Guest Editor: T. P. Yoon Beilstein J. Org. Chem.2016,12, 2293–2297.https://doi.org/10.3762/bjoc.12.222 Received 21 Aug 2016,
Accepted 19 Oct 2016,
Published 01 Nov 2016
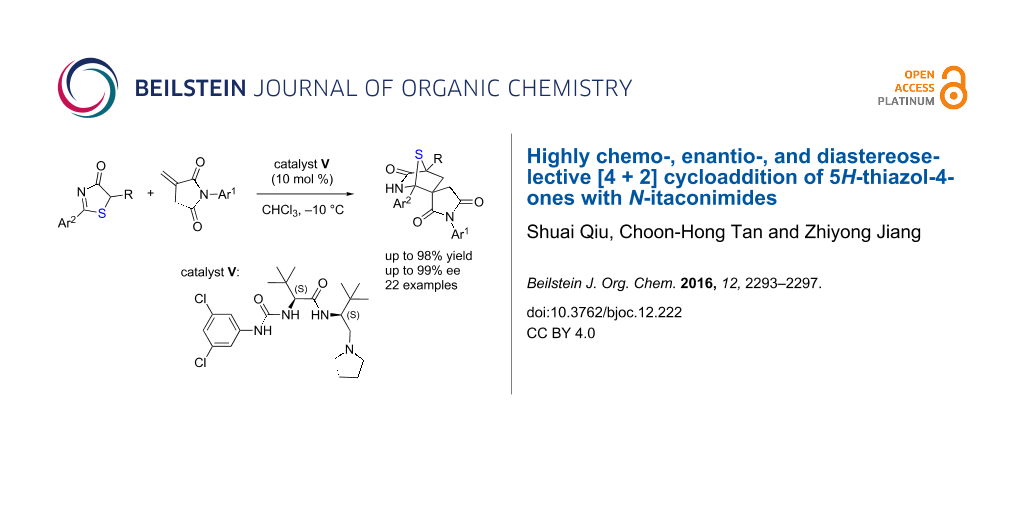
A dipeptide-based urea-amide tertiary amine (DP-UAA) was shown to be an effective Brønsted base catalyst for the first asymmetric annulation reaction between 5H-thiazol-4-ones and N-itaconimides. High levels of enantioselectivity (up to 99% ee) and diastereoselectivity (>19:1 dr) were obtained for a series of spirocyclic 1,4-sulfur-bridged piperidinone-based succinimides.
Sulfur-containing tetrasubstituted carbon stereocenters are widely present in a number of natural and non-natural products with significant biological activities [1]. In the past few decades, diverse competent asymmetric strategies have been established to access these chiral entities [1-4]. Among them, catalytic asymmetric functionalization of S-containing prochiral carbon centers can be considered to be one of the most efficient and expedient approach [1-4]. The development of novel S-containing substrates has therefore attracted the attention of chemists [1-4]. For example in 2013, Palomo and co-workers introduced 5H-thiazol-4-ones as a new class of sulfur-containing pro-nucleophiles in a highly enantio- and diastereoselective conjugate addition to nitroalkenes, providing α,α-disubstituted α-mercapto carboxylic acids [5]. Since then, several asymmetric variants using 5H-thiazol-4-ones as nucleophiles have been disclosed; such as amination [6], allylation [7], conjugate addition to enones [8], and γ-addition with allenoates [9]. All these examples focused on nucleophilic addition reactions of the C5 atom of 5H-thiazol-4-ones.
Recently, we described an organocatalytic asymmetric [4 + 2] cyclization of 5H-thiazol-4-ones with a series of activated alkenes, including nitroalkenes, 4-oxo-4-arylbutenones, 4-oxo-4-arylbutenoates and methyleneindolinones [10]. This work elucidated the feasibility of the electrophilic addition to the C2 position of 5H-thiazol-4-ones. More importantly, it provided a direct and convenient approach to synthesize three kinds of biologically important chiral 1,4-sulfur-bridged piperidinones and their related derivatives [10]. In order to develop novel chiral S-containing polycyclic scaffolds, the development of [4 + 2] annulations of 5H-thiazol-4-ones with unusual activated alkenes still remains highly desirable.
Succinimides are present in many biologically significant molecules and are investigated as potential pharmacophores in the research of drug discovery [11,12]. Our group has recently devised N-itaconimides for the assembly of succinimide frameworks [13-18]. As an extension of these works, herein, we report an asymmetric [4 + 2] annulation reaction of 5H-thiazol-4-ones with N-itaconimides. The method features excellent chemo-, enantio, and diastereoselectivities, thus leading to a series of chiral spirocyclic 1,4-sulfur-bridged piperidinone-based succinimides with excellent results.
Results and Discussion
Our studies were initiated by examining a model reaction between 5H-thiazol-4-one 1a and N-phenyl itaconimide 2a (Table 1). The reaction was first evaluated in toluene at 25 °C and using L-tert-leucine-based thiourea−tertiary amine I as the catalyst (Table 1, entry 1), with excellent catalytic efficacy as demonstrated in a series of asymmetric reactions [18]. It was found that the reaction was completed after 48 hours, affording the desired [4 + 2] annulation adduct 3a in 55% yield with 64% ee. A significant amount of conjugate addition adduct led to the unsatisfactory chemoselectivity and thus the moderate yield. When the H-bond donor was changed from thiourea to urea (catalyst II), it did not provide better results (Table 1, entry 2) [17,19]. In the [4 + 2] annulation of 5H-thiazol-4-ones with nitroalkenes, dipeptide-based thiourea−amide−tertiary amine III (DP-TAA) was devised and demonstrated as a competent catalyst to furnish excellent chemo- and stereoselectivity [10]. Therefore, we examined catalyst III for this reaction (Table 1, entry 3); annulation adduct 3a was obtained in 60% yield with 70% ee. The increased in enantioselectivity indicates the potential of dipeptide-based tertiary amine for this type of reaction. By modifying the thiourea moiety of III to urea lead us to catalyst DP-UAA IV, which could further increase the enantioselectivity (Table 1, entry 4). Subsequently, we screened the solvent effect with IV as the catalyst (Table 1, entries 5–7), and the results revealed that chloroform was the best reaction medium regarding the enantioselectivity (Table 1, entry 7). By changing the reaction temperature (Table 1, entries 8 and 9), we found that the yield of 3a was decreased to 60% but the ee value was increased to 92% at −10 °C (Table 1, entry 9). To improve the chemoselectivity, we synthesized a series of DP-UAAs through tuning of the substituent groups of the urea. We were pleased to find that the reaction rate could be tremendously increased by utilizing DP-UAA V as the catalyst, and 3a was obtainable with high enantioselectivity, high chemoselectivity and excellent yield of 98% (Table 1, entry 10).
Table 1:
Optimization of reaction conditionsa.
Entry
Catalyst
Solvent
T (°C)
t (h)
Yield (%)b
ee (%)c
1
I
toluene
25
48
55
64
2
II
toluene
25
48
50
62
3
III
toluene
25
48
60
70
4
IV
toluene
25
48
62
74
5
IV
CH2Cl2
25
48
68
77
6
IV
Et2O
25
48
50
76
7
IV
CHCl3
25
48
72
86
8
IV
CHCl3
0
48
70
89
9
IV
CHCl3
−10
60
60
92
10
V
CHCl3
−10
18
98
93
aThe reaction was performed in a 0.05 mmol scale; byield was isolated by flash column; cee was determined by HPLC.
With the optimal reaction conditions in hand, we examined the substrate scope of the enantioselective [4 + 2] cycloaddition between 5H-thiazol-4-ones 1 and N-itaconimides 2, catalyzed by DP-UAA V (Scheme 1). Firstly, with 1a as the model 5H-thiazol-4-one substrate, a series of N-itaconimides containing various N-aryl groups were transformed to the corresponding [4 + 2] annulation adducts 3a–i in 82−98% yield with 90−96% ee. We then investigated substrates with diverse aromatic (3j–o) and heteroaromatic (3p–t) groups on the 2-position of 5H-thiazol-4-ones, and found the reactions completed within 12−72 hours, affording the corresponding annulation adducts 3j–t in 80−98% yield with 90−99% ee. Altering the R group on 5H-thiazol-4-ones for a benzyl (3u) or phenyl (3v) substituent also presented 3u and 3v in high yields and excellent enantioselectivities. The absolute configurations of annulation adducts 3 were assigned based on X-ray crystallographic analysis of a single crystal of 3r[20].
Scheme 1:
Substrate scope of the [4 + 2] annulation. Reaction conditions: 1 (0.1 mmol), 2 (0.15 mmol), V (0.01 mmol), CHCl3 (1.0 mL) at −10 °C. All drs are >20:1; ees were determined via chiral HPLC analysis. a20 mol % of V was used, T = 0 °C.
Scheme 1:
Substrate scope of the [4 + 2] annulation. Reaction conditions: 1 (0.1 mmol), 2 (0.15 mmol), V (0.0...
Through an analysis of the absolute configuration of adduct 3, it is proposed that a plausible reaction mechanism should be similar to the DP-TAA-catalyzed [4 + 2] annulation between 5H-thiazol-4-ones and nitroalkenes [10]. In this stepwise process, the use of 3,5-dichlorophenyl as the substituent group of the urea in catalyst V would remarkably increase the free energy difference between R- and S-selection in the first Michael addition step, and also decreases the free energy of the second formal Mannich reaction, thus improving the reaction rate and chemoselectivity of the [4 + 2] cycloaddition.
To extend the usefulness of this reaction, we demonstrate that the adduct can be efficiently reduced with borane. As shown in Scheme 2, 3a was readily reduced to afford a spirocyclic sulfur-bridged N-heterocycle 4 in 70% yield and without compromising its ee value.
In conclusion, we have developed the first asymmetric reaction of 5H-thiazol-4-ones with N-itaconimides. By employing a DP-UAA catalyst, the reaction undergoes a [4 + 2] annulation process with excellent chemoselectivity and with a broad substrate scope, affording a series of valuable chiral spirocyclic 1,4-sulfur-bridged piperidinone-based succinimides in high yields (up to 98%) and excellent enantio- and diastereoselectivities (up to 99% ee and >19:1 dr). Further investigations involving new [4 + 2] annulation of 5H-thiazol-4-ones using DP-TAAs and DP UAAs are currently ongoing and will be reported in due course.
Experimental
Representative procedure for the synthesis of 3a: N-Phenyl itaconimide 2a (0.15 mmol, 1.5 equiv) and catalyst V (0.01 mmol, 0.1 equiv) were dissolved in chloroform (1.0 mL) and stirred at −10 °C for 10 min. This is followed by the addition of 5H-thiazol-4-one 1a (0.1 mmol, 1.0 equiv). The reaction mixture was stirred at −10 °C and monitored by TLC. Upon complete consumption of 1a, the reaction mixture was directly loaded onto a short silica gel column, followed by gradient elution with DCM/MeOH mixture (500:1−200:1 ratio). Removing the solvent in vacuo, afforded product 3a.
Supporting Information
Supporting Information File 1:
Experimental information and spectroscopic data.
Financial support from the National Science Foundation of China (Nos. 21672052, 21072044), the Program for Innovative Research Team from the University of Henan Province (14IRTSTHN006), and Henan University is greatly appreciated.
References
Yu, J.-S.; Huang, H.-M.; Ding, P.-G.; Hu, X.-S.; Zhou, F.; Zhou, J. ACS Catal.2016,6, 5319–5344. doi:10.1021/acscatal.6b01496
Return to citation in text:
[1]
[2]
[3]
[4]
Enders, D.; Lüttgen, K.; Narine, A. A. Synthesis2007, 959–980. doi:10.1055/s-2007-965968
Return to citation in text:
[1]
[2]
[3]
Clayden, J.; MacLellan, P. Beilstein J. Org. Chem.2011,7, 582–595. doi:10.3762/bjoc.7.68
Return to citation in text:
[1]
[2]
[3]
Diosdado, S.; Etxabe, J.; Izquierdo, J.; Landa, A.; Mielgo, A.; Olaizola, I.; López, R.; Palomo, C. Angew. Chem., Int. Ed.2013,52, 11846–11851. doi:10.1002/anie.201305644
Return to citation in text:
[1]
Diosdado, S.; López, R.; Palomo, C. Chem. – Eur. J.2014,20, 6526–6531. doi:10.1002/chem.201304877
Return to citation in text:
[1]
Chen, W.; Hartwig, J. F. J. Am. Chem. Soc.2014,136, 377–382. doi:10.1021/ja410650e
Return to citation in text:
[1]
Badiola, E.; Fiser, B.; Gómez-Bengoa, E.; Mielgo, A.; Olaizola, I.; Urruzuno, I.; García, J. M.; Odriozola, J. M.; Razkin, J.; Oiarbide, M.; Palomo, C. J. Am. Chem. Soc.2014,136, 17869–17881. doi:10.1021/ja510603w
Return to citation in text:
[1]
Wang, T.; Yu, Z.; Hoon, D. L.; Huang, K.-W.; Lan, Y.; Lu, Y. Chem. Sci.2015,6, 4912–4922. doi:10.1039/C5SC01614B
Return to citation in text:
[1]
Zhu, B.; Qiu, S.; Li, J.; Coote, M. L.; Lee, R.; Jiang, Z. Chem. Sci.2016,7, 6060–6067. doi:10.1039/C6SC02039A
Return to citation in text:
[1]
[2]
[3]
[4]
Katritzky, A. R.; Yao, J.; Qi, M.; Chou, Y.; Sikora, D. J.; Davis, S. Heterocycles1998,48, 2677–2691. doi:10.3987/REV-98-506
Return to citation in text:
[1]
Ballini, R.; Bosica, G.; Cioci, G.; Fiorini, D.; Petrini, M. Tetrahedron2003,59, 3603–3608. doi:10.1016/S0040-4020(03)00508-8
Return to citation in text:
[1]
Leow, D.; Lin, S.; Chittimalla, S. K.; Fu, X.; Tan, C.-H. Angew. Chem., Int. Ed.2008,47, 5641–5645. doi:10.1002/anie.200801378
Return to citation in text:
[1]
Badiola, E.; Fiser, B.; Gómez-Bengoa, E.; Mielgo, A.; Olaizola, I.; Urruzuno, I.; García, J. M.; Odriozola, J. M.; Razkin, J.; Oiarbide, M.; Palomo, C. J. Am. Chem. Soc.2014,136, 17869–17881. doi:10.1021/ja510603w


![[Graphic 1]](/bjoc/content/inline/1860-5397-12-222-i3.svg?max-width=637&scale=1.0)
![[1860-5397-12-222-i1]](/bjoc/content/inline/1860-5397-12-222-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-12-222-i2]](/bjoc/content/inline/1860-5397-12-222-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)