimethyl acetal (FDA), FeCl3 and that organic small molecules can produce very low cost materials with high yield [28]. Hyper-cross-linking prevents the close packing of polymeric chains in this type of material to impart the intrinsic porosity. Hyper-cross-linked polymers have been applied in the field of gas storage, catalysis, separation and recently also in CO2 capture [29-32]. The increasing environmental pollution due to carbon dioxide, urges us to develop new materials with high stability, which are cost-effective and demonstrate a high efficiency in CO2 capture. Based on the interaction of Lewis basic sites with carbon dioxide it has been observed that porous materials functionalised with –NH2 groups or –OH groups exhibit a selective uptake of CO2 in contrast to other gases [33,34] (Scheme 1). Inspired by this we have designed and synthesized two hydroxy-functionalised hyper-cross-linked microporous organic polymers for selective CO2 capture at room temperature. Both compounds (HCP-91 and HCP-94) were synthesized via hyper-cross-linked C–C coupling of hydroxyl-functionalised aromatic rings by using a Friedel–Craftys reaction. At different temperatures (273 K and 298 K) gas (CO2, N2, H2 and O2) adsorption experiments were carried out for both compounds. HCP-91 and HCP-94 showed selective CO2 capture at both temperatures over other flue gases.
![[1860-5397-12-185-i1]](/bjoc/content/inline/1860-5397-12-185-i1.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Schematic representation of selective CO2 capture in a porous material.
Scheme 1: Schematic representation of selective CO2 capture in a porous material.
Results and Discussion
For the synthesis of HCP-91 and HCP-94, we used 4-phenylphenol and 9-(hydroxymethyl)anthracene, respectively (Figure 1). HCP-91 and HCP-94 have been synthesized by using a Friedel–Crafts alkylation reaction. The thus obtained as-synthesized compounds were washed repeatedly with dimethylformamide (DMF), methanol, water, chloroform, dichloromethane and tetrahydrofuran (THF) to obtain phase-pure hyper-cross-linked polymers. Both compounds were immersed in a CHCl3–THF (1:1) mixture and kept for 4–5 days to exchange the high boiling solvents occluded inside the framework with low boiling CHCl3 and THF. The solvent-exchanged phases of HCP-91 and HCP-94 were then heated at ≈100 °C under vacuum to get the guest-free desolvated phases of the respective compounds. Infrared (IR) spectroscopy was done first to characterize the constituents of both compounds. A broad peak at ≈3000–3500 cm−1 and two sharp peaks at ≈1465 and ≈1527 cm−1 can be observed in HCP-91 corresponding to the stretching frequencies of –OH groups and aromatic C=C double bonds, respectively (Figure 2a). Similar to the HCP-91, peaks corresponding to –OH groups and aromatic C=C double bonds were found at ≈3300–3500 cm−1 and ≈1643 and 1500 cm−1, respectively (Figure 2a). Meanwhile a thermogravimetric analysis (TGA) was performed with both as-synthesized and desolvated phases for HCP-91 and HCP-94. Because of the occluded solvents in the as-synthesized phases of HCP-91 and HCP-94, an initial weight loss of ≈8% and ≈10% was observed in the TGA, respectively (Figures S1 and S2 in Supporting Information File 1). Upon desolvation guest-free phases were obtained and in the TG curve a negligible loss was obtained up to ≈350 °C and ≈250 °C for HCP-91 and HCP-94, respectively (Figures S1 and S2). As confirmation of the local structures of the compounds, we performed solid state 13C NMR measurements (Figures S3 and S4). To investigate the morphology of both compounds we performed a field emission scanning electron microscope (FESEM) study. The morphology of HCP-91 can be described as agglomerated particles consisting of small particles without any distinct shape (Figure 2c and Figure S5). But in case of HCP-94, a clear capsule-type of morphology was found in the FESEM (Figure 2d and Figure S6).
![[1860-5397-12-185-1]](/bjoc/content/figures/1860-5397-12-185-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: a) General synthesis scheme for hyper-cross-linked polymers (HCPs) and b) synthesis schemes for HCP-91 and HCP-94.
Figure 1: a) General synthesis scheme for hyper-cross-linked polymers (HCPs) and b) synthesis schemes for HCP...
![[1860-5397-12-185-2]](/bjoc/content/figures/1860-5397-12-185-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: a) Infra-red spectra of HCP-91 (dark yellow) and HCP-94 (purple); b) N2 adsorption isotherms for HCP-91 (wine red) and HCP-94 (green) at 77 K; c) SEM image of HCP-91 and d) SEM image of HCP-94.
Figure 2: a) Infra-red spectra of HCP-91 (dark yellow) and HCP-94 (purple); b) N2 adsorption isotherms for HC...
After all characterizations and proper desolvation of both compounds, we investigated their porosity. First, we measured the N2 adsorption at 77 K. The N2 uptake for HCP-91 was found to be 595 mL/g, whereas that for HCP-94 was 342 mL/g (Figure 2b). Both low temperature N2 adsorption isotherms were of type-I category and a hysteresis was observed in desorption profiles. The hysteresis in the desorption curves can be explained in terms of a network swelling in the presence of condensed nitrogen [34]. The Howarth–Kawazoe pore-size distributions were calculated from low-temperature N2 adsorption data. HCP-91 and HCP-94 exhibit pore sizes of 0.59 and 0.46 nm, respectively (Supporting Information File 1, Figures S7 and S8). According to recent literature, both compounds belong to the ultra-microporous material domain as pore sizes for the above mentioned compounds are lesser than 0.7 nm [35]. Carbon dioxide uptakes of 365 mL/g and 224 mL/g for HCP-91 and HCP-94, respectively, were observed when the CO2 adsorption was carried out at 195 K (Figure 3a). The hysteresis in the CO2 desorption profile in case of both compounds accounts for the interaction between hydroxy groups and CO2 molecules [33,34]. Since both compounds are ultra-microporous in nature, BET (Brunauer–Emmett–Teller) surface areas were calculated from the CO2 adsorption profile at 195 K (Supporting Information File 1, Table S1).
![[1860-5397-12-185-3]](/bjoc/content/figures/1860-5397-12-185-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: a) CO2 adsorption isotherms for HCP-91 (purple) and HCP-94 (green) at 195 K; b) adsorption isotherms of CO2 (wine red), N2 (dark yellow), H2 (green) and O2 (blue) for HCP-91 at 273 K; c) adsorption isotherms of CO2 (purple), N2 (orange), H2 (green) and O2 (blue) for HCP-91 at 298 K; d) adsorption isotherms of CO2 (green), N2 (blue), H2 (dark yellow) and O2 (orange) for HCP-94 at 273 K; e) adsorption isotherms of CO2 (wine red), N2 (dark yellow), H2 (green) and O2 (blue) for HCP-94 at 298 K and f) Qst plots for HCP-91 (dark yellow) and HCP-94 (purple).
Figure 3: a) CO2 adsorption isotherms for HCP-91 (purple) and HCP-94 (green) at 195 K; b) adsorption isotherm...
The effective CO2 uptake at 195 K encouraged us to perform a CO2 adsorption study at room temperature. HCP-91 and HCP-94 both exhibit an adequate amount of carbon dioxide uptake at 273 K and 298 K (Figures S9 and S10, Supporting Information File 1). At 273 K the CO2 uptake was 74 mL/g for HCP-91 and 65 mL/g for HCP-94 at 1 bar (Figure 3b,d). In the case of CO2 adsorption at 298 K a similar uptake has been observed for both compounds: 43 mL/g (HCP-91) and 45 mL/g (HCP-94) at 1 bar (Figure 3c,e). The uptake amounts of CO2 at room temperature and 1 bar are comparable with other well performing microporous polymer compounds. Meanwhile adsorption of other gases like nitrogen, hydrogen and oxygen (constituents of air) were performed at 273 K and 298 K and 1 bar. Interestingly very negligible amounts of uptake were obtained for each of them (Figure 3b–e). This type of CO2 separation over other flue gases at room temperature can be attributed to the high interaction of carbon dioxide with the framework. Both hyper-cross-linked polymers have hydroxy groups which are polar in nature. On the other hand CO2 molecules have a quadrupole moment, which renders a positive charge density over the carbon atom. So the polar hydroxy groups can offer a strong dipole-quadrupole moment interaction with carbon dioxide molecules. For a better understanding of the interaction between CO2 and our HCPs materials, we calculated the isosteric heat of adsorption (Qst) for CO2. Heat of adsorptions for both compounds has been calculated from CO2 adsorption data at 273 K and 298 K by using the Clausius–Clapeyron equation (Figure 3f) [33]. The Qst values for HCP-91 and HCP-94 were found to be 30.7 kJ mol−1 and 32 kJ mol−1, respectively. According to the previous reports, this high isosteric heat of adsorption values for both the materials indicates the strong interaction of it with CO2 molecules.
Conclusion
In this report, we have synthesized two hyper-cross-linked ultra-microporous organic polymers (HCP-91 and HCP-94) by following a cost-effective and easy synthesis route. One step Friedel–Crafts syntheses were carried out by using hydroxy-functionalized organic building blocks. Both compounds were characterised thoroughly by IR spectroscopy, TG analysis, solid state 13C NMR technique, FESEM and adsorption measurements. An efficient selective carbon dioxide capture was obtained for both compounds over other flue gases. High Qst values for both compounds ascribed the strong dipole–quadrupole interaction between polar –OH groups and CO2 molecules. We believe that this result will stimulate further design and fabrication of such low cost materials to be used as carbon dioxide capture materials.
Supporting Information
The Supporting Information contains the experimental section, thermo-gravimetric analysis curves, solid state 13C NMR, FESEM images, pore size distribution plots and room temperature CO2 adsorption plots.
| Supporting Information File 1: Experimental and analytical data. | ||
| Format: PDF | Size: 1.7 MB | Download |
References
-
Forster, P.; Ramaswamy, V.; Artaxo, P.; Berntsen, T.; Betts, R.; Fahey, D. W.; Haywood, J.; Lean, J.; Lowe, D. C.; Myhre, G.; Nganga, J.; Prinn, R.; Raga, G.; Schulz, M.; Van Dorland, R. Climate Change 2007: The Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change; 2007.
Return to citation in text: [1] -
Rochelle, G. T. Science 2009, 325, 1652–1654. doi:10.1126/science.1176731
Return to citation in text: [1] -
Bae, Y.-S.; Snurr, R. Q. Angew. Chem., Int. Ed. 2011, 50, 11586–11596. doi:10.1002/anie.201101891
Return to citation in text: [1] -
Slater, A. G.; Cooper, A. I. Science 2015, 348. doi:10.1126/science.aaa8075
Return to citation in text: [1] -
Li, J.-R.; Kuppler, R. J.; Zhou, H.-C. Chem. Soc. Rev. 2009, 38, 1477–1504. doi:10.1039/b802426j
Return to citation in text: [1] -
Mason, J. A.; Sumida, K.; Herm, Z. R.; Krishna, R.; Long, J. R. Energy Environ. Sci. 2011, 4, 3030–3040. doi:10.1039/c1ee01720a
Return to citation in text: [1] -
Lu, W.; Yuan, D.; Sculley, J.; Zhao, D.; Krishna, R.; Zhou, H.-C. J. Am. Chem. Soc. 2011, 133, 18126–18129. doi:10.1021/ja2087773
Return to citation in text: [1] -
Keskin, S.; van Heest, T. M.; Sholl, D. S. ChemSusChem 2010, 3, 879–891. doi:10.1002/cssc.201000114
Return to citation in text: [1] -
Jiang, J.; Zhao, Y.; Yaghi, O. M. J. Am. Chem. Soc. 2016, 138, 3255–3265. doi:10.1021/jacs.5b10666
Return to citation in text: [1] -
Thomas, A. Angew. Chem., Int. Ed. 2010, 49, 8328–8344. doi:10.1002/anie.201000167
Return to citation in text: [1] -
McKeown, N. B.; Budd, P. M. Macromolecules 2010, 43, 5163–5176. doi:10.1021/ma1006396
Return to citation in text: [1] -
Dawson, R.; Stöckel, E.; Holst, J. R.; Adams, D. J.; Cooper, A. I. Energy Environ. Sci. 2011, 4, 4239–4245. doi:10.1039/c1ee01971f
Return to citation in text: [1] -
Patel, H. A.; Je, S. H.; Park, J.; Chen, D. P.; Jung, Y.; Yavuz, C. T.; Coskun, A. Nat. Commun. 2013, 4, No. 1357. doi:10.1038/ncomms2359
Return to citation in text: [1] -
DeBlase, C. R.; Silberstein, K. E.; Truong, T.-T.; Abruña, H. D.; Dichtel, W. R. J. Am. Chem. Soc. 2013, 135, 16821–16824. doi:10.1021/ja409421d
Return to citation in text: [1] -
Stegbauer, L.; Schwinghammer, K.; Lotsch, B. V. Chem. Sci. 2014, 5, 2789–2793. doi:10.1039/C4SC00016A
Return to citation in text: [1] -
Wang, J.; Sng, W.; Yi, G.; Zhang, Y. Chem. Commun. 2015, 51, 12076–12079. doi:10.1039/C5CC04702A
Return to citation in text: [1] -
Gopalakrishnan, D.; Dichtel, W. R. J. Am. Chem. Soc. 2013, 135, 8357–8362. doi:10.1021/ja402668e
Return to citation in text: [1] -
Karmakar, A.; Kumar, A.; Chaudhari, A. K.; Samanta, P.; Desai, A. V.; Krishna, R.; Ghosh, S. K. Chem. – Eur. J. 2016, 22, 4931–4937. doi:10.1002/chem.201600109
Return to citation in text: [1] -
Waller, P. J.; Gándara, F.; Yaghi, O. M. Acc. Chem. Res. 2015, 48, 3053–3063. doi:10.1021/acs.accounts.5b00369
Return to citation in text: [1] -
Xu, Y.; Jin, S.; Xu, H.; Nagai, A.; Jiang, D. Chem. Soc. Rev. 2013, 42, 8012–8031. doi:10.1039/c3cs60160a
Return to citation in text: [1] -
Ben, T.; Pei, C.; Zhang, D.; Xu, J.; Deng, F.; Jing, X.; Qiu, S. Energy Environ. Sci. 2011, 4, 3991–3999. doi:10.1039/c1ee01222c
Return to citation in text: [1] -
Bojdys, M. J.; Jeromenok, J.; Thomas, A.; Antonietti, M. Adv. Mater. 2010, 22, 2202–2205. doi:10.1002/adma.200903436
Return to citation in text: [1] -
Ben, T.; Ren, H.; Ma, S.; Cao, D.; Lan, J.; Jing, X.; Wang, W.; Xu, J.; Deng, F.; Simmons, J. M.; Qiu, S.; Zhu, G. Angew. Chem., Int. Ed. 2009, 48, 9457–9460. doi:10.1002/anie.200904637
Return to citation in text: [1] -
Lu, W.; Sculley, J. P.; Yuan, D.; Krishna, R.; Wei, Z.; Zhou, H.-C. Angew. Chem., Int. Ed. 2012, 51, 7480–7484. doi:10.1002/anie.201202176
Return to citation in text: [1] -
Li, B.; Gong, R.; Wang, W.; Huang, X.; Zhang, W.; Li, H.; Hu, C.; Tan, B. Macromolecules 2011, 44, 2410–2414. doi:10.1021/ma200630s
Return to citation in text: [1] -
Luo, Y.; Li, B.; Wang, W.; Wu, K.; Tan, B. Adv. Mater. 2012, 24, 5703–5707. doi:10.1002/adma.201202447
Return to citation in text: [1] -
Dawson, R.; Ratvijitvech, T.; Corker, M.; Laybourn, A.; Khimyak, Y. Z.; Cooper, A. I.; Adams, D. J. Polym. Chem. 2012, 3, 2034–2038. doi:10.1039/c2py20136d
Return to citation in text: [1] -
Msayib, K. J.; McKeown, N. B. J. Mater. Chem. A 2016, 4, 10110–10113. doi:10.1039/C6TA03257E
Return to citation in text: [1] -
Woodward, R. T.; Stevens, L. A.; Dawson, R.; Vijayaraghavan, M.; Hasell, T.; Silverwood, I. P.; Ewing, A. V.; Ratvijitvech, T.; Exley, J. D.; Chong, S. Y.; Blanc, F.; Adams, D. J.; Kazarian, S. G.; Snape, C. E.; Drage, T. C.; Cooper, A. I. J. Am. Chem. Soc. 2014, 136, 9028–9035. doi:10.1021/ja5031968
Return to citation in text: [1] -
Bhunia, S.; Banerjee, B.; Bhaumik, A. Chem. Commun. 2015, 51, 5020–5023. doi:10.1039/C4CC09872B
Return to citation in text: [1] -
Mondal, J.; Kundu, S. K.; Ng, W. K. H.; Singuru, R.; Borah, P.; Hirao, H.; Zhao, Y.; Bhaumik, A. Chem. – Eur. J. 2015, 21, 19016–19027. doi:10.1002/chem.201504055
Return to citation in text: [1] -
Li, H.; Meng, B.; Chai, S.-H.; Liu, H.; Dai, S. Chem. Sci. 2016, 7, 905–909. doi:10.1039/C5SC04034E
Return to citation in text: [1] -
Jing, X.; Zou, D.; Cui, P.; Ren, H.; Zhu, G. J. Mater. Chem. A 2013, 1, 13926–13931. doi:10.1039/c3ta13115g
Return to citation in text: [1] [2] [3] -
Dawson, R.; Stevens, L. A.; Drage, T. C.; Snape, C. E.; Smith, M. W.; Adams, D. J.; Cooper, A. I. J. Am. Chem. Soc. 2012, 134, 10741–10744. doi:10.1021/ja301926h
Return to citation in text: [1] [2] [3] -
Kim, K. C.; Yoon, T.-U.; Bae, Y.-S. Microporous Mesoporous Mater. 2016, 224, 294–301. doi:10.1016/j.micromeso.2016.01.003
Return to citation in text: [1]
| 1. | Forster, P.; Ramaswamy, V.; Artaxo, P.; Berntsen, T.; Betts, R.; Fahey, D. W.; Haywood, J.; Lean, J.; Lowe, D. C.; Myhre, G.; Nganga, J.; Prinn, R.; Raga, G.; Schulz, M.; Van Dorland, R. Climate Change 2007: The Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change; 2007. |
| 7. | Lu, W.; Yuan, D.; Sculley, J.; Zhao, D.; Krishna, R.; Zhou, H.-C. J. Am. Chem. Soc. 2011, 133, 18126–18129. doi:10.1021/ja2087773 |
| 8. | Keskin, S.; van Heest, T. M.; Sholl, D. S. ChemSusChem 2010, 3, 879–891. doi:10.1002/cssc.201000114 |
| 33. | Jing, X.; Zou, D.; Cui, P.; Ren, H.; Zhu, G. J. Mater. Chem. A 2013, 1, 13926–13931. doi:10.1039/c3ta13115g |
| 34. | Dawson, R.; Stevens, L. A.; Drage, T. C.; Snape, C. E.; Smith, M. W.; Adams, D. J.; Cooper, A. I. J. Am. Chem. Soc. 2012, 134, 10741–10744. doi:10.1021/ja301926h |
| 6. | Mason, J. A.; Sumida, K.; Herm, Z. R.; Krishna, R.; Long, J. R. Energy Environ. Sci. 2011, 4, 3030–3040. doi:10.1039/c1ee01720a |
| 33. | Jing, X.; Zou, D.; Cui, P.; Ren, H.; Zhu, G. J. Mater. Chem. A 2013, 1, 13926–13931. doi:10.1039/c3ta13115g |
| 3. | Bae, Y.-S.; Snurr, R. Q. Angew. Chem., Int. Ed. 2011, 50, 11586–11596. doi:10.1002/anie.201101891 |
| 4. | Slater, A. G.; Cooper, A. I. Science 2015, 348. doi:10.1126/science.aaa8075 |
| 5. | Li, J.-R.; Kuppler, R. J.; Zhou, H.-C. Chem. Soc. Rev. 2009, 38, 1477–1504. doi:10.1039/b802426j |
| 34. | Dawson, R.; Stevens, L. A.; Drage, T. C.; Snape, C. E.; Smith, M. W.; Adams, D. J.; Cooper, A. I. J. Am. Chem. Soc. 2012, 134, 10741–10744. doi:10.1021/ja301926h |
| 35. | Kim, K. C.; Yoon, T.-U.; Bae, Y.-S. Microporous Mesoporous Mater. 2016, 224, 294–301. doi:10.1016/j.micromeso.2016.01.003 |
| 25. | Li, B.; Gong, R.; Wang, W.; Huang, X.; Zhang, W.; Li, H.; Hu, C.; Tan, B. Macromolecules 2011, 44, 2410–2414. doi:10.1021/ma200630s |
| 26. | Luo, Y.; Li, B.; Wang, W.; Wu, K.; Tan, B. Adv. Mater. 2012, 24, 5703–5707. doi:10.1002/adma.201202447 |
| 27. | Dawson, R.; Ratvijitvech, T.; Corker, M.; Laybourn, A.; Khimyak, Y. Z.; Cooper, A. I.; Adams, D. J. Polym. Chem. 2012, 3, 2034–2038. doi:10.1039/c2py20136d |
| 29. | Woodward, R. T.; Stevens, L. A.; Dawson, R.; Vijayaraghavan, M.; Hasell, T.; Silverwood, I. P.; Ewing, A. V.; Ratvijitvech, T.; Exley, J. D.; Chong, S. Y.; Blanc, F.; Adams, D. J.; Kazarian, S. G.; Snape, C. E.; Drage, T. C.; Cooper, A. I. J. Am. Chem. Soc. 2014, 136, 9028–9035. doi:10.1021/ja5031968 |
| 30. | Bhunia, S.; Banerjee, B.; Bhaumik, A. Chem. Commun. 2015, 51, 5020–5023. doi:10.1039/C4CC09872B |
| 31. | Mondal, J.; Kundu, S. K.; Ng, W. K. H.; Singuru, R.; Borah, P.; Hirao, H.; Zhao, Y.; Bhaumik, A. Chem. – Eur. J. 2015, 21, 19016–19027. doi:10.1002/chem.201504055 |
| 32. | Li, H.; Meng, B.; Chai, S.-H.; Liu, H.; Dai, S. Chem. Sci. 2016, 7, 905–909. doi:10.1039/C5SC04034E |
| 19. | Waller, P. J.; Gándara, F.; Yaghi, O. M. Acc. Chem. Res. 2015, 48, 3053–3063. doi:10.1021/acs.accounts.5b00369 |
| 20. | Xu, Y.; Jin, S.; Xu, H.; Nagai, A.; Jiang, D. Chem. Soc. Rev. 2013, 42, 8012–8031. doi:10.1039/c3cs60160a |
| 21. | Ben, T.; Pei, C.; Zhang, D.; Xu, J.; Deng, F.; Jing, X.; Qiu, S. Energy Environ. Sci. 2011, 4, 3991–3999. doi:10.1039/c1ee01222c |
| 22. | Bojdys, M. J.; Jeromenok, J.; Thomas, A.; Antonietti, M. Adv. Mater. 2010, 22, 2202–2205. doi:10.1002/adma.200903436 |
| 23. | Ben, T.; Ren, H.; Ma, S.; Cao, D.; Lan, J.; Jing, X.; Wang, W.; Xu, J.; Deng, F.; Simmons, J. M.; Qiu, S.; Zhu, G. Angew. Chem., Int. Ed. 2009, 48, 9457–9460. doi:10.1002/anie.200904637 |
| 24. | Lu, W.; Sculley, J. P.; Yuan, D.; Krishna, R.; Wei, Z.; Zhou, H.-C. Angew. Chem., Int. Ed. 2012, 51, 7480–7484. doi:10.1002/anie.201202176 |
| 33. | Jing, X.; Zou, D.; Cui, P.; Ren, H.; Zhu, G. J. Mater. Chem. A 2013, 1, 13926–13931. doi:10.1039/c3ta13115g |
| 34. | Dawson, R.; Stevens, L. A.; Drage, T. C.; Snape, C. E.; Smith, M. W.; Adams, D. J.; Cooper, A. I. J. Am. Chem. Soc. 2012, 134, 10741–10744. doi:10.1021/ja301926h |
| 13. | Patel, H. A.; Je, S. H.; Park, J.; Chen, D. P.; Jung, Y.; Yavuz, C. T.; Coskun, A. Nat. Commun. 2013, 4, No. 1357. doi:10.1038/ncomms2359 |
| 14. | DeBlase, C. R.; Silberstein, K. E.; Truong, T.-T.; Abruña, H. D.; Dichtel, W. R. J. Am. Chem. Soc. 2013, 135, 16821–16824. doi:10.1021/ja409421d |
| 15. | Stegbauer, L.; Schwinghammer, K.; Lotsch, B. V. Chem. Sci. 2014, 5, 2789–2793. doi:10.1039/C4SC00016A |
| 16. | Wang, J.; Sng, W.; Yi, G.; Zhang, Y. Chem. Commun. 2015, 51, 12076–12079. doi:10.1039/C5CC04702A |
| 17. | Gopalakrishnan, D.; Dichtel, W. R. J. Am. Chem. Soc. 2013, 135, 8357–8362. doi:10.1021/ja402668e |
| 18. | Karmakar, A.; Kumar, A.; Chaudhari, A. K.; Samanta, P.; Desai, A. V.; Krishna, R.; Ghosh, S. K. Chem. – Eur. J. 2016, 22, 4931–4937. doi:10.1002/chem.201600109 |
| 9. | Jiang, J.; Zhao, Y.; Yaghi, O. M. J. Am. Chem. Soc. 2016, 138, 3255–3265. doi:10.1021/jacs.5b10666 |
| 10. | Thomas, A. Angew. Chem., Int. Ed. 2010, 49, 8328–8344. doi:10.1002/anie.201000167 |
| 11. | McKeown, N. B.; Budd, P. M. Macromolecules 2010, 43, 5163–5176. doi:10.1021/ma1006396 |
| 12. | Dawson, R.; Stöckel, E.; Holst, J. R.; Adams, D. J.; Cooper, A. I. Energy Environ. Sci. 2011, 4, 4239–4245. doi:10.1039/c1ee01971f |
| 28. | Msayib, K. J.; McKeown, N. B. J. Mater. Chem. A 2016, 4, 10110–10113. doi:10.1039/C6TA03257E |
© 2016 Samanta et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)