Abstract
New triazine-based tricarboxylic acid linkers were prepared as elongated relatives of triazinetribenzoic acid (TATB). Additionally, functional groups (NO2, NH2, OMe, OH) were introduced for potential post-synthetic modification (PSM) of MOFs. Functionalized tris(4-bromoaryl)triazine “cores” (3a,3b) were obtained by unsymmetric trimerization mixing one equivalent of an acid chloride (OMe or NO2 substituted) with two equivalents of an unsubstituted nitrile. Triple Suzuki coupling of the cores 3 with suitable phenyl- and biphenylboronic acid derivatives provided elongated tricarboxylic acid linkers as carboxylic acids 17 and 20 or their esters 16 and 19. Reduction of the nitro group and cleavage of the methoxy group gave the respective amino and hydroxy-substituted triazine linkers.
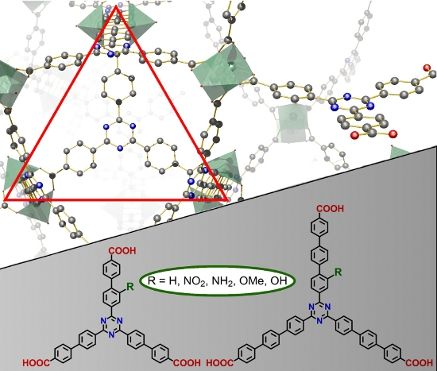
Graphical Abstract
Introduction
A typical building block for many metal–organic frameworks (MOFs) [1-4] carries two functional groups such as carboxylic acids or heterocycles resulting in a bridging ligand for the metal-containing „edges“ of the porous structures. Linkers with more than two ligating sites (carboxylic acids or heterocycles) have also been used successfully. Tri- to hexatopic linkers are also known. The prototype for a trivalent linker is 1,3,5-benzenetricarboxylic acid. By elongating the „arms“ of the linkers, larger pores can be achieved, and often the MOF structures containing extended linkers resemble those obtained with the smaller parent linker (isoreticular structures, see for instance [5]).
In the case of the tritopic linker 1,3,5-benzenetricarboxylic acid (BTC, forming HKUST-1 [6] in the presence of copper ions), an obvious elongated building block is BTB (1,3,5-benzenetribenzoate, Figure 1, left) forming for instance MOF-14 [7]. But this type of elongation has a drawback: the direct vicinity of the aryl groups does not allow planarity due to the repulsion of the ortho-hydrogen atoms in the biphenyl subunits. An exchange of the central benzene ring by a 1,3,5-triazine (1,3,5-triazine-2,4,6-tribenzoate, TATB, Figure 1, right), however, permits planarity. The respective MOF from TATB and copper is PCN-6 [8]. While the orientation of the carboxylate plane relative to the central six-membered ring is in the same plane in the TATB based MOF PCN-6, the BTB analogue MOF-14 shows a tilt angle of ca. 35°. B3LYP//6-31G* calculations of the isolated linkers in the gas phase result in dihedral angles of 38.5° for BTB and 0.006° for TATB, respectively (for further information see Supporting Information File 1). A detailed comparison of triarylbenzene and triaryl-1,3,5-triazine based MOFs and a discussion on the differences in sterical hindrance in benzene and 1,3,5-triazine based structures has been undertaken for tetrazol terminated linkers [9].
![[1860-5397-12-219-1]](/bjoc/content/figures/1860-5397-12-219-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Steric repulsion between ortho-hydrogen atoms in benzene-1,3,5-tribenzoic acid (BTB) leads to a non-planar structure (left). The exchange of CH units by nitrogen atoms in the central six-membered ring allows a planar structure: 1,3,5-triazine-2,4,6-tribenzoic acid (TATB, right). The two structures in the lower half have been calculated with B3LYP//6-31G*.
Figure 1: Steric repulsion between ortho-hydrogen atoms in benzene-1,3,5-tribenzoic acid (BTB) leads to a non...
The properties of MOFs are determined by the nature of the linker, the nature of the metal-containing secondary building unit (SBU) and of course by the pore size. Additional properties are expected if other functional groups were present in the pores. There are two general strategies to introduce additional functional groups into a MOF: (i) by post-synthetic modification, i.e., a reaction of an assembled MOF with some added reagent, or (ii) by the use of modified linkers in the synthesis of the MOF hoping that functionalized linkers lead to isoreticular structures.
It is obvious that the chance for isoreticular structures decreases with the extend with which the linker is altered. Therefore, in the case of tritopic linkers, it makes a difference whether one additional functional group is introduced per linker or one per „arm“. Also the issue of planarity discussed above is related to the amount of additional substituents in the linker.
Indeed, when TATB 1a was substituted with only one substituent per linker, syntheses of isoreticular analogues of MOFs were possible. Structures which are isoreticular to PCN-6 could be synthesized with nitro and amino-substituted TATBs 1b–d [10] (Figure 2).
![[1860-5397-12-219-2]](/bjoc/content/figures/1860-5397-12-219-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Mono-substituted TATB linkers 1b–d were successfully employed in the isoreticular syntheses of PCN-6 MOFs [10].
Figure 2: Mono-substituted TATB linkers 1b–d were successfully employed in the isoreticular syntheses of PCN-...
How can this strategy be extended to even larger monofunctionalized triazine linkers? Obviously by the introduction of an additional benzene ring into each arm. Since some decades, palladium-catalyzed cross coupling is the method of choice when aryl–aryl bonds should be constructed, for instance by applying the Suzuki–Miyaura coupling [11]. A retrosynthetic analysis (Figure 3) of extended mono-functionalized triazinetricarboxylic acids 2 calls for tris(4-bromoaryl)-1,3,5-triazines 3 and boronic acids 4 with an additional carboxylic acid functionality. The respective methyl carboxylate of boronic acid 4 with n = 1, i.e. 15 (see below, Figure 7), is commercially available. In case of the elongated methyl carboxylate of 4 with n = 2, the pinacol boronate 18 (Figure 8) has been described in the literature [12].
![[1860-5397-12-219-3]](/bjoc/content/figures/1860-5397-12-219-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Retrosynthetic analysis for extended TATBs 2: triple Suzuki coupling between tribromotriazines 3 and boronic acids 4.
Figure 3: Retrosynthetic analysis for extended TATBs 2: triple Suzuki coupling between tribromotriazines 3 an...
Results and Discussion
Symmetric 1,3,5-triazines such as 3a are usually synthesized by trimerization of respective nitriles [13]. Unsymmetric 1,3,5-triazines 3 can be made by combining one equivalent of an acid chloride with two equivalents of a nitrile [14-16] (for a recently described alternative access to unsymmetrical 1,3,5-triazines; see [17]). In the presence of a suitable Lewis acid such as antimony(V) chloride, the acid chloride condenses with the nitriles to form an oxadiazinium salt from which the triazine can be obtained by reaction with ammonia (Figure 4).
![[1860-5397-12-219-4]](/bjoc/content/figures/1860-5397-12-219-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Synthesis of unsymmetrically substituted 2,4,6-tris(bromoaryl)-1,3,5-triazines 3 from one equivalent of a substituted benzoyl chloride 5 and two equivalents of benzonitrile 6. As intermediates, oxadiazinium salts 7 are formed. The reaction with ammonia yields the desired triazines 3 (overall yields: 53% (3b), 61% (3c).
Figure 4: Synthesis of unsymmetrically substituted 2,4,6-tris(bromoaryl)-1,3,5-triazines 3 from one equivalen...
The respective syntheses have successfully been carried out with nitro and methoxy-substituted benzoyl chlorides 5b and 5c. The resulting triazines 3b and 3c can then be coupled to boronic acids or boronates to give substituted and elongated triazine-based linkers. Furthermore, the reduction of the nitro group or cleavage of the methoxy group will give additional substituted linkers, amino and hydroxy-substituted ones (see below).
The syntheses of larger linkers 2 therefore need: (i) substituted 4-bromobenzoic acid chlorides 5, 4-bromobenzonitrile (6) and the methoxycarbonyl-substituted phenylboronic acid 15 (n = 1) or pinacol biphenylboronate 18 (n = 2).
Syntheses
Four functional groups were chosen as additional substituents in the extended triazine linkers 2: nitro, methoxy, amino and hydroxy groups. An amino group can be obtained from a nitro group by reduction, and a hydroxy group by cleavage of a methoxy group. Therefore, it was sufficient to synthesize the nitro and methoxy-substituted acid chlorides 5 for the unsymmetric trimerization of the desired tribromotriazines 3.
The synthesis of 4-bromo-3-nitrobenzoyl chloride (5b) was straightforward. Commercially available 4-bromobenzoic acid (8) was nitrated according to a known procedure [18]. The reaction with thionyl chloride provided then acid chloride 5b (Figure 5).
![[1860-5397-12-219-5]](/bjoc/content/figures/1860-5397-12-219-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Synthesis of 4-bromo-3-nitrobenzoyl chloride (5b). Conditions: a) HNO3/H2SO4, 3 h 0 °C, 2 h, room temperature, 98%, b) SOCl2, 2 h reflux, not isolated.
Figure 5: Synthesis of 4-bromo-3-nitrobenzoyl chloride (5b). Conditions: a) HNO3/H2SO4, 3 h 0 °C, 2 h, room t...
In order to synthesize the respective methoxy compound 5c, commercially available 3-hydroxybenzoic acid (10) was used as starting material (Figure 6). Bromination introduced a bromine atom into the 4 position (11) [19], then the phenol was turned into its methyl ether. This methylation can be performed with the free carboxylic acid 11 [20] or with ester 12 [21,22]. In order to generate the methoxy-substituted benzoyl chloride 5c, methoxy ester 13 was cleaved first [23] and the resulting acid 14 was reacted with thionyl chloride. All steps were known from the literature and have been carried out on a multigram scale.
![[1860-5397-12-219-6]](/bjoc/content/figures/1860-5397-12-219-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Syntheses of 4-bromo-3-methoxybenzoyl chloride (5c). Conditions: a) Br2, EtOH/HOAc, 30 min, room temperature, 52%, b) MeOH/H2SO4, MeOH, 16 h, reflux, 94%, c) Me2SO4, K2CO3, acetone, 3 h, reflux, quant., d) MeI, K2CO3, DMF, 16 h, room temperature, 83%, e) NaOH, H2O, MeOH, 6 h, room temperature, 66%, f) SOCl2, DMF (cat.), 2 h, reflux, not isolated.
Figure 6: Syntheses of 4-bromo-3-methoxybenzoyl chloride (5c). Conditions: a) Br2, EtOH/HOAc, 30 min, room te...
Cyclotrimerization of one equivalent of an acid chloride 5b or 5c with two equivalents of 4-bromobenzonitrile (6) was achieved using the Lewis acid antimony(V) chloride as depicted in Figure 4. The resulting colourful oxadiazinium salts 7 were mixed with aqueous ammonia solution resulting in the formation of the desired triazines 3b and 3c in 53 and 61% yield, respectively.
Following the synthetic plan of Figure 3, the tribromides 3b and 3c were used in the Suzuki couplings. In this and respective following synthetic steps, optimization of the reaction conditions was very important because each of the reactions had to be performed three times with each triazine. Thus the total yield of the final product depends on the yield of the single step to the power of three. In a triple Suzuki coupling, 4-(methoxycarbonyl)phenylboronic acid (15) as the coupling partner yielded elongated and monofunctionalized 1,3,5-triazines 16b and 16c carrying methyl ester groups at the end of each „arm“ (Figure 7). The yields of 90% (16b) and 73% (16c) correspond to 97% and 90% yield per coupling step, respectively.
![[1860-5397-12-219-7]](/bjoc/content/figures/1860-5397-12-219-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Triple Suzuki–Miyaura coupling between tribromotriazines 3 and boronic acid 15 and subsequent hydrolyses of the esters 16. Conditions: a) Pd(PPh3)4, K3PO4, dioxane, H2O, 48 h, reflux, 16b: 90%, 16c: 73%, b) Pd/C, H2, 5 bar, CH2Cl2, 5 d, room temperature, 16d: 77%. c) 17b: LiOH/H2O/dioxane, 48 h, room temp, 96%; 17c: LiOH/H2O/THF, 24 h, 60 °C, quant.; 17d: H2O/THF, 24 h, 60 °C, quant., d) pyridinium hydrochloride, 12 h, 200 °C, 17e: 52%.
Figure 7: Triple Suzuki–Miyaura coupling between tribromotriazines 3 and boronic acid 15 and subsequent hydro...
By selective reduction, the nitro-substituted triester 16b could be transferred into its amino derivative. The reaction time and the hydrogen pressure had to be optimized. By heterogeneous catalytic hydrogenation at 5 bar with a Pd/C catalyst, aminotriazine 16d was obtained in 77% yield after 5 days. Hydrolyses of all three methyl esters 16b–d provided the tricarboxylic acids 17b–d in 96% to quantitative yields. Also the methoxy group could be cleaved to yield a hydroxy-substituted elongated triazine. In this case, four methoxy, three ester and one ether group had to be cleaved leading to increasingly less soluble products (carboxylic acids are less soluble than the respective esters). In order to improve the yield, a methoxy cleaving procedure which has been found to be effective in other demanding demethylations [24] was used. In liquid pyridinium chloride (which melts at 144 °C), apparently all intermediates were sufficiently soluble and consequently all four methoxy groups of 16c could be cleaved in a one-pot reaction. The tetrafold cleavage yielded hydroxytriazine 17e in 52% yield. All tricarboxylic acids 17 are considerably less soluble in most solvents when compared to the respective methyl esters 16. Nevertheless, they are sufficiently soluble in, for instance, DMSO or in base to allow proper analyses and future use in solvothermal syntheses of MOFs.
Tribromotriazines 3 are not only good starting materials for the syntheses of biphenyl-substituted triazine linkers 16 and 17. Also longer terphenyl-substituted linkers can be obtained if the same approach is chosen but a biphenylboronic acid derivative such as boronate 18 [12] is used instead of the phenyl compound 15.
As in the case of the biphenyl derivatives 16 and 17, the nitro and the methoxy-substituted triazines 3b and 3c were used to obtain the terphenyl-based nitro and methoxy-substituted triesters 19b and 19c (Figure 8). But also the non-substituted precursor 3a was coupled leading to the unsubstituted parent compound 19a, a triester which has not been described yet. Direct hydrolyses of triesters 19a and 19b gave the carboxylic acids 20a and 20b.
![[1860-5397-12-219-8]](/bjoc/content/figures/1860-5397-12-219-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Triple Suzuki coupling between tribromotriazines 3 and boronate 18. Conditions: a) 19a: Pd(PPh3)4, K3PO4, dioxane/H2O, 2 d, 120 °C, 24%; 19b: Pd(dppf)Cl2, K3PO4, dioxane/H2O, 3 d, 120 °C, 16%; 19c: Pd(PPh3)4, K3PO4, dioxane/H2O, 2 d, 100 °C, 81%; b) LiOH/H2O/THF, 24 h, 120 °C, 20a: 99%; c) NaOH/H2O/dioxane, 24 h, 120 °C, 20b: 79%; d) pyridinium hydrochloride, 12 h, 200 °C, 20e: 62%.
Figure 8: Triple Suzuki coupling between tribromotriazines 3 and boronate 18. Conditions: a) 19a: Pd(PPh3)4, K...
Finally, a hydroxy-functionalized terphenyl-based linker was synthesized from the methoxy-substituted triester 19c. As successfully applied for 16c, molten pyridinium chloride was used as the cleaving reagent and all four methoxy groups of 19c could be cleaved leading to the hydroxy-substituted triacid 20e in 62% yield.
All three triacids 20a, b and e precipitate from water with decreasing pH. In DMSO, they are soluble and consequently, NMR analytics have been carried out in DMSO-d6.
Conclusion
In conclusion, 13 new elongated relatives of TATB 16, 17, 19 and 20 have been synthesized in batch sizes up to several grams, of which 11 were mono-substituted. Syntheses, purifications and solubilities are satisfying in the case of the biphenyl derivatives 16 and 17. The handling of the terphenyl derivatives 19, and especially 20, becomes more difficult due to a lower solubility. If the solublities were not sufficient for the solvothermal syntheses of MOFs, “discrete” methylation [25] could find a remedy.
Supporting Information
| Supporting Information File 1: Experimental details. | ||
| Format: PDF | Size: 2.9 MB | Download |
References
-
Zhou, H.-C. J.; Kitagawa, S., Eds. Metal–Organic Frameworks (MOFs). Chem. Soc. Rev. 2014, 43, 5415–6176. doi:10.1039/C4CS90059F
Return to citation in text: [1] -
Furukawa, H.; Cordova, K. E.; O’Keeffe, M.; Yaghi, O. M. Science 2013, 341, 1230444. doi:10.1126/science.1230444
Return to citation in text: [1] -
Farusseng, D. Metal-Organic Frameworks; Wiley-VCH: Weinheim, Germany, 2011. doi:10.1002/9783527635856
Return to citation in text: [1] -
Férey, G. Chem. Soc. Rev. 2008, 37, 191–214. doi:10.1039/B618320B
Return to citation in text: [1] -
Furukawa, H.; Go, Y. B.; Ko, N.; Park, Y. K.; Uribe-Romo, F. J.; Kim, J.; O’Keeffe, M.; Yaghi, O. M. Inorg. Chem. 2011, 50, 9147–9152. doi:10.1021/ic201376t
Return to citation in text: [1] -
Chui, S. S.-Y.; Lo, S. M.-F.; Charmant, J. P. H.; Orpen, A. G.; Williams, I. D. Science 1999, 283, 1148–1150. doi:10.1126/science.283.5405.1148
Return to citation in text: [1] -
Chen, B.; Eddaoudi, M.; Hyde, S. T.; O'Keeffe, M.; Yaghi, O. M. Science 2001, 291, 1021–1023. doi:10.1126/science.1056598
Return to citation in text: [1] -
Ma, S.; Sun, D.; Ambrogio, M.; Fillinger, J. A.; Parkin, S.; Zhou, H.-C. J. Am. Chem. Soc. 2007, 129, 1858–1859. doi:10.1021/ja067435s
Return to citation in text: [1] -
Dincă, M.; Dailly, A.; Tsay, C.; Long, J. R. Inorg. Chem. 2008, 47, 11–13. doi:10.1021/ic701917w
Return to citation in text: [1] -
Mühlbauer, E.; Klinkebiel, A.; Beyer, O.; Auras, F.; Wuttke, S.; Lüning, U.; Bein, T. Microporous Mesoporous Mater. 2015, 216, 51–55. doi:10.1016/j.micromeso.2015.06.007
Return to citation in text: [1] [2] -
Miyaura, N.; Suzuki, A. J. Chem. Soc., Chem. Commun. 1979, 866–867. doi:10.1039/c39790000866
Return to citation in text: [1] -
Barnard, R. A.; Dutta, A.; Schnobrich, J. K.; Morrison, C. N.; Ahn, S.; Matzger, A. J. Chem. – Eur. J. 2015, 21, 5954–5961. doi:10.1002/chem.201406332
Return to citation in text: [1] [2] -
Cook, A. H.; Jones, D. G. J. Chem. Soc. 1941, 278–282. doi:10.1039/jr9410000278
Return to citation in text: [1] -
Eitner, P.; Krafft, F. Ber. Dtsch. Chem. Ges. 1892, 25, 2263–2269. doi:10.1002/cber.18920250218
Return to citation in text: [1] -
Meerwein, H.; Laasch, P.; Mersch, R.; Spille, J. Chem. Ber. 1956, 89, 209–224. doi:10.1002/cber.19560890207
Return to citation in text: [1] -
Schmidt, R. R. Chem. Ber. 1965, 98, 334–345. doi:10.1002/cber.19650980203
Return to citation in text: [1] -
Tiwari, A. R.; Akash, T.; Bhanage, B. M. Org. Biomol. Chem. 2015, 13, 10973–10976. doi:10.1039/C5OB01835H
Return to citation in text: [1] -
Bhat, L.; Mohapatra, P. P.; Bhat, S. R. Compositions, Synthesis, and Methods of Using Quinolinone Based Atypical Antipsychotic Agents. U.S. Pat. Appl. US20080293736 A1, Nov 27, 2008.
Return to citation in text: [1] -
Li, X.; Longenecker, K. L.; Pei, Z.; Sham, H. L.; Wiedeman, P. E. Pyrrolidine-2-carbonitrile derivatives and their use as inhibitors of dipeptidyl peptidase-iv (dpp-iv). WO Pat. Appl. WO2005023762 A1, March 17, 2005.
Return to citation in text: [1] -
Burrows, A. D.; Frost, C. G.; Mahon, M. F.; Richardson, C. Angew. Chem., Int. Ed. 2008, 47, 8482–8486. doi:10.1002/anie.200802908
Angew. Chem. 2008, 120, 8610–8614. doi:10.1002/ange.200802908
Return to citation in text: [1] -
Govindachari, T. R.; Viswanathan, N.; Ravindranath, K. R.; Anjaneyulu, B. Indian J. Chem. 1973, 11, 1081–1083.
Return to citation in text: [1] -
Failli, A.; Quagliato, D.; Andrae, P.; Heffernan, G.; Coghlan, R.; Shen, E. Pyrrolobenzodiazepine arylcarboxamides and derivatives thereof as follicle-stimulating hormone receptor antagonists. U.S. Pat. Appl. US20060199806 A1, Sept 7, 2006.
Return to citation in text: [1] -
Fukaya, Y.; Mihara, Y.; Morizono, D.; Ohtake, Y.; Oishi, T.; Shoji, T.; Takashima, Y. 2,3,4,5-tetrahydro-1H-1,5-benzodiazepine derivative and medicinal composition. WO Pat. Appl. WO2006051851 A1, May 18, 2006.
Return to citation in text: [1] -
Dawson, C. R.; Kurtz, A. P. J. Med. Chem. 1971, 14, 729–732. doi:10.1021/jm00290a015
Return to citation in text: [1] -
Köhl, I.; Lüning, U. Synthesis 2014, 46, 2376–2382. doi:10.1055/s-0033-1339028
Return to citation in text: [1]
| 24. | Dawson, C. R.; Kurtz, A. P. J. Med. Chem. 1971, 14, 729–732. doi:10.1021/jm00290a015 |
| 21. | Govindachari, T. R.; Viswanathan, N.; Ravindranath, K. R.; Anjaneyulu, B. Indian J. Chem. 1973, 11, 1081–1083. |
| 22. | Failli, A.; Quagliato, D.; Andrae, P.; Heffernan, G.; Coghlan, R.; Shen, E. Pyrrolobenzodiazepine arylcarboxamides and derivatives thereof as follicle-stimulating hormone receptor antagonists. U.S. Pat. Appl. US20060199806 A1, Sept 7, 2006. |
| 23. | Fukaya, Y.; Mihara, Y.; Morizono, D.; Ohtake, Y.; Oishi, T.; Shoji, T.; Takashima, Y. 2,3,4,5-tetrahydro-1H-1,5-benzodiazepine derivative and medicinal composition. WO Pat. Appl. WO2006051851 A1, May 18, 2006. |
| 1. | Zhou, H.-C. J.; Kitagawa, S., Eds. Metal–Organic Frameworks (MOFs). Chem. Soc. Rev. 2014, 43, 5415–6176. doi:10.1039/C4CS90059F |
| 2. | Furukawa, H.; Cordova, K. E.; O’Keeffe, M.; Yaghi, O. M. Science 2013, 341, 1230444. doi:10.1126/science.1230444 |
| 3. | Farusseng, D. Metal-Organic Frameworks; Wiley-VCH: Weinheim, Germany, 2011. doi:10.1002/9783527635856 |
| 4. | Férey, G. Chem. Soc. Rev. 2008, 37, 191–214. doi:10.1039/B618320B |
| 8. | Ma, S.; Sun, D.; Ambrogio, M.; Fillinger, J. A.; Parkin, S.; Zhou, H.-C. J. Am. Chem. Soc. 2007, 129, 1858–1859. doi:10.1021/ja067435s |
| 19. | Li, X.; Longenecker, K. L.; Pei, Z.; Sham, H. L.; Wiedeman, P. E. Pyrrolidine-2-carbonitrile derivatives and their use as inhibitors of dipeptidyl peptidase-iv (dpp-iv). WO Pat. Appl. WO2005023762 A1, March 17, 2005. |
| 7. | Chen, B.; Eddaoudi, M.; Hyde, S. T.; O'Keeffe, M.; Yaghi, O. M. Science 2001, 291, 1021–1023. doi:10.1126/science.1056598 |
| 20. |
Burrows, A. D.; Frost, C. G.; Mahon, M. F.; Richardson, C. Angew. Chem., Int. Ed. 2008, 47, 8482–8486. doi:10.1002/anie.200802908
Angew. Chem. 2008, 120, 8610–8614. doi:10.1002/ange.200802908 |
| 6. | Chui, S. S.-Y.; Lo, S. M.-F.; Charmant, J. P. H.; Orpen, A. G.; Williams, I. D. Science 1999, 283, 1148–1150. doi:10.1126/science.283.5405.1148 |
| 17. | Tiwari, A. R.; Akash, T.; Bhanage, B. M. Org. Biomol. Chem. 2015, 13, 10973–10976. doi:10.1039/C5OB01835H |
| 5. | Furukawa, H.; Go, Y. B.; Ko, N.; Park, Y. K.; Uribe-Romo, F. J.; Kim, J.; O’Keeffe, M.; Yaghi, O. M. Inorg. Chem. 2011, 50, 9147–9152. doi:10.1021/ic201376t |
| 18. | Bhat, L.; Mohapatra, P. P.; Bhat, S. R. Compositions, Synthesis, and Methods of Using Quinolinone Based Atypical Antipsychotic Agents. U.S. Pat. Appl. US20080293736 A1, Nov 27, 2008. |
| 11. | Miyaura, N.; Suzuki, A. J. Chem. Soc., Chem. Commun. 1979, 866–867. doi:10.1039/c39790000866 |
| 13. | Cook, A. H.; Jones, D. G. J. Chem. Soc. 1941, 278–282. doi:10.1039/jr9410000278 |
| 10. | Mühlbauer, E.; Klinkebiel, A.; Beyer, O.; Auras, F.; Wuttke, S.; Lüning, U.; Bein, T. Microporous Mesoporous Mater. 2015, 216, 51–55. doi:10.1016/j.micromeso.2015.06.007 |
| 14. | Eitner, P.; Krafft, F. Ber. Dtsch. Chem. Ges. 1892, 25, 2263–2269. doi:10.1002/cber.18920250218 |
| 15. | Meerwein, H.; Laasch, P.; Mersch, R.; Spille, J. Chem. Ber. 1956, 89, 209–224. doi:10.1002/cber.19560890207 |
| 16. | Schmidt, R. R. Chem. Ber. 1965, 98, 334–345. doi:10.1002/cber.19650980203 |
| 10. | Mühlbauer, E.; Klinkebiel, A.; Beyer, O.; Auras, F.; Wuttke, S.; Lüning, U.; Bein, T. Microporous Mesoporous Mater. 2015, 216, 51–55. doi:10.1016/j.micromeso.2015.06.007 |
| 12. | Barnard, R. A.; Dutta, A.; Schnobrich, J. K.; Morrison, C. N.; Ahn, S.; Matzger, A. J. Chem. – Eur. J. 2015, 21, 5954–5961. doi:10.1002/chem.201406332 |
| 9. | Dincă, M.; Dailly, A.; Tsay, C.; Long, J. R. Inorg. Chem. 2008, 47, 11–13. doi:10.1021/ic701917w |
| 12. | Barnard, R. A.; Dutta, A.; Schnobrich, J. K.; Morrison, C. N.; Ahn, S.; Matzger, A. J. Chem. – Eur. J. 2015, 21, 5954–5961. doi:10.1002/chem.201406332 |
| 25. | Köhl, I.; Lüning, U. Synthesis 2014, 46, 2376–2382. doi:10.1055/s-0033-1339028 |
© 2016 Klinkebiel et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)