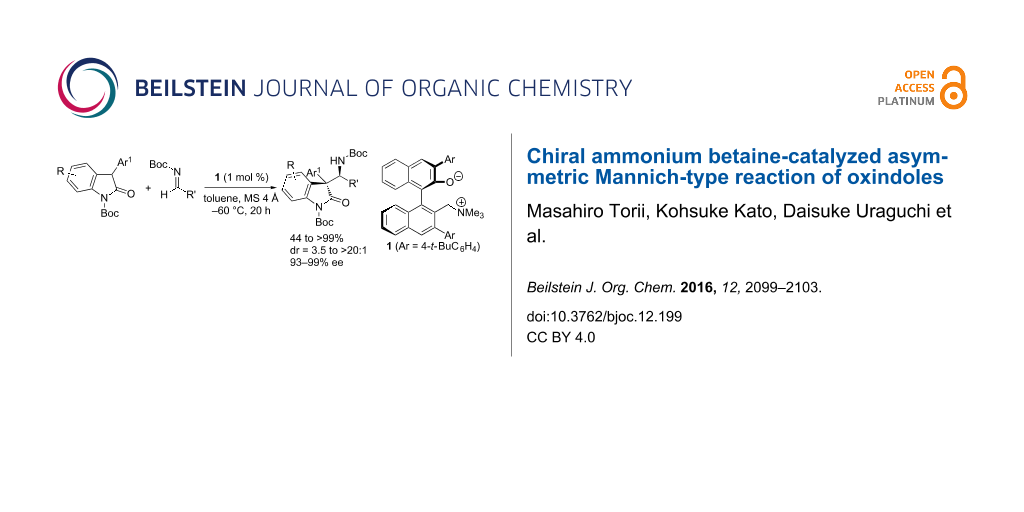
Abstract
A highly diastereo- and enantioselective Mannich-type reaction of 3-aryloxindoles with N-Boc aldimines was achieved under the catalysis of axially chiral ammonium betaines. This catalytic method provides a new tool for the construction of consecutive quaternary and tertiary stereogenic carbon centers on biologically intriguing molecular frameworks with high fidelity.

Graphical Abstract
Introduction
Chiral indole alkaloids possessing C-3 quaternary indoline frameworks are an important class of biologically relevant molecules, and numerous efforts have been made for the development of reliable synthetic methodologies to enable the installation of the C-3 stereogenic center [1-4]. Among them, the direct stereoselective functionalization of 3-monosubstituted oxindoles is a straightforward method for accessing a wide array of chiral indoline skeletons [5-8]. The most common strategy in this approach is to utilize an oxindole enolate as a nucleophile, because facile deprotonation from the C-3 carbon is ensured by the inductive effect of the α-carbonyl group and by the enolate stability arising from the aromatic character. Accordingly, a number of catalytic methods are available for the asymmetric functionalization of oxindole enolates with various different electrophiles. However, successful examples of Mannich-type reactions with imines are surprisingly limited despite allowing efficient construction of vicinal quaternary and tertiary stereocenters [9-22]. In particular, the application of 3-aryl substituted oxindoles seems problematic; hence, the full potential of this useful carbon–carbon bond formation is yet to be realized [12,14].
Ammonium betaines are defined as intramolecular ion-pairing quaternary ammonium salts. In 2008, we employed this structurally distinct molecular scaffold for designing a novel bifunctional organic base catalyst [23], namely axially chiral ammonium betaines of type 1 (Figure 1) [24,25], and uncovered their extraordinary catalytic performance [26-35]. The salient feature of 1 is that, upon abstracting a proton from a pro-nucleophile, the resulting conjugate acid, 1·H, has the ability to recognize the nucleophilic anion through cooperative electrostatic (ionic) and hydrogen-bonding interactions, thereby precisely controlling the stereochemical outcome of the subsequent bond-forming event. Taking advantage of this unique property, we have developed a series of highly stereoselective transformations, and disclose herein the effectiveness of 1 in solving a challenging problem regarding the rigorous control of the relative and absolute stereochemistry in the asymmetric Mannich-type reaction of 3-aryloxindoles.
![[1860-5397-12-199-1]](/bjoc/content/figures/1860-5397-12-199-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
As an initial attempt, the reaction of N-Boc 3-phenyloxindole (2a) with benzaldehyde-derived N-Boc imine 3a [36] was conducted in the presence of a catalytic amount of chiral ammonium betaine 1a (5 mol %) in toluene with 4 Å molecular sieves (MS 4 Å) at −60 °C. Bond formation occurred smoothly and, after 24 h of stirring, the desired Mannich adduct 4aa was isolated as a mixture of diastereomers in 90% yield. Although the diastereomeric ratio was moderate (dr = 7.3:1), the enantiomeric excess (ee) of the major isomer was determined to be 98% (Table 1, entry 1). The investigation then focused on the effects of the catalyst structure, primarily on diastereocontrol, which revealed the importance of steric bulk at the periphery of aromatic substituents at the 3,3’-positions of both naphthyl units (Ar), rather than their electronic attributes (Table 1, entries 2–6). For instance, while 4-trifluoromethylphenyl-substituted betaine 1b had no positive impact on the reaction profile (Table 1, entry 2), the use of 1c, bearing a 4-tert-butylphenyl group, delivered a critical improvement in diastereoselectivity, affording 4aa quantitatively and establishing consecutive quaternary and tertiary stereocenters with almost complete fidelity (Table 1, entry 3). Further examination of the reactions under the influence of 1d, having 3,5-bis(trifluoromethyl)phenyl groups, and 1e, bearing 3,5-bis(tert-butyl)phenyl groups, showed similar tendencies, but a considerable decrease in reactivity and selectivity was observed when using 1d (Table 1, entries 4 and 5). On the other hand, however, the introduction of 2,4,6-trimethylphenyl appendages (1f), which extended steric hindrance over the catalytically active sites, eroded the catalytic activity and diastereocontrol (Table 1, entry 6). These observations demonstrated the superior capability of 1c in facilitating this stereoselective Mannich-type transformation, for which the loading was reduced to 1 mol % without sacrificing reaction efficiency (Table 1, entry 7). It is noteworthy that the present system is scalable; the reaction with 1.0 g of 2a reached completion within 20 h to afford 4aa with a similar degree of stereoselectivity (Table 1, entry 8), and subsequent recrystallization furnished 0.83 g of essentially stereochemically pure 4aa.
Table 1: Optimization of catalyst structure.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-199-i1.svg?max-width=637&scale=1.0)
|
||||
| entry | Ar (1) | yield (%)b | drc | ee (%)d |
|---|---|---|---|---|
| 1 | Ph (1a) | 90 | 7.3:1 | 98/28 |
| 2 | 4-CF3C6H4 (1b) | >99 | 7.3:1 | 98/6 |
| 3 | 4-t-BuC6H4 (1c) | >99 | >20:1 | 99/– |
| 4 | 3,5-(CF3)2C6H3 (1d) | 54 | 1:1.3 | 98/−35 |
| 5 | 3,5-t-Bu2C6H3 (1e) | >99 | 10:1 | 98/– |
| 6 | 2,4,6-Me3C6H2 (1f) | 73 | 1.8:1 | 98/63 |
| 7 | 1ce | 92 | >20:1 | 97/– |
| 8 | 1ce,f | >99 | >20:1 | 98/– |
aUnless otherwise noted, reactions were conducted with 0.1 mmol of 2a, 0.12 mmol of 3a, and 5 mol % of 1 in toluene (1.0 mL) containing 100.0 mg of MS 4 Å at −60 °C for 24 h. bIsolated yield was indicated. cThe diastereomeric ratio was determined by 1H NMR (400 MHz) analysis of crude aliquot. dEnantiomeric excess was analyzed by chiral stationary phase HPLC (DAICEL CHIRALPAK AD-3). Absolute configuration of 4aa was assigned by analogy to 4ca (see Figure 2). e1 mol % of 1c was used. fThe reaction was performed on a 1.0 gram scale regarding 2a.
Having identified 1c as an optimal catalyst, the substrate scope of this asymmetric Mannich protocol was explored. As seen in representative results summarized in Table 2, excellent enantioselectivities were generally attained irrespective of the steric and electronic properties of both oxindoles 2 and N-Boc aldimines 3, but reactivity and diastereoselectivity sometimes fluctuated depending on the structure of these substrates. While significant variation in the imine substituents was feasible, the introduction of electron-withdrawing groups at the meta-position slightly reduced diastereoselectivity (Table 2, entries 1–4). Sterically demanding 2-tolualdehyde-derived imine 3f served as a good electrophile and the corresponding Mannich adduct 4af was isolated as virtually a single stereoisomer (Table 2, entry 5). 3-Thiophenyl aldimine 3g was also well tolerated, but a substantial decrease in diastereoselectivity was observed in the reaction with 2-furyl aldimine 3h, owing to the requisite higher reaction temperature (Table 2, entries 6 and 7). Catalysis with 1c was also applicable to aliphatic imines, which required prolonged reactions and slightly higher catalyst loadings to achieve adequate conversions; the desired adducts, 4ai and 4aj, were obtained with high enantioselectivities and moderate diastereoselectivities (Table 2, entries 8 and 9). With respect to oxindoles 2, the electronic nature of the 3-aryl moiety affected the diastereoselection; the incorporation of electron-deficient aromatics proved beneficial and the presence of electron-rich aryl components seemed detrimental (Table 2, entries 10–14). However, the diastereoselectivity was robust with regard to electronic differences in the oxindole core, and both 5-fluoro- and methoxy-substituted 2g and 2h were efficiently converted into 4ga and 4ha with rigorous relative and absolute stereocontrol (Table 2, entries 15 and 16). The absolute configuration of 4ca was unequivocally determined by X-ray crystallographic analysis (Figure 2), and the stereochemistry of the remaining examples was assumed to be analogous.
Table 2: Substrate scope.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-199-i2.svg?max-width=637&scale=1.0)
|
||||||
| entry | Ar1, R (2) | R’ (3) | yield (%)b | drc | ee (%)d | prod. |
|---|---|---|---|---|---|---|
| 1 | Ph, H (2a) | 4-MeOC6H4 (3b) | 96 | >20:1 | 99 | 4ab |
| 2 | Ph, H (2a) | 4-ClC6H4 (3c) | 96 | >20:1 | 99 | 4ac |
| 3 | Ph, H (2a) | 3-MeOC6H4 (3d) | 92 | >20:1 | 97 | 4ad |
| 4 | Ph, H (2a) | 3-BrC6H4 (3e) | >99 | 14:1 | 99 | 4ae |
| 5 | Ph, H (2a) | 2-MeC6H4 (3f) | 95 | >20:1 | 99 | 4af |
| 6 | Ph, H (2a) | 3-thiophenyl (3g) | 90 | >20:1 | 99 | 4ag |
| 7e | Ph, H (2a) | 2-furyl (3h) | 86 | 9:1 | 97 | 4ah |
| 8f | Ph, H (2a) | Ph(CH2)2 (3i) | 55 | 5:1 | 98/75 | 4ai |
| 9g | Ph, H (2a) | Me(CH2)7 (3j) | 44 | 3.5:1 | 93/60 | 4aj |
| 10 | 4-MeOC6H4, H (2b) | Ph (3a) | 96 | 12:1 | 99 | 4ba |
| 11 | 4-ClC6H4, H (2c) | Ph (3a) | 92 | >20:1 | 97 | 4ca |
| 12 | 3-MeOC6H4, H (2d) | Ph (3a) | 89 | 4:1 | 98/81 | 4da |
| 13 | 3-MeC6H4 (2e) | Ph (3a) | 87 | 13:1 | 99 | 4ea |
| 14 | 3-CF3C6H4, H (2f) | Ph (3a) | 80 | >20:1 | 99 | 4fa |
| 15 | Ph, 5-F (2g) | Ph (3a) | 85 | >20:1 | 97 | 4ga |
| 16 | Ph, 5-MeO (2h) | Ph (3a) | 89 | >20:1 | 96 | 4ha |
aUnless otherwise noted, reactions were performed on 0.2 mmol scale with 1.2 equiv of 3a in the presence of 1c (1 mol %) and MS 4 Å (100.0 mg) in toluene (1.0 mL) at −60 °C for 24 h. bIsolated yield was reported. cThe diastereomeric ratio was determined by 1H NMR (400 MHz) analysis of crude aliquot. dEnantiomeric excess of the major isomer was indicated except for entries 8, 9, and 12, which was analyzed by chiral stationary phase HPLC. Absolute configuration of 4ca was determined by single crystal X-ray diffraction analysis (Figure 2) and that of other 4 was assumed to be analogous. eThe reaction was conducted at –40 °C for 110 h. fThe reaction was stirred for 117 h. gThe reaction time was 72 h.
![[1860-5397-12-199-2]](/bjoc/content/figures/1860-5397-12-199-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ORTEP diagram of 4ca (Ellipsoids displayed at 50% probability. Calculated hydrogen atoms except it attaches to stereogenic carbon are omitted for clarity. Black: carbon, Red: oxygen, Blue: nitrogen, Green: chlorine).
Figure 2: ORTEP diagram of 4ca (Ellipsoids displayed at 50% probability. Calculated hydrogen atoms except it ...
Conclusion
In summary, we have clearly demonstrated that chiral ammonium betaine 1c acts as a uniquely effective catalyst in promoting a Mannich-type reaction between 3-aryloxindoles and N-Boc aldimines with high levels of diastereo- and enantioselectivity under mild conditions. This study greatly expands the scope of this mode of stereoselective Mannich-type reaction, which involve the generation of vicinal quaternary and tertiary stereocenters. Further investigations into the potential utility of ammonium betaine catalysis are underway in our laboratory.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, copies of NMR charts and HPLC traces, and X-ray data. | ||
| Format: PDF | Size: 3.0 MB | Download |
| Supporting Information File 2: Crystallographic information file of compound 4ca. | ||
| Format: CIF | Size: 367.6 KB | Download |
References
-
Marti, C.; Carreira, E. M. Eur. J. Org. Chem. 2003, 2209. doi:10.1002/ejoc.200300050
Return to citation in text: [1] -
Galliford, C. V.; Scheidt, K. A. Angew. Chem., Int. Ed. 2007, 46, 8748. doi:10.1002/anie.200701342
Return to citation in text: [1] -
Trost, B. M.; Brennan, M. K. Synthesis 2009, 3003. doi:10.1055/s-0029-1216975
Return to citation in text: [1] -
Singh, G. S.; Desta, Z. Y. Chem. Rev. 2012, 112, 6104. doi:10.1021/cr300135y
Return to citation in text: [1] -
Zhou, F.; Liu, Y.-L.; Zhou, J. Adv. Synth. Catal. 2010, 352, 1381. doi:10.1002/adsc.201000161
Return to citation in text: [1] -
Dalpozzo, R.; Bartoli, G.; Bencivenni, G. Chem. Soc. Rev. 2012, 41, 7247. doi:10.1039/c2cs35100e
Return to citation in text: [1] -
Cao, Z.-Y.; Wang, Y.-H.; Zeng, X.-P.; Zhou, J. Tetrahedron Lett. 2014, 55, 2571. doi:10.1016/j.tetlet.2014.01.084
Return to citation in text: [1] -
Ziarani, G. M.; Gholamzadeh, P.; Lashgari, N.; Hajiabbasi, P. ARKIVOC 2013, No. i, 470.
Return to citation in text: [1] -
Ting, A.; Schaus, S. E. Eur. J. Org. Chem. 2007, 5797. doi:10.1002/ejoc.200700409
Return to citation in text: [1] -
Verkade, J. M. M.; van Hemert, L. J. C.; Quaedflieg, P. J. L. M.; Rutjes, F. P. J. T. Chem. Soc. Rev. 2008, 37, 29. doi:10.1039/B713885G
Return to citation in text: [1] -
Cai, X.-H.; Xie, B. ARKIVOC 2013, No. i, 264.
Return to citation in text: [1] -
Tian, X.; Jiang, K.; Peng, J.; Du, W.; Chen, Y.-C. Org. Lett. 2008, 10, 3583. doi:10.1021/ol801351j
Return to citation in text: [1] [2] -
Cheng, L.; Liu, L.; Jia, H.; Wang, D.; Chen, Y.-J. J. Org. Chem. 2009, 74, 4650. doi:10.1021/jo9006688
Return to citation in text: [1] -
He, R.; Ding, C.; Maruoka, K. Angew. Chem., Int. Ed. 2009, 48, 4559. doi:10.1002/anie.200901277
Return to citation in text: [1] [2] -
Jin, Y.; Chen, D.; Zhang, X. R. Chirality 2014, 26, 801. doi:10.1002/chir.22358
Return to citation in text: [1] -
Shimizu, S.; Tsubogo, T.; Xu, P.; Kobayashi, S. Org. Lett. 2015, 17, 2006. doi:10.1021/acs.orglett.5b00749
Return to citation in text: [1] -
Shimasaki, Y.; Koshino, S.; Hayashi, Y. Chem. Lett. 2016, 45, 30. doi:10.1246/cl.150916
Return to citation in text: [1] -
Trost, B. M.; Jiang, C. Synthesis 2006, 369. doi:10.1055/s-2006-926302
Return to citation in text: [1] -
Cozzi, P. G.; Hilgraf, R.; Zimmermann, N. Eur. J. Org. Chem. 2007, 5969. doi:10.1002/ejoc.200700318
Return to citation in text: [1] -
Bella, M.; Gasperi, T. Synthesis 2009, 1583. doi:10.1055/s-0029-1216796
Return to citation in text: [1] -
Minko, Y.; Marek, I. Chem. Commun. 2014, 50, 12597. doi:10.1039/C4CC04391J
Return to citation in text: [1] -
Vetica, F.; de Figueiredo, R. M.; Orsini, M.; Tofani, D.; Gasperi, T. Synthesis 2015, 47, 2139. doi:10.1055/s-0034-1378742
Return to citation in text: [1] -
Uraguchi, D.; Koshimoto, K.; Ooi, T. J. Am. Chem. Soc. 2008, 130, 10878. doi:10.1021/ja8041004
Return to citation in text: [1] -
Uraguchi, D.; Koshimoto, K.; Ooi, T. Chem. Commun. 2010, 46, 300. doi:10.1039/B916627K
Return to citation in text: [1] -
Uraguchi, D.; Koshimoto, K.; Sanada, C.; Ooi, T. Tetrahedron: Asymmetry 2010, 21, 1189. doi:10.1016/j.tetasy.2010.04.006
Return to citation in text: [1] -
Uraguchi, D.; Oyaizu, K.; Ooi, T. Chem. – Eur. J. 2012, 18, 8306. doi:10.1002/chem.201201259
Return to citation in text: [1] -
Uraguchi, D.; Oyaizu, K.; Noguchi, H.; Ooi, T. Chem. – Asian J. 2015, 10, 334. doi:10.1002/asia.201402943
Return to citation in text: [1] -
Oyaizu, K.; Uraguchi, D.; Ooi, T. Chem. Commun. 2015, 51, 4437. doi:10.1039/C4CC10261D
Return to citation in text: [1] -
Zhang, W.-Q.; Cheng, L.-F.; Yu, J.; Gong, L.-Z. Angew. Chem., Int. Ed. 2012, 51, 4085. doi:10.1002/anie.201107741
Return to citation in text: [1] -
Claraz, A.; Landelle, G.; Oudeyer, S.; Levacher, V. Eur. J. Org. Chem. 2013, 7693. doi:10.1002/ejoc.201301345
Return to citation in text: [1] -
Wang, Y.-B.; Sun, D.-S.; Zhou, H.; Zhang, W.-Z.; Lu, X.-B. Green Chem. 2014, 16, 2266. doi:10.1039/c3gc42346h
Return to citation in text: [1] -
Tsutsumi, Y.; Yamakawa, K.; Yoshida, M.; Ema, T.; Sakai, T. Org. Lett. 2010, 12, 5728. doi:10.1021/ol102539x
Return to citation in text: [1] -
Guillerm, B.; Lemaur, V.; Cornil, J.; Lazzaroni, R.; Dubois, P.; Coulembier, O. Chem. Commun. 2014, 50, 10098. doi:10.1039/C4CC03347G
Return to citation in text: [1] -
Uraguchi, D.; Koshimoto, K.; Miyake, S.; Ooi, T. Angew. Chem., Int. Ed. 2010, 49, 5567. doi:10.1002/anie.201002315
Return to citation in text: [1] -
Uraguchi, D.; Koshimoto, K.; Ooi, T. J. Am. Chem. Soc. 2012, 134, 6972. doi:10.1021/ja3022939
Return to citation in text: [1] -
Vesely, J.; Rios, R. Chem. Soc. Rev. 2014, 43, 611. doi:10.1039/C3CS60321K
Return to citation in text: [1]
| 1. | Marti, C.; Carreira, E. M. Eur. J. Org. Chem. 2003, 2209. doi:10.1002/ejoc.200300050 |
| 2. | Galliford, C. V.; Scheidt, K. A. Angew. Chem., Int. Ed. 2007, 46, 8748. doi:10.1002/anie.200701342 |
| 3. | Trost, B. M.; Brennan, M. K. Synthesis 2009, 3003. doi:10.1055/s-0029-1216975 |
| 4. | Singh, G. S.; Desta, Z. Y. Chem. Rev. 2012, 112, 6104. doi:10.1021/cr300135y |
| 23. | Uraguchi, D.; Koshimoto, K.; Ooi, T. J. Am. Chem. Soc. 2008, 130, 10878. doi:10.1021/ja8041004 |
| 12. | Tian, X.; Jiang, K.; Peng, J.; Du, W.; Chen, Y.-C. Org. Lett. 2008, 10, 3583. doi:10.1021/ol801351j |
| 14. | He, R.; Ding, C.; Maruoka, K. Angew. Chem., Int. Ed. 2009, 48, 4559. doi:10.1002/anie.200901277 |
| 9. | Ting, A.; Schaus, S. E. Eur. J. Org. Chem. 2007, 5797. doi:10.1002/ejoc.200700409 |
| 10. | Verkade, J. M. M.; van Hemert, L. J. C.; Quaedflieg, P. J. L. M.; Rutjes, F. P. J. T. Chem. Soc. Rev. 2008, 37, 29. doi:10.1039/B713885G |
| 11. | Cai, X.-H.; Xie, B. ARKIVOC 2013, No. i, 264. |
| 12. | Tian, X.; Jiang, K.; Peng, J.; Du, W.; Chen, Y.-C. Org. Lett. 2008, 10, 3583. doi:10.1021/ol801351j |
| 13. | Cheng, L.; Liu, L.; Jia, H.; Wang, D.; Chen, Y.-J. J. Org. Chem. 2009, 74, 4650. doi:10.1021/jo9006688 |
| 14. | He, R.; Ding, C.; Maruoka, K. Angew. Chem., Int. Ed. 2009, 48, 4559. doi:10.1002/anie.200901277 |
| 15. | Jin, Y.; Chen, D.; Zhang, X. R. Chirality 2014, 26, 801. doi:10.1002/chir.22358 |
| 16. | Shimizu, S.; Tsubogo, T.; Xu, P.; Kobayashi, S. Org. Lett. 2015, 17, 2006. doi:10.1021/acs.orglett.5b00749 |
| 17. | Shimasaki, Y.; Koshino, S.; Hayashi, Y. Chem. Lett. 2016, 45, 30. doi:10.1246/cl.150916 |
| 18. | Trost, B. M.; Jiang, C. Synthesis 2006, 369. doi:10.1055/s-2006-926302 |
| 19. | Cozzi, P. G.; Hilgraf, R.; Zimmermann, N. Eur. J. Org. Chem. 2007, 5969. doi:10.1002/ejoc.200700318 |
| 20. | Bella, M.; Gasperi, T. Synthesis 2009, 1583. doi:10.1055/s-0029-1216796 |
| 21. | Minko, Y.; Marek, I. Chem. Commun. 2014, 50, 12597. doi:10.1039/C4CC04391J |
| 22. | Vetica, F.; de Figueiredo, R. M.; Orsini, M.; Tofani, D.; Gasperi, T. Synthesis 2015, 47, 2139. doi:10.1055/s-0034-1378742 |
| 5. | Zhou, F.; Liu, Y.-L.; Zhou, J. Adv. Synth. Catal. 2010, 352, 1381. doi:10.1002/adsc.201000161 |
| 6. | Dalpozzo, R.; Bartoli, G.; Bencivenni, G. Chem. Soc. Rev. 2012, 41, 7247. doi:10.1039/c2cs35100e |
| 7. | Cao, Z.-Y.; Wang, Y.-H.; Zeng, X.-P.; Zhou, J. Tetrahedron Lett. 2014, 55, 2571. doi:10.1016/j.tetlet.2014.01.084 |
| 8. | Ziarani, G. M.; Gholamzadeh, P.; Lashgari, N.; Hajiabbasi, P. ARKIVOC 2013, No. i, 470. |
| 26. | Uraguchi, D.; Oyaizu, K.; Ooi, T. Chem. – Eur. J. 2012, 18, 8306. doi:10.1002/chem.201201259 |
| 27. | Uraguchi, D.; Oyaizu, K.; Noguchi, H.; Ooi, T. Chem. – Asian J. 2015, 10, 334. doi:10.1002/asia.201402943 |
| 28. | Oyaizu, K.; Uraguchi, D.; Ooi, T. Chem. Commun. 2015, 51, 4437. doi:10.1039/C4CC10261D |
| 29. | Zhang, W.-Q.; Cheng, L.-F.; Yu, J.; Gong, L.-Z. Angew. Chem., Int. Ed. 2012, 51, 4085. doi:10.1002/anie.201107741 |
| 30. | Claraz, A.; Landelle, G.; Oudeyer, S.; Levacher, V. Eur. J. Org. Chem. 2013, 7693. doi:10.1002/ejoc.201301345 |
| 31. | Wang, Y.-B.; Sun, D.-S.; Zhou, H.; Zhang, W.-Z.; Lu, X.-B. Green Chem. 2014, 16, 2266. doi:10.1039/c3gc42346h |
| 32. | Tsutsumi, Y.; Yamakawa, K.; Yoshida, M.; Ema, T.; Sakai, T. Org. Lett. 2010, 12, 5728. doi:10.1021/ol102539x |
| 33. | Guillerm, B.; Lemaur, V.; Cornil, J.; Lazzaroni, R.; Dubois, P.; Coulembier, O. Chem. Commun. 2014, 50, 10098. doi:10.1039/C4CC03347G |
| 34. | Uraguchi, D.; Koshimoto, K.; Miyake, S.; Ooi, T. Angew. Chem., Int. Ed. 2010, 49, 5567. doi:10.1002/anie.201002315 |
| 35. | Uraguchi, D.; Koshimoto, K.; Ooi, T. J. Am. Chem. Soc. 2012, 134, 6972. doi:10.1021/ja3022939 |
| 24. | Uraguchi, D.; Koshimoto, K.; Ooi, T. Chem. Commun. 2010, 46, 300. doi:10.1039/B916627K |
| 25. | Uraguchi, D.; Koshimoto, K.; Sanada, C.; Ooi, T. Tetrahedron: Asymmetry 2010, 21, 1189. doi:10.1016/j.tetasy.2010.04.006 |
© 2016 Torii et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)