

Abstract
In the field of organosulfur chemistry, sulfenylating agents are an important key in C–S bond formation strategies. Among various organosulfur precursors, N-sulfenylsuccinimide/phthalimide derivatives have shown highly electrophilic reactivity for the asymmetric synthesis of many organic compounds. Hence, in this review article, we focus on the application of these alternative sulfenylating reagents in organic transformations.
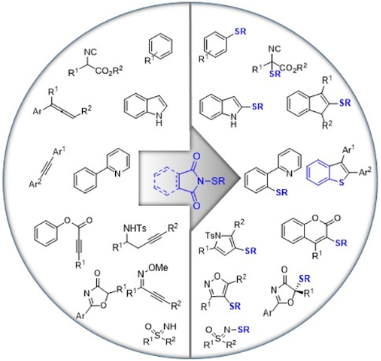
Graphical Abstract
Introduction
Sulfur-containing compounds are of high importance in organic synthesis, medicinal chemistry, and materials science [1-5]. For example, they are used in the treatment of cancer [6-8], inflammation [9-11], human immunodeficiency virus [12,13], Alzheimer’s and Parkinson’s diseases [14,15]. Scheme 1 shows selected examples of sulfur-containing pharmaceutical molecules. Considering the synthetic applications of sulfur-based compounds, a large number of researchers have noted that these scaffolds have promising potential for the research and development of new biomedicines.
![[1860-5397-19-106-i1]](/bjoc/content/inline/1860-5397-19-106-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Sulfur-containing bioactive molecules.
Scheme 1: Sulfur-containing bioactive molecules.
In the sulfenylation of organic compounds, the sulfenylating agents are important factors, and the commonly utilized chemicals include thiols [16-18], disulfides [19-22], sulfenyl halides [23-25], sulfonamides [26], sulfenate esters [27,28], and methyl(bismethylthio)sulfonium salts [29,30]. Among various organic molecules, aryl sulfides are recognized as functional materials and indispensable synthetic intermediates in drug discovery [31-33]. Because of their value, constructing C–S bonds has attracted significant attention via metal-catalyzed cross-coupling reactions and metal-free C–S bond formation [34-37]. Direct sulfenylation of the C–H bonds of unactivated aryls or aromatic sulfenylation by electrophilic aromatic substitution (SEAr) has also recently received attention [38].
In recent years, N-(aryl/alkylsulfenyl)succinimides and N-(arylsulfenyl)phthalimides have been widely employed as new alternative sulfenylating reagents in the field of organic synthesis. These compounds are readily accessible, safe, and more stable than toxic, unstable, and foul-smelling thiols. These electrophilic sulfur sources have deserved particular interest for the C–S bond formation via the reaction with various nucleophiles. Their preparation is usually a two-step procedure, involving a treatment of the thiol with sulfuryl chloride in the presence of Et3N and the addition of the resulting solution to a mixture of succinimide/phthalimide and Et3N in the next step [39,40].
According to the irreplaceable role of sulfur-based frameworks in materials science and the pharmaceutical area, there is a force for researchers to identify sustainable methodologies for efficient C–S bond coupling under mild reaction conditions for achieving these distinguished compounds. Recently, several reviews about sulfenylating reagents have been reported [41-43]. To the best of our knowledge there are no review articles focusing on the application of N-(sulfenyl)succinimides/phthalimides in sulfenylation reactions. In this context, we describe various sulfenylation reactions, such as electrophilic aromatic substitution, ring-opening, dehydrogenative cross-coupling, and direct sulfenylation reactions, which are classified into three categories: sulfenylation catalyzed by i) transition metal catalysts, ii) organocompound catalysts, and iii) catalyst-free sulfenylation.
Review
Sulfenylation of organic compounds by N-(sulfenyl)succinimides/phthalimides
Metal-catalyzed sulfenylation by N-(sulfenyl)succinimides/phthalimides
In 2012, Chen and co-workers found that in the reaction of N-(organothio)succinimides 1 and sodium sulfinates 2 using a Lewis acid in ionic liquids (ILs) and water as a green solvent system leads to the formation of thiosulfonates 3 (Scheme 2) [44]. Among different Lewis acid catalysts, such as Cu(OTf)2, Mg(OTf)2, Zn(OTf)2, Sc(OTf)3, Eu(OTf)3, and Yb(OTf)3, it was found that Sc(OTf)3 gave higher product yield. In addition, the combination of Sc(OTf)3/ILs displayed good recyclability in this transformation.
![[1860-5397-19-106-i2]](/bjoc/content/inline/1860-5397-19-106-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scandium-catalyzed synthesis of thiosulfonates.
Scheme 2: Scandium-catalyzed synthesis of thiosulfonates.
In 2014, Anbarasan and Saravanan succeeded in synthesizing various diaryl(alkyl) sulfides 5 through the sulfenylation of unactivated arenes 4 with an electrophilic sulfur reagent in the presence of a palladium catalyst (Scheme 3) [45]. In the second phase, dibenzothiophene derivatives 6 were obtained via subsequent intramolecular arylation of aryl sulfides by using the catalyst and the base. A catalytic cycle is shown in Scheme 4. Firstly, electrophilic Pd(TFA)2 generated from Pd(OAc)2 and TFA, which (by C–H functionalization of arene 4) led to intermediate II. Oxidative insertion of intermediate II into the N–S bond of 1 afforded intermediate III. Reductive elimination of Pd from III gave product 5 and species IV. Finaly, Pd(II) species were reproduced by ligand exchange to restart the next cycle (Scheme 4).
![[1860-5397-19-106-i3]](/bjoc/content/inline/1860-5397-19-106-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Palladium-catalyzed aryl(alkyl)thiolation of unactivated arenes.
Scheme 3: Palladium-catalyzed aryl(alkyl)thiolation of unactivated arenes.
![[1860-5397-19-106-i4]](/bjoc/content/inline/1860-5397-19-106-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Catalytic cycle for Pd-catalyzed aryl(alkyl)thiolation of unactivated arenes.
Scheme 4: Catalytic cycle for Pd-catalyzed aryl(alkyl)thiolation of unactivated arenes.
In 2014, Fu and co-workers described a facile method for the C–H thiolation of phenols 7 with 1-(substituted phenylthio)pyrrolidine-2,5-diones 1 using FeCl3 or BF3·OEt2 as a catalyst (Scheme 5) [46]. A wide variety of thiolated phenols 8 were produced under mild reaction conditions without using any base, ligand, or additive. For both substrates, 7 and 1 aryl rings containing electron-donating groups exhibited a higher reactivity than electron-withdrawing groups, and the thiolation occurred mainly at the para position to the hydroxy group in phenols.
![[1860-5397-19-106-i5]](/bjoc/content/inline/1860-5397-19-106-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Iron- or boron-catalyzed C–H arylthiation of substituted phenols.
Scheme 5: Iron- or boron-catalyzed C–H arylthiation of substituted phenols.
In 2016, the azidoarylthiation of various alkenes 9 by trimethylsilyl azide (10) and N-(organothio)succinimide 1 to the corresponding products containing ortho-sited azide and sulfide moieties 11 was performed by Fu et al. (Scheme 6) [47]. Iron(III) chloride was used as a catalyst for this coupling reaction without the need of any ligand and additive. Screening for other metal salts, such as Cu(OAc)2, Pd(OAc)2, AgOAc or CuI was not successful, although FeS·7H2O, FeS, Fe2(SO4)3·H2O, FeSO4, and Fe(acac)3 resulted in inferior chemical yields. Employment of 2,2,6,6-tetramethylpiperidinyl-1-oxyl (TEMPO) as a radical trapper inhibited the reaction, which proved that a radical process was involved. The reaction was initiated by a single electron transfer (SET) process from the sulfur atom to Fe3+ to generate Fe2+ and radical cation I. Subsequent cleavage of the N–S bond led to cation II and radical III. Interaction of III with Fe2+ regenerated the Fe3+ species and IV. At the same time, electrophilic addition of II to alkene 9 yielded intermediate V, which was subjected to the nucleophilic attack of TMSN3 to deliver product 11 (Scheme 7).
![[1860-5397-19-106-i6]](/bjoc/content/inline/1860-5397-19-106-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Iron-catalyzed azidoalkylthiation of alkenes.
Scheme 6: Iron-catalyzed azidoalkylthiation of alkenes.
![[1860-5397-19-106-i7]](/bjoc/content/inline/1860-5397-19-106-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Plausible mechanism for iron-catalyzed azidoalkylthiation of alkenes.
Scheme 7: Plausible mechanism for iron-catalyzed azidoalkylthiation of alkenes.
Tian and Chang et al. could synthesize 3‑sulfenylated coumarin compounds 13 by using N-sulfanylsuccinimides 1 under a Lewis acid catalysis system (Scheme 8) [48]. Additionally, oxidation of 3-sulfenylated coumarins utilizing (diacetoxyiodo)benzene (PIDA) and meta-chloroperbenzoic acid (m-CPBA) toward 3-sulfinylated and 3-sulfonylated product, respectively, were performed in this work. A plausible mechanism involves the treatment of 1 with BF3·Et2O toward cation I, which reacted with the C–C triple bond in 12 to give sulfonium intermediate II. Intramolecular nucleophilic addition of the phenoxy ring of 12 to the activated C–C triple bond afforded intermediate III, followed by deprotonation to deliver product 13 (Scheme 9). When substrate 12 had an OMe group on the phenoxy ring, ipso sulfenylcyclization, or sulfenylation of the phenoxy ring occurred according to the different positions of the OMe group. The preparation of α,α-bisthiofunctionalized butenolides through a bis-sulfenylation methodology was reported by Zhou and Yuan et al. [49]. For this purpose, they applied N-(alkyl(aryl)sulfanyl)succinimides or N-(phenylsulfanyl)phthalimides using a catalytic amount of Et3N. Moreover, mono-sulfenylation of α-methyl-γ-phenyl-substituted butenolide at α-position was carried out in the presence of Et3N as well as quinine organocatalyst and products were obtained in high yields.
![[1860-5397-19-106-i8]](/bjoc/content/inline/1860-5397-19-106-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: BF3·Et2O‑mediated electrophilic cyclization of aryl alkynoates.
Scheme 8: BF3·Et2O‑mediated electrophilic cyclization of aryl alkynoates.
![[1860-5397-19-106-i9]](/bjoc/content/inline/1860-5397-19-106-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Tentative mechanism for BF3·Et2O‑mediated electrophilic cyclization of aryl alkynoates.
Scheme 9: Tentative mechanism for BF3·Et2O‑mediated electrophilic cyclization of aryl alkynoates.
In addition to the use of N-(alkyl/arylthio)succinimides in the sulfenylation of organic compounds, N-(alkyl/arylthio)phthalimides are also considered good candidates for this purpose. In 2017, Sahoo and co-workers established a method for intramolecular annulation of N-(arylthio)phthalimides 14 and N-(arylthio)succinimides 1 with alkynes 15 in the presence of AlCl3 as an efficient Lewis acid catalyst (Scheme 10) [50]. In the procedure, oxidative cleavage of one S–N bond and 1,2-sulfur migration afforded π-conjugated 6-substituted 2,3-diarylbenzo[b]thiophenes 16. A plausible mechanism is shown in Scheme 11. The coordination of AlCl3 with the phthalimide/succinimide unit of 1 or 14, caused polarization of the S–N bond and produced an electrophilic intermediate I. Through the nucleophilic attack of the alkyne on I, cation II was generated, leaving Al-coordinated phthalimide/succinimide III. Finally, 4-endo-trig spirocyclization of II rendered the unstable intermediate IV, which underwent a ring expansion and 1,2-sulfur migration and subsequent deprotonation/aromatization to deliver product 16. Another work in the use of AlCl3 for cyclization of N‑arylpropynamides 17 with N‑sulfanylsuccinimides 1 was described by Gao and Zhou et al. (Scheme 12) [51]. Annulation in the presence of AlCl3 led to 3‑sulfenylquinolin-2-ones 18, while the addition of methanol into the reaction mixture gave 3-sulfenylazaspiro[4,5]trienones 19 as the target products. On the other hand, when free N–H alkynamides 20 were treated with N-sulfanylsuccinimides 1 in the presence of AlCl3, the coupling chlorinated product 21 was detected, which with POCl3 gave the cyclized product 22. Also, the synthesis of benzo[b]thieno[2,3-c]quinolone 24 as an anticancer molecule was demonstrated in this approach (Scheme 13).
![[1860-5397-19-106-i10]](/bjoc/content/inline/1860-5397-19-106-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Construction of 6-substituted benzo[b]thiophenes.
Scheme 10: Construction of 6-substituted benzo[b]thiophenes.
![[1860-5397-19-106-i11]](/bjoc/content/inline/1860-5397-19-106-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Plausible mechanism for construction of 6-substituted benzo[b]thiophenes.
Scheme 11: Plausible mechanism for construction of 6-substituted benzo[b]thiophenes.
![[1860-5397-19-106-i12]](/bjoc/content/inline/1860-5397-19-106-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: AlCl3‑catalyzed cyclization of N‑arylpropynamides with N‑sulfanylsuccinimides.
Scheme 12: AlCl3‑catalyzed cyclization of N‑arylpropynamides with N‑sulfanylsuccinimides.
![[1860-5397-19-106-i13]](/bjoc/content/inline/1860-5397-19-106-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Synthetic utility of AlCl3‑catalyzed cyclization of N‑arylpropynamides with N‑sulfanylsuccinimides.
Scheme 13: Synthetic utility of AlCl3‑catalyzed cyclization of N‑arylpropynamides with N‑sulfanylsuccinimides.
An intermolecular sulfenoamination of alkenes 9 with sulfonamides 25 as the nitrogen source and N-thiosuccinimides 1 as the sulfur source was reported by Gao and Liu et al. (Scheme 14) [52]. Highly regio- and diastereoselective β-sulfonylamino sulfides 26 were obtained from alkenes 9, N-thiosuccinimides 1, and sulfonamides 25 in the presence of 20 mol % BF3·Et2O. While the transformation in the presence of N-(2-bromophenylthio)succinimide 1’ and copper catalyst led to intermolecular sulfenoamination of alkenes and subsequent C–N coupling to produce dihydrobenzothiazine structures 27 in a one-pot manner. Furthermore, deprotection of the amine unit by K2CO3 and Na metal was performed in this work.
![[1860-5397-19-106-i14]](/bjoc/content/inline/1860-5397-19-106-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Sulfenoamination of alkenes with sulfonamides and N-sulfanylsuccinimides.
Scheme 14: Sulfenoamination of alkenes with sulfonamides and N-sulfanylsuccinimides.
In 2018, Anbarasan and Chaitanya developed an efficient approach for the C–H bond functionalization of aryl compounds containing a directing group using N-(thioaryl)phthalimides 14 in the presence of a palladium catalyst (Scheme 15) [53]. The thiolation occurred in the presence of Pd(OAc)2 and acetic acid (AcOH) as a Brønsted acid, whereas i(a)midation was achieved by using Pd(OAc)2 as catalyst and Cu(OAc)2 as a Lewis acid. A possible mechanism for this chemodivergent C–H activation is depicted in Scheme 16. First, Pd catalyzed the formation of palladacycle I. Oxidative addition of AcOH activated the N–S bond in II, which reacted with I to obtain IV, followed by C–S reductive elimination to give the thiolated product 30 or 31. On the other hand, the interaction of I with Cu(OAc)2 activated the N–S bond in III to afford IV, which was subjected to C–N reductive elimination to deliver the imidated product 32.
![[1860-5397-19-106-i15]](/bjoc/content/inline/1860-5397-19-106-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Lewis acid/Brønsted acid controlled Pd-catalyzed functionalization of aryl C(sp2)–H bonds.
Scheme 15: Lewis acid/Brønsted acid controlled Pd-catalyzed functionalization of aryl C(sp2)–H bonds.
![[1860-5397-19-106-i16]](/bjoc/content/inline/1860-5397-19-106-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Possible mechanism for Lewis acid/Brønsted acid controlled Pd-catalyzed functionalization of aryl C(sp2)–H bonds.
Scheme 16: Possible mechanism for Lewis acid/Brønsted acid controlled Pd-catalyzed functionalization of aryl C...
In 2018, an Fe-catalyzed carbosulfenylation and carboselenylation 33 of alkenes with N-(thio/seleno)phthalimides 14 was introduced by Lv and Li (Scheme 17) [54]. The use of Lewis acids, such as AlCl3, ZnCl2, InCl3, Fe(OTf)2 and Fe(acac)3 was not beneficial. However, BF3·OEt2, SnCl4, and TMSOTf resulted in good chemical yields. In the transformation, the selectivity of the endo or exo cyclization depended on the atom number of the chain between alkene and arene, leading to the formation of 6-, 7-, or 8-membered rings. In addition to N-(thio)phthalimides, benzenesulfenyl chloride as a sulfenylating source gave the target product in 93% yield. Knochel and co-workers found that copper acetate can catalyze the cross-coupling reaction between (hetero)aryl, alkyl and benzylic zinc halides 36 with N-thiophthalimides 14 (Scheme 18) [55]. Various metal catalysts, including CrCl2, CoCl2, NiCl2, MnCl2, FeCl2, Fe(acac)3 and copper salts such as Cu(OAc)2, CuBr2, CuBr, CuCl2, and CuCN·2LiCl were evaluated in this coupling reaction, in which Cu(OAc)2 showed highest product yields. Moreover, phthalimides with SCF3, SCN, and SePh groups also worked well in this approach. Because of the low reactivity of these phthalimides, 10 mol % of catalyst was required. Cross-coupling reaction of sulfoximines 44 with N‑(arylthio)succinimides 1 catalyzed by a nanomaterial containing hexagonal boron nitride (h-BN) and γ-cyclodextrin-supported copper(II) acetate (h-BN@γ-CD@Cu(OAc)2) was developed by Guo and Wu et al. (Scheme 19) [56]. Employment of a reusable heterogeneous nanomaterial, mild reaction conditions, avoiding the use of any additive, or base, and water/EtOH as a green solvent system were the advantages of this new method. N-Sulfenyl sulfoximines 45 were synthesized as coupling products in moderate to excellent yields.
![[1860-5397-19-106-i17]](/bjoc/content/inline/1860-5397-19-106-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: FeCl3-catalyzed carbosulfenylation of unactivated alkenes.
Scheme 17: FeCl3-catalyzed carbosulfenylation of unactivated alkenes.
![[1860-5397-19-106-i18]](/bjoc/content/inline/1860-5397-19-106-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Copper-catalyzed electrophilic thiolation of organozinc halides.
Scheme 18: Copper-catalyzed electrophilic thiolation of organozinc halides.
![[1860-5397-19-106-i19]](/bjoc/content/inline/1860-5397-19-106-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: h-BN@Copper(II) nanomaterial catalyzed cross-coupling reaction of sulfoximines and N‑(arylthio)succinimide.
Scheme 19: h-BN@Copper(II) nanomaterial catalyzed cross-coupling reaction of sulfoximines and N‑(arylthio)succ...
In 2019, Gao and Yang et al. disclosed a new protocol for the synthesis of 4‑aryl/alkylsulfenylisoxazoles 48 from sulfenylation of 2-alkyn-1-one O-methyloximes 46 with N-sulfenylsuccinimides 1 (Scheme 20) [57]. The transformation proceeded via an electrophilic cyclization and sulfenylation promoted by AlCl3. Dialkyl disulfides 47 were also well tolerated in this Lewis acid-mediated sulfenylation reaction in solvent-free conditions at room temperature. In the same year, a three-component reaction between highly substituted cyclopropanes 49, sulfonamides 25 and N-(arylthio)succinimides 1 or N-(arylseleno)succinimides 1’’ was developed under a Lewis acid catalysis system. This reaction involves ring-opening of the substituted cyclopropane 49, amination at the C1-site, and thiolation at the C3-site. In the transformation, sulfonamide acted as a nucleophile, chalcogensuccinimide as an electrophile, and cyclopropane as a zwitterion component (Scheme 21) [58].
![[1860-5397-19-106-i20]](/bjoc/content/inline/1860-5397-19-106-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: AlCl3‑mediated cyclization and sulfenylation of 2‑alkyn-1-one O‑methyloximes.
Scheme 20: AlCl3‑mediated cyclization and sulfenylation of 2‑alkyn-1-one O‑methyloximes.
![[1860-5397-19-106-i21]](/bjoc/content/inline/1860-5397-19-106-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Lewis acid-promoted 2-substituted cyclopropane 1,1-dicarboxylates with sulfonamides and N-(arylthio)succinimides.
Scheme 21: Lewis acid-promoted 2-substituted cyclopropane 1,1-dicarboxylates with sulfonamides and N-(arylthio...
In 2020, a Lewis acid-mediated cyclization of β,γ-unsaturated oximes 51 and hydrazones 52 with N-(arylsulfenyl)succinimide 1 and N-(arylseleno)succinimide 1’’ was extended for the formation of isoxazoles 53 and dihydropyrazoles 54 as products (Scheme 22) [59]. A credible pathway for the production of isoxazole 53 is illustrated in Scheme 23. The interaction of 1 with BF3·Et2O resulted in intermediate I that is in balance with I’. Cleavage of the N–S bond of I afforded cationic species PhS+ II, which activated the double bond of 51 to give the three-membered ring III. Afterwards, intermediate IV was formed by an intramolecular ring opening of III (path I) and presumably produced IV' by path II, which through deprotonation delivered products 53 and 53' respectively. In the meantime, another Lewis acid-promoted construction of 4-chalcogenylated pyrazoles 57 and 59 was carried out starting from α,β-alkynic hydrazones 55 (Scheme 24) [60]. In the procedure, α,β-alkynic hydrazones were subjected to S- or Se-electrophiles 56 and cyclization reaction. Additionally, NCS and ArSH produced the S-electrophile for the cyclization reaction with arylpropynal hydrazones. Also, the reaction of 1-(1,3-diphenylprop-2-yn-1-ylidene)-2-phenylhydrazine 58 as the substrate with N-sulfenylsuccinimides 1 afforded fully substituted pyrazoles 59 in up to 98% yield.
![[1860-5397-19-106-i22]](/bjoc/content/inline/1860-5397-19-106-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Lewis acid-mediated cyclization of β,γ-unsaturated oximes and hydrazones with N-(arylthio/seleno)succinimides.
Scheme 22: Lewis acid-mediated cyclization of β,γ-unsaturated oximes and hydrazones with N-(arylthio/seleno)su...
![[1860-5397-19-106-i23]](/bjoc/content/inline/1860-5397-19-106-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 23: Credible pathway for Lewis acid-mediated cyclization of β,γ-unsaturated oximes with N-(arylthio)succinimide.
Scheme 23: Credible pathway for Lewis acid-mediated cyclization of β,γ-unsaturated oximes with N-(arylthio)suc...
![[1860-5397-19-106-i24]](/bjoc/content/inline/1860-5397-19-106-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Synthesis of 4-chalcogenyl pyrazoles via chalcogenation/cyclization of α,β-alkynic hydrazones.
Scheme 24: Synthesis of 4-chalcogenyl pyrazoles via chalcogenation/cyclization of α,β-alkynic hydrazones.
In 2021, a solvent-controllable approach for the construction of 3-thiolated pyrroles 61 and pyrrolines 62 from propargylic tosylamides 60 and N-thiosuccinimides 1 was described by Gao′s group (Scheme 25) [61]. When AlCl3 as the Lewis acid catalyst and nitromethane as the solvent were used, a series of 3-thiolated pyrrole products 61 were detected, and 3-thiolated pyrrolines 62 were obtained by changing the reaction solvent to MeCN. Also, organic fluorophore compounds such as benzothienopyrrole and bis-thiolated boron dipyrromethene can be achieved from 3-thiolated pyrroles. Mechanistic studies showed that the oxidative species HNO and HCHO were generated through a Nef reaction in MeNO2 under acidic conditions. In the meantime, 1 was activated by AlCl3 to form sulfenium cation I, which induced an intramolecular cyclization of 60 to produce pyrroline 62. In MeCN solvent, 3-thiolated pyrroline 62 was stable and could be isolated, but in MeNO2, in the presence of the HNO species, the pyrroline structure could oxidize and aromatize to the pyrrole ring 61 (Scheme 26).
![[1860-5397-19-106-i25]](/bjoc/content/inline/1860-5397-19-106-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Controllable synthesis of 3-thiolated pyrroles and pyrrolines.
Scheme 25: Controllable synthesis of 3-thiolated pyrroles and pyrrolines.
![[1860-5397-19-106-i26]](/bjoc/content/inline/1860-5397-19-106-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: Possible mechanism for controllable synthesis of 3-thiolated pyrroles and pyrrolines.
Scheme 26: Possible mechanism for controllable synthesis of 3-thiolated pyrroles and pyrrolines.
In 2021, Anbarasan and co-workers were able to obtain a diverse range of sulfenylated products 64 in a Co-catalyzed C2-sulfenylation and C2,C3-disulfenylation of indole derivatives with N-(arylsulfenyl)succinimide 1 (Scheme 27) [62]. The reaction involves the formation of active cobalt species I from the interaction of KOAc with the cobalt pre-catalyst. Treatment of I with 63 resulted in the five-membered cobaltocycle complex II. Next, coordination of 1 to II gave III, followed by intramolecular nucleophilic trapping of the electrophilic SAr unit to furnish C2-sulfenylated product 65 and Co-complex IV. At last, active cobalt species I regenerated from IV in the presence of AcOH. It should be noted that when R = H, C2-sulfenylated product 65 may be sulfenylated via a thermal electrophilic aromatic substitution to provide C2,C3-disulfenylated product 66 (Scheme 28). In the same year, Sutherland and Dodds disclosed a protocol for the C–H bond thioarylation of electron-rich arenes 4 like anisoles, acetanilides, phenols, and N-heterocycles in the presence of Fe(III) Lewis acid and ionic liquid [BMIM]NTf2 as an effective catalysis system (Scheme 29) [63]. Kinetic studies in this cross coupling-reaction indicated that N-(arylthio)succinimides 1 with electron-deficient arene 4 undergoe thioarylation catalyzed by Fe(NTf2)3. Related molecules bearing an electron-rich arene showed an autocatalytic pathway that is enhanced due to the Lewis basic character of the final product.
![[1860-5397-19-106-i27]](/bjoc/content/inline/1860-5397-19-106-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: Co-catalyzed C2-sulfenylation and C2,C3-disulfenylation of indole derivatives.
Scheme 27: Co-catalyzed C2-sulfenylation and C2,C3-disulfenylation of indole derivatives.
![[1860-5397-19-106-i28]](/bjoc/content/inline/1860-5397-19-106-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: Plausible catalytic cycle for Co-catalyzed C2-sulfenylation and C2,C3-disulfenylation of indoles.
Scheme 28: Plausible catalytic cycle for Co-catalyzed C2-sulfenylation and C2,C3-disulfenylation of indoles.
![[1860-5397-19-106-i29]](/bjoc/content/inline/1860-5397-19-106-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 29: C–H thioarylation of electron-rich arenes by iron(III) triflimide catalysis.
Scheme 29: C–H thioarylation of electron-rich arenes by iron(III) triflimide catalysis.
Reddy and co-workers developed a simple method for the preparation of 1,2-thiosulfonylethenes 71 and 1,1-dithioethenes 69 in the presence of a nickel catalyst (Scheme 30) [64]. Various alkynyl bromides 68 as starting materials reacted with thiosulfonates 70 and N-arylthiosuccinimides 1 as thiolating reagents. 1,2-Thiosulfonylethenes 71 were obtained via vicinal thiosulfonylation. However, in the case of 1,1-dithioethenes 69, germinal disulfenylation occurred. In addition, 1,2-difunctionalization of indole-derived 1,1-bromoalkenes 72 was also investigated in the presence of Cs2CO3 without the need of a metal catalyst. The synthetic applicability of the procedure was demonstrated by a gram-scale synthesis of the 1,2-thiosulfonylethene product. A possible mechanism for the formation of 1,2-thiosulfonylethenes is shown in Scheme 31. Initially, homolytic cleavage of thiosulfonate 70 generated PhS· and PhSO2· radicals. The reduction of Ni(II) to Ni(0) in the presence of Cs2CO3 and the reaction with 68 formed alkynyl-Ni species I. Then, the PhS· radical reacted with I to generate alkenyl radical II, which can react with the PhSO2· radical to obtain intermediate III. Radical II underwent oxidation with PhSO2· to form alkenyl cation IV and PhSO2−. At last, H-abstraction from DMF delivered product 71 and the Ni(0) species to continue the catalytic cycle.
![[1860-5397-19-106-i30]](/bjoc/content/inline/1860-5397-19-106-i30.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 30: Difunctionalization of alkynyl bromides with thiosulfonates and N-arylthio succinimides.·
Scheme 30: Difunctionalization of alkynyl bromides with thiosulfonates and N-arylthio succinimides.·
![[1860-5397-19-106-i31]](/bjoc/content/inline/1860-5397-19-106-i31.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 31: Suggested mechanism for difunctionalization of alkynyl bromides with thiosulfonates and N-arylthio succinimides.
Scheme 31: Suggested mechanism for difunctionalization of alkynyl bromides with thiosulfonates and N-arylthio ...
In 2022, Gao and co-workers introduced a new protocol for the preparation of thioesters, acyl disulfides, ketones, and amides starting from N-thiohydroxy succinimide esters (NTSEs) 1’’’, which can serve as the acylthiolating and acylating source (Scheme 32) [65]. First, they synthesized a series of N-thiohydroxy succinimide esters by treating potassium thiolates with N-chlorosuccinimide in MeCN at room temperature for 20 min. N-Thiohydroxysuccinimide esters were obtained in up to high yields (21–83%). In the next phase, they performed the reaction of NTSEs with different nucleophiles, according to hard acyl and soft acylthio electrophilic sites contained in the NTSEs to selectively transfer the acyl or acylthio moieties. Arylboronic acids 74 and amines 76 were suitable for the acyl transfer and led to ketones 78 and amides 80 as the desired products. While, Grignard reagents 75 and thiols 77 acted as soft nucleophiles and resulted in thioesters 79 and acyl disulfides 81, respectively. It should be noted that the use of a palladium catalyst was essential for the cross-coupling reaction between 1’’’ and 74. Also, the presence of ZnCl2 could facilitate the cleavage of the N–S bond. In the case of amines and thiols, there was no need for a metal catalyst for the formation of S–N and S–S bonds. A plausible mechanism for the metal-catalyzed acylation and acylthiolation is illustrated in Scheme 33. Firstly, oxidative addition of palladium to the C–S bond of NTSE 1’’’ afforded intermediate I. The transmetalation from boron to palladium led to intermediate III, followed by reductive elimination to yield ketone 78. In the acylthiolation cycle, the azaphilic ZnCl2 activated NTSE 1’’’ via N–Zn coordination to facilitate the leaving ability of succinimide. Then, nucleophilic substitution of arylmagnesium bromide 75 to intermediate IV provided thioester 79.
![[1860-5397-19-106-i32]](/bjoc/content/inline/1860-5397-19-106-i32.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 32: Synthesis of thioesters, acyl disulfides, ketones, and amides by N-thiohydroxy succinimide esters.
Scheme 32: Synthesis of thioesters, acyl disulfides, ketones, and amides by N-thiohydroxy succinimide esters.
![[1860-5397-19-106-i33]](/bjoc/content/inline/1860-5397-19-106-i33.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 33: Proposed mechanism for metal-catalyzed selective acylation and acylthiolation.
Scheme 33: Proposed mechanism for metal-catalyzed selective acylation and acylthiolation.
In 2022, Gao and co-workers demonstrated bisulfenylation/cyclization of homopropargylic azides 82 with N-thiosuccinimides 1 in the presence of AlCl3 as the catalyst, 3,4-bisthiolated pyrroles 83 were obtained as the desired products in moderate to high yields (Scheme 34) [66]. The reaction involves the Lewis acid-catalyzed first thiolation and intramolecular cyclization of propargyl azides the removal of N2 and a proton. Subsequently, monothiolated perroles were subjected to the second thiolation process to prepare 3,4-bisthiolated pyrroles. Cyclic voltammetry and DFT calculations revealed that the 3,4-bisthiolated pyrroles 83 contained higher HOMO orbital energies, and lower band gaps compared to the unsubstituted parent 2,5-diphenylpyrrole.
![[1860-5397-19-106-i34]](/bjoc/content/inline/1860-5397-19-106-i34.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 34: AlCl3-catalyzed synthesis of 3,4-bisthiolated pyrroles.
Scheme 34: AlCl3-catalyzed synthesis of 3,4-bisthiolated pyrroles.
Organocatalyzed sulfenylation by N-(sulfenyl)succinimides/phthalimides
In 2004, direct sulfenylation of a series of aldehydes and ketones 84 with N-(phenylthio)phthalimide (14) by using an organocatalyst was reported by Wang and co-workers (Scheme 35) [67]. Several orgnocatalysts, such as piperidine, and pyrrolidine derivatives were evaluated for the coupling reaction, in which pyrrolidine trifluoromethanesulfonamide A was selected as the best catalyst for this purpose. It is noteworthy that the use of diphenyl disulfide as a sulfenylating agent was not effective in this protocol. N-(Aryl/alkylthio)phthalimide as an efficient sulfenylating reagent could also react with indoles to produce 3-thioindoles in the presence of 0.5 mol % of MgBr2, as a Lewis acid [68]. Moreover, sulfenylation of ketoximes and secondary nitro compounds toward N-arenesulfenyl ketimines occurred by applying N-(phenylthio)phthalimide [69].
![[1860-5397-19-106-i35]](/bjoc/content/inline/1860-5397-19-106-i35.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 35: α-Sulfenylation of aldehydes and ketones.
Scheme 35: α-Sulfenylation of aldehydes and ketones.
In 2011, Shi et al. developed a method for the sulfenylation of unsaturated alcohols 86 by N-(benzylthio)succinimide 1 access to tetrahydrofurans 87 and tetrahydropyrans 88 (Scheme 36) [70]. In this protocol, by controlling acid catalyst (camphorsulfonic acid (CSA) or trifluoromethanesulfonic acid (TfOH)), two different products were achieved and tetrahydrofurans 87 could be converted to tetrahydropyrans 88 by stereoselective rearrangement. In the same year, Zhu and Cheng et al. developed a convenient approach for the thiolation of β-keto phosphonates 89 by using N-(arylthio)phthalimides 14 under α,α-diaryl-ʟ-prolinols B organocatalytic system (Scheme 37) [71].
![[1860-5397-19-106-i36]](/bjoc/content/inline/1860-5397-19-106-i36.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 36: Acid-catalyzed sulfetherification of unsaturated alcohols.
Scheme 36: Acid-catalyzed sulfetherification of unsaturated alcohols.
![[1860-5397-19-106-i37]](/bjoc/content/inline/1860-5397-19-106-i37.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 37: Enantioselective sulfenylation of β-keto phosphonates.
Scheme 37: Enantioselective sulfenylation of β-keto phosphonates.
Sulfenylation of 3-aryloxindoles 91 with N-(arylsulfenyl)phthalimides 14 as the electrophilic sulfur reagents resulted in thiolated products 92 up to 99% ee, in the presence of quinidine as the organocatalyst (Scheme 38) [72]. For the study of enantioselectivity of products, different N-substituted oxindoles with H, Me, phenyl, and benzyl groups were investigated. As the size of N-protecting groups increased, the percentage of enantioselectivity decreased, where in the case of NH-oxindoles, the product was achieved with only 6% ee. Another sulfenylation at the 3-position of unprotected oxindoles with N-(phenylthio)phthalimide was reported by Feng et al. [73]. A chiral N,N′-dioxide-Sc(OTf)3 complex as a catalyst and a Brønsted base were applied in the procedure. It is interesting to note that in such a method, sulfenylation of NH-oxindoles resulted in the thiolated products with excellent enantioselectivities (up to 99% ee).
![[1860-5397-19-106-i38]](/bjoc/content/inline/1860-5397-19-106-i38.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 38: Organocatalyzed sulfenylation of 3‑substituted oxindoles.
Scheme 38: Organocatalyzed sulfenylation of 3‑substituted oxindoles.
In 2013, sulfenylation and chlorination of β-ketoesters 93, and 95 with N-(arylthio)phthalimide 14 and N-chlorophthalimide (96) under phase-transfer conditions was developed by Maruoka and co-workers (Scheme 39) [74]. The presence of chiral bifunctional catalysts C and D with the amide, or sulfonamide moieties could improve the enantioselectivity. Also, the heterogeneous medium coming from H2O and toluene was beneficial for the progress of the transformation.
![[1860-5397-19-106-i39]](/bjoc/content/inline/1860-5397-19-106-i39.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 39: Sulfenylation and chlorination of β-ketoesters.
Scheme 39: Sulfenylation and chlorination of β-ketoesters.
In 2014, Denmark and Chi successfully synthesized a wide variety of pyrrolidines 99, piperidines 100, and azepanes via intramolecular sulfenoamination of olefins 98 (Scheme 40) [75]. The reduction of endo to exo ratio was either related to the electron density of the alkene or the steric effect of a substituent. The tether lengths could affect the cyclization. For example, the two-carbon-tethered substrate completely showed endo selectivity, while the four-carbon-tethered substrate exclusively led to azepane. A possible mechanism was suggested for this Lewis base catalysis system. Methanesulfonic acid (MsOH) activated reagent 14, which coordinated with the Lewis base (S)-E, to form complex I. Then, the transfer of the sulfenium ion to the alkene resulted in chiral thiiranium ion II. Capture of the thiiranium ion by the tosylamide and deprotonation led to the final product 99 or 100 (Scheme 41). Through the coupling reaction of N-(aryl/alkylthio)succinimides 1 with 5H-oxazol-4-ones 101 in the presence of an organocatalyst named cinchona alkaloid-derived squaramide F, a series of α-sulfenylated products 102 were obtained in moderate to excellent yields with good to excellent enantioselectivities (Scheme 42) [76]. It should be noted that the authors did not define the exact role of the organocatalyst in the reaction mechanism.
![[1860-5397-19-106-i40]](/bjoc/content/inline/1860-5397-19-106-i40.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 40: Intramolecular sulfenoamination of olefins.
Scheme 40: Intramolecular sulfenoamination of olefins.
![[1860-5397-19-106-i41]](/bjoc/content/inline/1860-5397-19-106-i41.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 41: Plausible mechanism for intramolecular sulfenoamination of olefins.
Scheme 41: Plausible mechanism for intramolecular sulfenoamination of olefins.
![[1860-5397-19-106-i42]](/bjoc/content/inline/1860-5397-19-106-i42.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 42: α-Sulfenylation of 5H-oxazol-4-ones.
Scheme 42: α-Sulfenylation of 5H-oxazol-4-ones.
Transition-metal-free C–H sulfenylation of electron-rich arenes 103 by N-(alkyl/arylthio)succinimides 1 led to aryl sulfides 104 (Scheme 43) [77]. The cross-coupling reaction involves protonation of the succinimide moiety by trifluoroacetic acid (TFA) to create electrophilic thio intermediate I. Nucleophilic attack of arene 103 on I led to target product 104. Also, TFA-catalyzed C–H sulfenylation at the C2-position of protected and unprotected indoles 105 to form 2-thioindoles 106 (Scheme 44) [78]. The reaction initiated with TFA-promoted electrophilic addition of 1 to 105 towards C3-sulfenylated indole I, which was protonated by TFA, led to intermediate II. Then, CF3CO2SR, which was produced in the previous step, as a sulfenylating reagent, reacted with I to form the 3,3-bis-sulfide indolenium III. The migration of a sulfide group to the C2-site of indole, generated 2,3-disubstituted indole V. Protonation of V resulted in indolenium intermediate VI. Finally, desulfenylation of VI by anion CF3CO2, afforded 2-thioindole 107 (Scheme 45).
![[1860-5397-19-106-i43]](/bjoc/content/inline/1860-5397-19-106-i43.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 43: Metal-free C–H sulfenylation of electron-rich arenes.
Scheme 43: Metal-free C–H sulfenylation of electron-rich arenes.
![[1860-5397-19-106-i44]](/bjoc/content/inline/1860-5397-19-106-i44.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 44: TFA-promoted C–H sulfenylation indoles.
Scheme 44: TFA-promoted C–H sulfenylation indoles.
![[1860-5397-19-106-i45]](/bjoc/content/inline/1860-5397-19-106-i45.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 45: Proposed mechanism for TFA-promoted C–H sulfenylation indoles.
Scheme 45: Proposed mechanism for TFA-promoted C–H sulfenylation indoles.
The enantioselective synthesis of a broad spectrum of 3-thio-3-pyrrolyloxindoles 109 and 3-seleno-3-pyrrolyloxindoles 110 via sulfenylation and selenenylation of 3-pyrrolyloxindoles 108 was described by Yuan′s research group in 2015 (Scheme 46) [79]. By testing several alkaloids as organocatalysts for the transformation, cinchonidine G proved to be the best catalyst for C–H sulfenylation and selenenylation of substrates in toluene at −20 or 0 °C. The reaction occurred in shorter times in the presence of N-(arylsulfanyl)succinimide, while the coupling reaction using N-(alkylsulfanyl)succinimide and N-(heteroarylsulfanyl)succinimide longed several days. The gram-scale synthesis demonstrated the practicality of this method. In the same year, sulfenylation of different types of S-based nucleophiles 111 and 113 with N-(organosulfanyl)succinimide 1 catalyzed by dihydroquinine as an easily available organocatalyst was reported by Zhou et al. (Scheme 47) [80]. This is the first example of the preparation of chiral dithioketals. The presence of the OH group was essential in dihydroquinine H. By changing OH into a OMe group, the enantioselectivity and the product yield were reduced. Although, the authors did not further explain the catalytic pathway.
![[1860-5397-19-106-i46]](/bjoc/content/inline/1860-5397-19-106-i46.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 46: Organocatalyzed sulfenylation and selenenylation of 3-pyrrolyloxindoles.
Scheme 46: Organocatalyzed sulfenylation and selenenylation of 3-pyrrolyloxindoles.
![[1860-5397-19-106-i47]](/bjoc/content/inline/1860-5397-19-106-i47.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 47: Organocatalyzed sulfenylation of S-based nucleophiles.
Scheme 47: Organocatalyzed sulfenylation of S-based nucleophiles.
The use of organocatalysts in sulfenylation of N-heterocyclic compounds was investigated by Gustafson′s group in 2017 (Scheme 48) [81]. In their work, a series of conjugate Lewis base Brønsted acid organocatalysts were evaluated for sulfenylation on C3, or C2 position of N-heterocycles 115, including indoles, peptides, pyrrole, and 1-methyl-1H-pyrrolo[2,3-b]pyridine. The authors hypothesized a mechanism for the activation of N-sulfanylsuccinimides 1 or 14 by conjugate Lewis base Brønsted acid catalyst I, leading to the formation of an electrophilic sulfenium source (Scheme 49). The use of dimeric cinchona alkaloid J as another organocatalyst for α-sulfenylation of deconjugated butyrolactam substrates 117 with N-(arylsulfanyl)succinimides 1 demonstrated in Mukherjee′s work (Scheme 50) [82]. In the method, functionalized γ-lactams 102 were produced in aqueous media with high enantioselectivities. However, N-(alkylsulfanyl)succinimides and α-isobutyl containing butyrolactam did not work in this reaction. Another work by Denmark on intramolecular sulfenylation of alkenes 119 with phenols by using N-(arylthio)phthalimide 14 as a sulfur source was reported in the same year (Scheme 51) [83]. Benzopyrans 120 and benzoxepins were obtained in the presence of a Lewis base catalyst in good to high yields.
![[1860-5397-19-106-i48]](/bjoc/content/inline/1860-5397-19-106-i48.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 48: Conjugate Lewis base Brønsted acid-catalyzed sulfenylation of N-heterocycles.
Scheme 48: Conjugate Lewis base Brønsted acid-catalyzed sulfenylation of N-heterocycles.
![[1860-5397-19-106-i49]](/bjoc/content/inline/1860-5397-19-106-i49.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 49: Mechanism for activation of N-sulfanylsuccinimide by conjugate Lewis base Brønsted acid catalyst.
Scheme 49: Mechanism for activation of N-sulfanylsuccinimide by conjugate Lewis base Brønsted acid catalyst.
![[1860-5397-19-106-i50]](/bjoc/content/inline/1860-5397-19-106-i50.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 50: Sulfenylation of deconjugated butyrolactams.
Scheme 50: Sulfenylation of deconjugated butyrolactams.
![[1860-5397-19-106-i51]](/bjoc/content/inline/1860-5397-19-106-i51.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 51: Intramolecular sulfenofunctionalization of alkenes with phenols.
Scheme 51: Intramolecular sulfenofunctionalization of alkenes with phenols.
In 2018, Liang and Chen et al. extended 1,3-difunctionalizations of a series of Morita–Baylis–Hillman carbonates from isatins by using a Lewis base catalytic system (Scheme 52) [84]. Screening several organocatalysts showed that the 1,3-oxo-ethynylation of starting materials with silylethynyl-1,2-benziodoxol-3(1H)-ones 123 was obtained by using catalyst N, while 1,3-aminosulfenylation with N-(aryl/alkylthio)imides 1 or 14 occurred in the presence of catalyst L. Meanwhile, Zhou and Chen′s research team was able to synthesize a broad range of enantioenriched naphthalenone structures 126 by utilizing another organocatalyst (Scheme 53) [85]. In the procedure, β‑naphthols 125 reacted with N-(arylthio)succinimide 1 or N-(arylthio)phthalimide 14 as the sulfenylating reagents in the presence of cinchona-derived thiourea O as a catalyst to afford the corresponding chiral naphthalenone products 126 under mild reaction conditions.
![[1860-5397-19-106-i52]](/bjoc/content/inline/1860-5397-19-106-i52.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 52: Organocatalytic 1,3-difunctionalizations of Morita–Baylis–Hillman carbonates.
Scheme 52: Organocatalytic 1,3-difunctionalizations of Morita–Baylis–Hillman carbonates.
![[1860-5397-19-106-i53]](/bjoc/content/inline/1860-5397-19-106-i53.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 53: Organocatalytic sulfenylation of β‑naphthols.
Scheme 53: Organocatalytic sulfenylation of β‑naphthols.
Another work from Anbarasan and Chaitanya on the use of N-(arylsulfanyl)succinimide 1 and N-(arylseleno)succinimide 1’’ in an oxychalcogenation process was reported in 2018 (Scheme 54) [86]. In this method, they succeeded in applying methanesulfonic acid (MsOH) as a promoter for oxythiolation and oxyselenation of o-vinylanilides 127 through the formation of three-membered cyclic sulfonium ion II followed by ring-opening of sulfonium ion and intramolecular cyclization. The use of a Lewis base/Brønsted acid catalysis system for the sulfenylation of aromatic substrates 4 was reported by Gustafson et al. (Scheme 55) [87]. In the method, catalyst P acted as a Lewis base, where TfOH acted as a Brønsted acid. It is worth noting that coupling reactions without Lewis base catalyst P occurred in much lower yields. The mechanistic investigations showed that electron-rich sulfenyl groups can participate in an autocatalytic mechanism due to their Lewis basic nature. However, the electron-poor ones exhibited less autocatalysis effect requiring the use of the Lewis base catalyst P.
![[1860-5397-19-106-i54]](/bjoc/content/inline/1860-5397-19-106-i54.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 54: Acid-promoted oxychalcogenation of o‑vinylanilides with N‑(arylthio/arylseleno)succinimides.
Scheme 54: Acid-promoted oxychalcogenation of o‑vinylanilides with N‑(arylthio/arylseleno)succinimides.
![[1860-5397-19-106-i55]](/bjoc/content/inline/1860-5397-19-106-i55.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 55: Lewis base/Brønsted acid dual-catalytic C–H sulfenylation of aryls.
Scheme 55: Lewis base/Brønsted acid dual-catalytic C–H sulfenylation of aryls.
In 2019, Denmark and Panger disclosed a novel method for the preparation of γ‑lactams 133 through the reaction of alkenes 132 with N-thiophthalimides 14 in the presence of Lewis base organocatalysts (Scheme 56) [88]. In this procedure, the cyclized products were obtained via the activation of the sulfur electrophile by a Lewis base to generate the thiiranium ion intermediate from the β,γ-unsaturated sulfonyl carboxamide. The attack of the sulfonamide nitrogen atom on this intermediate led to intramolecular cyclization. In 2020, electrophilic cyclization of allylic amides 134 using N-(phenylthio)succinimide 1 in the presence of camphorsulfonic acid (CSA) as a Brønsted acid and tetrabutylammonium chloride (TBAC) led to 5-[(phenylthio)methyl]oxazoline scaffolds 135 (Scheme 57) [89]. Combination of CSA/TBAC formed an efficient activator system for this sulfenylation/intramolecular cyclization.
![[1860-5397-19-106-i56]](/bjoc/content/inline/1860-5397-19-106-i56.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 56: Lewis base-catalyzed sulfenoamidation of alkenes.
Scheme 56: Lewis base-catalyzed sulfenoamidation of alkenes.
![[1860-5397-19-106-i57]](/bjoc/content/inline/1860-5397-19-106-i57.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 57: Cyclization of allylic amide using a Brønsted acid and tetrabutylammonium chloride.
Scheme 57: Cyclization of allylic amide using a Brønsted acid and tetrabutylammonium chloride.
In the same year, Zhao and co-workers reported the thiocarbocyclization of allenes 136 with N-(organothio)succinimides 1 as electrophilic aryl/alkylthio reagents for the assembly of indene-based sulfide molecules 137 (Scheme 58) [90]. The Lewis basicity nature of PhSePh as a catalyst and the presence of Lewis acid TMSOTf improved the chemical yields. It is interesting to note that the reaction carried out at a lower temperature because of the high reactivity of allene 136. When the reaction was performed at room temperature, no desired product was observed, and performing the reaction at 0 °C enhanced the regioselectivity but still in low yield. By further lowering the temperature to −60 °C the yield was increased. The authors suggested a possible mechanism for this organoselenium-catalyzed cyclization transformation involving activation of the electrophilic sulfur reagent by PhSePh with the assistance of TMSOTf to form transition state I. Intermediate II formed through capturing of sulfonium by selenium. Then, II reacted with 136 to give regioselective cyclic thiiranium ion III. Nucleophilic attack of the aromatic ring on the thiiranium ion moiety furnished products 137 and reproduced the selenide catalyst (Scheme 59).
![[1860-5397-19-106-i58]](/bjoc/content/inline/1860-5397-19-106-i58.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 58: Catalytic electrophilic thiocarbocyclization of allenes with N-thiosuccinimides.
Scheme 58: Catalytic electrophilic thiocarbocyclization of allenes with N-thiosuccinimides.
![[1860-5397-19-106-i59]](/bjoc/content/inline/1860-5397-19-106-i59.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 59: Suggested mechanism for electrophilic thiocarbocyclization of allenes with N-thiosuccinimides.
Scheme 59: Suggested mechanism for electrophilic thiocarbocyclization of allenes with N-thiosuccinimides.
Zhao and co-workers found that N-thiosuccinimides are also suitable promoters for the enantioselective hydrothiolation of alkenes at low temperatures (Scheme 60) [91]. The synthesis of chiral sulfides up to 97% ee was achieved in this method. A wide range of cyclic alkenes 138 and acyclic alkenes 52 and 9 were smoothly tolerated in this organocatalysis strategy. According to the proposed mechanism, initially, the organocatalyst activated the electrophilic sulfur species to form intermediate I with the assistance of the Lewis acid. Intermediate I reduced by Et3SiH 139 to give thiol. Through the reaction of thiol with I, disulfide as a byproduct was formed, and intermediate II was generated by the reaction of I with 138. Product 140 was obtained via direct hydride reduction of II by silane. On the other hand, most of II were converted to intermediate III, which underwent hydride reduction to render product 140 (Scheme 61). Another organocatalysis system was disclosed by Liu and co-workers for sulfenylation of α-fluoro-β-ketoamides 143 and azlactones 145 (Scheme 62) [92]. Besides α-fluoro-β-ketoamides, α-chloro-substituted ketoamide was also tolerated well in this transformation. Screening several chiral guanidines as the bifunctional catalyst revealed that these organocatalysts were suitable for the synthesis of α-fluoro/chloro-α-sulfenyl-β-ketoamides 144 and azlactone 146 skeletons. The presence of two heteroatom-bearing tetrasubstituted chiral carbon centers in a one-step fashion, avoiding the use of the heavy metal catalysts, and the performance of the reaction at ambient temperature are the prominent features of the protocol.
![[1860-5397-19-106-i60]](/bjoc/content/inline/1860-5397-19-106-i60.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 60: Chiral chalcogenide-catalyzed enantioselective hydrothiolation of alkenes.
Scheme 60: Chiral chalcogenide-catalyzed enantioselective hydrothiolation of alkenes.
![[1860-5397-19-106-i61]](/bjoc/content/inline/1860-5397-19-106-i61.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 61: Proposed mechanism for chalcogenide-catalyzed enantioselective hydrothiolation of alkenes.
Scheme 61: Proposed mechanism for chalcogenide-catalyzed enantioselective hydrothiolation of alkenes.
![[1860-5397-19-106-i62]](/bjoc/content/inline/1860-5397-19-106-i62.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 62: Organocatalytic sulfenylation for synthesis a diheteroatom-bearing tetrasubstituted carbon centre.
Scheme 62: Organocatalytic sulfenylation for synthesis a diheteroatom-bearing tetrasubstituted carbon centre.
Sahoo and co-workers found that sulfenylation of yne-tethered ynamide 147 with N-thiosuccinimides 1 was possible in the presence of only methanesulfonic acid in dichloromethane at room temperature (Scheme 63) [93]. The electrophilic activation of propargylalkyne 147 generated in situ a sulfonium cation 1-I. Afterwards, 6-endo-dig cyclization of polarized ketene-N,O-acetal to the alkyne β-carbon and trapping of the sulfonium cation at the alkyne-α-carbon afforded 5-(arylthio)-3,6-dihydropyridin-2(1H)-one 148. The coordination of a sulfonium electrophile to the C–C triple bond of 1-I occurred through cyclopropyl intermediate 1-I. The conversion of 1-I to 2-II was confirmed by mechanistic studies due to the stability of the benzyl carbocation, followed by 6-endo-dig cyclization. In this method, toxic transition metal catalysts, oxidants, or bases are not used, which made it economically and environmentally reliable.
![[1860-5397-19-106-i63]](/bjoc/content/inline/1860-5397-19-106-i63.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 63: Thiolative cyclization of yne-ynamides.
Scheme 63: Thiolative cyclization of yne-ynamides.
In 2023, Gao et al. developed a metal-free procedure for the synthesis of functionalized alkynyl disulfides 149 and acyl disulfides 151 under acid catalysis (Scheme 64) [94]. In this regard, they used N-alkynylthiophthalimides in the reaction with thiols to make a series of bioactive disulfides. Various simple thiols, cystines, peptides, drugs and saccharides reacted smoothly with N-alkynylthiophthalimides in the presence of TFA as a catalyst. Also, aliphatic and aromatic thiols reacted with N-alkynylthiophthalimide and sulfoxide 149 to obtain acyl disulfides 151 through alkynylthiolation and hydrative oxyarylation in the presence of TMSOTf.
![[1860-5397-19-106-i64]](/bjoc/content/inline/1860-5397-19-106-i64.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 64: Synthesis of alkynyl and acyl disulfides from reaction of thiols with N-alkynylthio phthalimides.
Scheme 64: Synthesis of alkynyl and acyl disulfides from reaction of thiols with N-alkynylthio phthalimides.
Catalyst-free sulfenylation by N-(sulfenyl)succinimides/phthalimides
In 2015, oxysulfenylation of styrene derivatives 9 utilizing 1-(arylthio)pyrrolidine-2,5-diones 1 and alkyl/benzyl alcohols 86 toward β-alkoxy sulfides was developed by Fu et al. (Scheme 65) [95]. In this metal-free method, diverse β-alkoxy sulfides were synthesized without the need to any catalyst, or additive. The reaction proceeded through the formation of carbonium ion intermediate I, which underwent electrophilic addition of alcohol to provide product 152. In the meantime, N-(arylthio)succinimide 1 as a thiolating reagent was used by another research team for the arylthiolation of arylamines 76 in acetonitrile as a solvent under metal-free conditions (Scheme 66) [96]. A broad spectrum of mono-, or diarylthiolated anilines 153 was obtained in low to excellent yields. Arylthiolation occurred predominantly at the para-position to the amino group, and when the para-position of aniline was occupied by another group, ortho-substituted products were identified.
![[1860-5397-19-106-i65]](/bjoc/content/inline/1860-5397-19-106-i65.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 65: Oxysulfenylation of alkenes with 1-(arylthio)pyrrolidine-2,5-diones and alcohols.
Scheme 65: Oxysulfenylation of alkenes with 1-(arylthio)pyrrolidine-2,5-diones and alcohols.
![[1860-5397-19-106-i66]](/bjoc/content/inline/1860-5397-19-106-i66.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 66: Arylthiolation of arylamines with (arylthio)-pyrrolidine-2,5-diones.
Scheme 66: Arylthiolation of arylamines with (arylthio)-pyrrolidine-2,5-diones.
Fu′s research group established an isothiocyanatoalkylthiation of styrenes 9 in the presence of isothiocyanate 154 and N-(organothio)succinimides 1 under catalyst-free conditions (Scheme 67) [97]. The reaction proceeded through the formation of a three-membered cyclic intermediate II by the cleavage of 1 under thermal conditions. Between the nitrogen or sulfur atom in TMSNCS, electrophilic attack of nitrogen on II led to thermodynamically favored product 155.
![[1860-5397-19-106-i67]](/bjoc/content/inline/1860-5397-19-106-i67.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 67: Catalyst-free isothiocyanatoalkylthiation of styrenes.
Scheme 67: Catalyst-free isothiocyanatoalkylthiation of styrenes.
In 2017, sulfenylation of (E)-β-chlorovinyl ketones 156 using N-(alkyl/arylthio)phthalimides 14 access to 3,4-dimercaptofuran skeletons 159 was presented by Kim and Oh et al. (Scheme 68) [98]. In the first step, by using Et3N and t-BuOK in the reaction of (E)-β-chlorovinyl ketones 156 and N-(phenylthio)phthalimide 14, a series of α,γ-dithioallenyl ketones 157 and α,α-dithiopropargyl ketones 158 were obtained in a different ratio. In the second phase, by adding copper chloride as a catalyst in the reaction medium, 3,4-dimercaptofurans 159 were formed via 1,2-sulfur migration.
![[1860-5397-19-106-i68]](/bjoc/content/inline/1860-5397-19-106-i68.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 68: Sulfenylation of (E)-β-chlorovinyl ketones toward 3,4-dimercaptofurans.
Scheme 68: Sulfenylation of (E)-β-chlorovinyl ketones toward 3,4-dimercaptofurans.
In 2018, Shen′s research team disclosed a new protocol for 1,2-thiofunctionalization of arylalkenes 160 with N-arylthiophthalimide 14 and various nucleophiles, including aryl ethers, carboxylic acids, indoles, and pyrroles in the presence of HCl (Scheme 69) [99]. The procedure utilized no toxic metal catalyst, or additive, which made it economically and environmentally reliable. According to the mechanism, two pathways occurred after the formation of intermediate I by the reaction of 14 with HX. In path I, intermediate I reacted with alkene 160 to give intermediate II, which underwent a nucleophilic attack of 161 to give the product 162 and regenerated HX. In path II, I reacted with nucleophile 161 to produce a byproduct, phthalimide, and HX (Scheme 70). The coupling reaction was influenced by nucleophilic properties and the steric effect of the nucleophile reagents. In the same year, the treatment of amines with N-thiophthalimides led to sulfenamides promoted by 2-ethoxyethanol under microwave irradiation [100]. Alkylamines, such as morpholine, cyclohexylamine, pyrrolidine, and tert-butylamine were participated in this coupling process. All reactions occurred in a shorter time with higher chemical yields compared to the traditional heating methods.
![[1860-5397-19-106-i69]](/bjoc/content/inline/1860-5397-19-106-i69.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 69: HCl-promoted intermolecular 1, 2-thiofunctionalization of aromatic alkenes.
Scheme 69: HCl-promoted intermolecular 1, 2-thiofunctionalization of aromatic alkenes.
![[1860-5397-19-106-i70]](/bjoc/content/inline/1860-5397-19-106-i70.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 70: Possible mechanism for HCl-promoted 1,2-thiofunctionalization of aromatic alkenes.
Scheme 70: Possible mechanism for HCl-promoted 1,2-thiofunctionalization of aromatic alkenes.
In the meanwhile, Qiu and Xu et al. reported the coupling reaction between diazo compounds 163 and N-sulfenylsuccinimides 1 under catalyst-, base-, and additive-free conditions (Scheme 71) [101]. The reaction proceeded via a radical pathway, in which a free carbene was generated under heating, followed by the formation of ylide, N–S bond cleavage, and C–N bond formation along with the release of N2.
![[1860-5397-19-106-i71]](/bjoc/content/inline/1860-5397-19-106-i71.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 71: Coupling reaction of diazo compounds with N-sulfenylsuccinimides.
Scheme 71: Coupling reaction of diazo compounds with N-sulfenylsuccinimides.
In 2019, Sun and co-workers introduced an unprecedented method for the synthesis of isothiourea derivatives via the activation of diaryl/alkyl disulfides 47 with N-halosuccinimides in the presence of TEMPO, followed by insertion of an isocyanide molecule 166 or other nucleophiles 161 (Scheme 72) [102]. By studying the spectroscopic evidence, the authors found that both sulfenyl halide 165 and N-sulfenylsuccinimide 1 intermediates were involved in the reaction. Removal of TEMPO as a radical initiator from the reaction mixture did not result in product formation so, it seems that the reaction moved through a radical route for the formation of sulfenyl halide I, and N-sulfenylsuccinimide II. The use of azobisisobutyronitrile (AIBN) instead of TEMPO also resulted in 85% yield of the product, while benzoyl peroxide (BPO) gave a low yield. Various nucleophiles 161, including ammonia, alkylamines, hydrazines, alcohols and alkoxides, indole, N-alkylpyrrole, N-substituted anilines, PhSH, and PhMgBr worked well under these conditions. Asymmetric thiolation of 4-substituted pyrazolone derivatives with N-thiophthalimides catalyzed by 1 mol % of chiral iminophosphorane organocatalyst was carried out under mild conditions [103]. Solvent control in the procedure can affect the yield of products due to the solubility of the catalysts. Various solvents, such as acetone, ethyl acetate, tetrahydrofuran, methanol, toluene, hexane, and n-pentane were employed, in which the products in non-polar hydrocarbon solvents like hexane and n-pentane were obtained in excellent efficiency and enantioselectivity.
![[1860-5397-19-106-i72]](/bjoc/content/inline/1860-5397-19-106-i72.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 72: Multicomponent reactions of disulfides with isocyanides and other nucleophiles.
Scheme 72: Multicomponent reactions of disulfides with isocyanides and other nucleophiles.
Song et al. found that the chemoselective α-sulfenylation and β-thiolation of α,β-unsaturated carbonyl compounds 168 can be achieved with N-thiophthalimides 14 and diaryl disulfides 47, respectively (Scheme 73) [104]. They remarked that the presence of B2pin2 was essential in the coupling reaction of disulfides with α,β-unsaturated carbonyl compounds 168. The sulfenylation involved a 1,4-addition of the phthalimide anion to the β-carbon of chalcone, followed by electrophilic sulfur attack and deprotonation. In the thiolation, in situ formation of thiophenol occurred, followed by thio-Michael addition of chalcone with thiophenol. N-Calcogenophthalimide also can be used to prepare thiophosphates, thiophosphinates and selenophosphates by reaction with the P(O)H moieties of H-phosphonates [105].
![[1860-5397-19-106-i73]](/bjoc/content/inline/1860-5397-19-106-i73.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 73: α-Sulfenylation and β-sulfenylation of α,β-unsaturated carbonyl compounds.
Scheme 73: α-Sulfenylation and β-sulfenylation of α,β-unsaturated carbonyl compounds.
Conclusion
To date, both metal-catalyzed and organocatalyzed C–S bond formations have been widely expanded. In particular, organocatalytic methodologies are effective for direct construction of stereogenic carbon centers bearing a sulfur atom. Although, significant efforts have been made to form enantioselective C–S bonds, the direct sulfenylation with more green, economical, and environmentally friendly sulfenylating reagents remains a challenge for organic chemists. N-(Sulfenyl)succinimides/phthalimides as new alternative sulfenylating reagents can meet this demand. In this context, we observed that most of the reactions have used unactivated C–H bonds, such as C(sp2)–H and C(sp3)–H bonds. In some reactions, chiral organocatalysts catalyzed asymmetric sulfenylation processes. In most cases, there is no need to use a metal catalyst, base, or additive. N-(Sulfenyl)succinimide/phthalimide acted as an active electrophilic sulfur source, acted in the reaction mechanisms. However, mechanistic studies need further exploration to define a valid reaction pathway. Therefore, we believe that the use of N-(sulfenyl)succinimide/phthalimide in chemical syntheses will be widely seen in the future.
References
-
Beletskaya, I. P.; Ananikov, V. P. Chem. Rev. 2011, 111, 1596–1636. doi:10.1021/cr100347k
Return to citation in text: [1] -
Liu, L.; Stelmach, J. E.; Natarajan, S. R.; Chen, M.-H.; Singh, S. B.; Schwartz, C. D.; Fitzgerald, C. E.; O'Keefe, S. J.; Zaller, D. M.; Schmatz, D. M.; Doherty, J. B. Bioorg. Med. Chem. Lett. 2003, 13, 3979–3982. doi:10.1016/j.bmcl.2003.08.059
Return to citation in text: [1] -
Fontecave, M.; Ollagnier-de-Choudens, S.; Mulliez, E. Chem. Rev. 2003, 103, 2149–2166. doi:10.1021/cr020427j
Return to citation in text: [1] -
El-Aasr, M.; Fujiwara, Y.; Takeya, M.; Ikeda, T.; Tsukamoto, S.; Ono, M.; Nakano, D.; Okawa, M.; Kinjo, J.; Yoshimitsu, H.; Nohara, T. J. Nat. Prod. 2010, 73, 1306–1308. doi:10.1021/np100105u
Return to citation in text: [1] -
Scott, K. A.; Njardarson, J. T. Analysis of US FDA-Approved Drugs Containing Sulfur Atoms. In Sulfur Chemistry; Jiang, X., Ed.; Topics in Current Chemistry Collections; Springer: Cham, 2019; pp 1–34. doi:10.1007/978-3-030-25598-5_1
Return to citation in text: [1] -
De Martino, G.; La Regina, G.; Coluccia, A.; Edler, M. C.; Barbera, M. C.; Brancale, A.; Wilcox, E.; Hamel, E.; Artico, M.; Silvestri, R. J. Med. Chem. 2004, 47, 6120–6123. doi:10.1021/jm049360d
Return to citation in text: [1] -
Eryilmaz, M. A.; Kozanhan, B.; Solak, I.; Çetinkaya, Ç. D.; Neselioglu, S.; Erel, Ö. J. Cancer Res. Ther. (Mumbai, India) 2019, 15, 1062–1066. doi:10.4103/jcrt.jcrt_553_17
Return to citation in text: [1] -
Dirican, N.; Dirican, A.; Sen, O.; Aynali, A.; Atalay, S.; Bircan, H. A.; Oztürk, O.; Erdogan, S.; Cakir, M.; Akkaya, A. Redox Rep. 2016, 21, 197–203. doi:10.1179/1351000215y.0000000027
Return to citation in text: [1] -
Liu, G.; Huth, J. R.; Olejniczak, E. T.; Mendoza, R.; DeVries, P.; Leitza, S.; Reilly, E. B.; Okasinski, G. F.; Fesik, S. W.; von Geldern, T. W. J. Med. Chem. 2001, 44, 1202–1210. doi:10.1021/jm000503f
Return to citation in text: [1] -
Altiparmak, I. H.; Erkus, M. E.; Sezen, H.; Demirbag, R.; Kaya, Z.; Sezen, Y.; Gunebakmaz, O.; Asoglu, R.; Besli, F.; Neselioglu, S.; Erel, O. Coron. Artery Dis. 2016, 27, 295–301. doi:10.1097/mca.0000000000000362
Return to citation in text: [1] -
Hoffman, S.; Nolin, J.; McMillan, D.; Wouters, E.; Janssen‐Heininger, Y.; Reynaert, N. J. Cell. Biochem. 2015, 116, 884–892. doi:10.1002/jcb.25017
Return to citation in text: [1] -
Stantchev, T. S.; Paciga, M.; Lankford, C. R.; Schwartzkopff, F.; Broder, C. C.; Clouse, K. A. Retrovirology 2012, 9, 97. doi:10.1186/1742-4690-9-97
Return to citation in text: [1] -
Markovic, I.; Stantchev, T. S.; Fields, K. H.; Tiffany, L. J.; Tomiç, M.; Weiss, C. D.; Broder, C. C.; Strebel, K.; Clouse, K. A. Blood 2004, 103, 1586–1594. doi:10.1182/blood-2003-05-1390
Return to citation in text: [1] -
Wang, Y.; Chackalamannil, S.; Hu, Z.; Clader, J. W.; Greenlee, W.; Billard, W.; Binch, H., III; Crosby, G.; Ruperto, V.; Duffy, R. A.; McQuade, R.; Lachowicz, J. E. Bioorg. Med. Chem. Lett. 2000, 10, 2247–2250. doi:10.1016/s0960-894x(00)00457-1
Return to citation in text: [1] -
Nielsen, S. F.; Nielsen, E. Ø.; Olsen, G. M.; Liljefors, T.; Peters, D. J. Med. Chem. 2000, 43, 2217–2226. doi:10.1021/jm990973d
Return to citation in text: [1] -
Lee, C.-F.; Liu, Y.-C.; Badsara, S. S. Chem. – Asian J. 2014, 9, 706–722. doi:10.1002/asia.201301500
Return to citation in text: [1] -
Gensch, T.; Klauck, F. J. R.; Glorius, F. Angew. Chem., Int. Ed. 2016, 55, 11287–11291. doi:10.1002/anie.201605193
Return to citation in text: [1] -
Kianmehr, E.; Doraghi, F.; Foroumadi, A. Synthesis 2022, 54, 2464–2472. doi:10.1055/s-0041-1737337
Return to citation in text: [1] -
Taniguchi, N. J. Org. Chem. 2006, 71, 7874–7876. doi:10.1021/jo060834l
Return to citation in text: [1] -
Movassagh, B.; Navidi, M. Tetrahedron Lett. 2008, 49, 6712–6714. doi:10.1016/j.tetlet.2008.09.071
Return to citation in text: [1] -
Trost, B. M.; Ochiai, M.; McDougal, P. G. J. Am. Chem. Soc. 1978, 100, 7103–7106. doi:10.1021/ja00490a072
Return to citation in text: [1] -
Yan, S.-Y.; Liu, Y.-J.; Liu, B.; Liu, Y.-H.; Zhang, Z.-Z.; Shi, B.-F. Chem. Commun. 2015, 51, 7341–7344. doi:10.1039/c5cc01436k
Return to citation in text: [1] -
Preuss, R.; Schmidt, R. R. Synthesis 1988, 694–697. doi:10.1055/s-1988-27673
Return to citation in text: [1] -
Nicolaou, K. C.; Seitz, S. P.; Sipio, W. J.; Blount, J. F. J. Am. Chem. Soc. 1979, 101, 3884–3893. doi:10.1021/ja00508a028
Return to citation in text: [1] -
Young, R. N.; Coombs, W.; Guindon, Y.; Rokach, J.; Ethier, D.; Hall, R. Tetrahedron Lett. 1981, 22, 4933–4936. doi:10.1016/s0040-4039(01)92385-8
Return to citation in text: [1] -
Denmark, S. E.; Kornfilt, D. J. P.; Vogler, T. J. Am. Chem. Soc. 2011, 133, 15308–15311. doi:10.1021/ja2064395
Return to citation in text: [1] -
Ramesh, S.; Franck, R. W. J. Chem. Soc., Chem. Commun. 1989, 960–962. doi:10.1039/c39890000960
Return to citation in text: [1] -
Furukawa, N.; Morishita, T.; Akasaka, T.; Oae, S. Tetrahedron Lett. 1979, 20, 3973–3976. doi:10.1016/s0040-4039(01)86480-7
Return to citation in text: [1] -
Capozzi, G.; Menichetti, S.; Nicastro, M.; Taddei, M. Heterocycles 1989, 29, 1703–1708. doi:10.3987/com-89-5010
Return to citation in text: [1] -
Capozzi, G.; Ottana', R.; Romeo, G. Heterocycles 1987, 26, 39–42. doi:10.3987/r-1987-01-0039
Return to citation in text: [1] -
Eichman, C. C.; Stambuli, J. P. Molecules 2011, 16, 590–608. doi:10.3390/molecules16010590
Return to citation in text: [1] -
Sinha, A. K.; Equbal, D.; Rastogi, S. K.; Kumar, S.; Kumar, R. Asian J. Org. Chem. 2022, 11, e202100744. doi:10.1002/ajoc.202100744
Return to citation in text: [1] -
Abedinifar, F.; Bahadorikhalili, S.; Larijani, B.; Mahdavi, M.; Verpoort, F. Appl. Organomet. Chem. 2022, 36, e6482. doi:10.1002/aoc.6482
Return to citation in text: [1] -
Iwasaki, M.; Nishihara, Y. Dalton Trans. 2016, 45, 15278–15284. doi:10.1039/c6dt02167k
Return to citation in text: [1] -
Yan, S.-Y.; Liu, Y.-J.; Liu, B.; Liu, Y.-H.; Shi, B.-F. Chem. Commun. 2015, 51, 4069–4072. doi:10.1039/c4cc10446c
Return to citation in text: [1] -
Li, B.; Chen, Z.; Cao, H.; Zhao, H. Org. Lett. 2018, 20, 3291–3295. doi:10.1021/acs.orglett.8b01168
Return to citation in text: [1] -
Kibriya, G.; Mondal, S.; Hajra, A. Org. Lett. 2018, 20, 7740–7743. doi:10.1021/acs.orglett.8b03549
Return to citation in text: [1] -
Vásquez-Céspedes, S.; Ferry, A.; Candish, L.; Glorius, F. Angew. Chem., Int. Ed. 2015, 54, 5772–5776. doi:10.1002/anie.201411997
Return to citation in text: [1] -
Gillis, H. M.; Greene, L.; Thompson, A. Synlett 2009, 112–116. doi:10.1055/s-0028-1087486
Return to citation in text: [1] -
Merricks, D.; Sammes, P. G.; Walker, E. R. H.; Henrick, K.; McPartlin, M. M. J. Chem. Soc., Perkin Trans. 1 1991, 2169–2176. doi:10.1039/p19910002169
Return to citation in text: [1] -
Mampuys, P.; McElroy, C. R.; Clark, J. H.; Orru, R. V. A.; Maes, B. U. W. Adv. Synth. Catal. 2020, 362, 3–64. doi:10.1002/adsc.201900864
Return to citation in text: [1] -
Wang, X.; Meng, J.; Zhao, D.; Tang, S.; Sun, K. Chin. Chem. Lett. 2023, 34, 107736. doi:10.1016/j.cclet.2022.08.016
Return to citation in text: [1] -
Wei, Y.-F.; Gao, W.-C.; Chang, H.-H.; Jiang, X. Org. Chem. Front. 2022, 9, 6684–6707. doi:10.1039/d2qo01447e
Return to citation in text: [1] -
Liang, G.; Chen, J.; Chen, J.; Li, W.; Chen, J.; Wu, H. Tetrahedron Lett. 2012, 53, 6768–6770. doi:10.1016/j.tetlet.2012.09.132
Return to citation in text: [1] -
Saravanan, P.; Anbarasan, P. Org. Lett. 2014, 16, 848–851. doi:10.1021/ol4036209
Return to citation in text: [1] -
Tian, H.; Zhu, C.; Yang, H.; Fu, H. Chem. Commun. 2014, 50, 8875–8877. doi:10.1039/c4cc03600j
Return to citation in text: [1] -
Yu, J.; Jiang, M.; Song, Z.; He, T.; Yang, H.; Fu, H. Adv. Synth. Catal. 2016, 358, 2806–2810. doi:10.1002/adsc.201600133
Return to citation in text: [1] -
Gao, W.-C.; Liu, T.; Zhang, B.; Li, X.; Wei, W.-L.; Liu, Q.; Tian, J.; Chang, H.-H. J. Org. Chem. 2016, 81, 11297–11304. doi:10.1021/acs.joc.6b02271
Return to citation in text: [1] -
Zhao, J.-Q.; Luo, S.-W.; Zhang, X.-M.; Xu, X.-Y.; Zhou, M.-Q.; Yuan, W.-C. Tetrahedron 2017, 73, 5444–5450. doi:10.1016/j.tet.2017.07.053
Return to citation in text: [1] -
Ramesh, E.; Guntreddi, T.; Sahoo, A. K. Eur. J. Org. Chem. 2017, 4405–4413. doi:10.1002/ejoc.201700607
Return to citation in text: [1] -
Gao, W.-C.; Liu, T.; Cheng, Y.-F.; Chang, H.-H.; Li, X.; Zhou, R.; Wei, W.-L.; Qiao, Y. J. Org. Chem. 2017, 82, 13459–13467. doi:10.1021/acs.joc.7b02498
Return to citation in text: [1] -
Liu, T.; Tian, J.; Gao, W.-C.; Chang, H.-H.; Liu, Q.; Li, X.; Wei, W.-L. Org. Biomol. Chem. 2017, 15, 5983–5992. doi:10.1039/c7ob01225j
Return to citation in text: [1] -
Chaitanya, M.; Anbarasan, P. Org. Lett. 2018, 20, 3362–3366. doi:10.1021/acs.orglett.8b01281
Return to citation in text: [1] -
Lv, L.; Li, Z. J. Org. Chem. 2018, 83, 10985–10994. doi:10.1021/acs.joc.8b01621
Return to citation in text: [1] -
Graßl, S.; Hamze, C.; Koller, T. J.; Knochel, P. Chem. – Eur. J. 2019, 25, 3752–3755. doi:10.1002/chem.201806261
Return to citation in text: [1] -
Lin, Y.; Guanghui, L. Ü.; Liu, Y.; Zheng, Y.; Nie, R.; Guo, L.; Wu, Y. Catal. Commun. 2018, 112, 68–73. doi:10.1016/j.catcom.2018.04.021
Return to citation in text: [1] -
Gao, W.-C.; Cheng, Y.-F.; Chang, H.-H.; Li, X.; Wei, W.-L.; Yang, P. J. Org. Chem. 2019, 84, 4312–4317. doi:10.1021/acs.joc.9b00256
Return to citation in text: [1] -
Augustin, A. U.; Jones, P. G.; Werz, D. B. Chem. – Eur. J. 2019, 25, 11620–11624. doi:10.1002/chem.201902160
Return to citation in text: [1] -
Yu, W.; Yang, S.; Wang, P.-L.; Li, P.; Li, H. Org. Biomol. Chem. 2020, 18, 7165–7173. doi:10.1039/d0ob01388a
Return to citation in text: [1] -
Yu, X.; Shang, Y.-Z.; Cheng, Y.-F.; Tian, J.; Niu, Y.; Gao, W.-C. Org. Biomol. Chem. 2020, 18, 1806–1811. doi:10.1039/d0ob00050g
Return to citation in text: [1] -
Tian, J.; Yuan, K.-N.; Liu, W.; Chang, H.-H.; Li, X.; Gao, W.-C. Chem. Commun. 2021, 57, 1943–1946. doi:10.1039/d0cc07988j
Return to citation in text: [1] -
Ghorai, J.; Kesavan, A.; Anbarasan, P. Chem. Commun. 2021, 57, 10544–10547. doi:10.1039/d1cc03760a
Return to citation in text: [1] -
Dodds, A. C.; Sutherland, A. J. Org. Chem. 2021, 86, 5922–5932. doi:10.1021/acs.joc.1c00448
Return to citation in text: [1] -
Kumari, A. H.; Kumar, J. J.; Krishna, G. R.; Reddy, R. J. Synthesis 2021, 53, 2850–2864. doi:10.1055/a-1482-2486
Return to citation in text: [1] -
Li, Y.-F.; Wei, Y.-F.; Tian, J.; Zhang, J.; Chang, H.-H.; Gao, W.-C. Org. Lett. 2022, 24, 5736–5740. doi:10.1021/acs.orglett.2c02160
Return to citation in text: [1] -
Tian, J.; Feng, K.; Yuan, K.-N.; Li, X.; Chang, H.-H.; Gao, W.-C. J. Org. Chem. 2022, 87, 2402–2409. doi:10.1021/acs.joc.1c02269
Return to citation in text: [1] -
Wang, W.; Li, H.; Wang, J.; Liao, L. Tetrahedron Lett. 2004, 45, 8229–8231. doi:10.1016/j.tetlet.2004.09.021
Return to citation in text: [1] -
Tudge, M.; Tamiya, M.; Savarin, C.; Humphrey, G. R. Org. Lett. 2006, 8, 565–568. doi:10.1021/ol052615c
Return to citation in text: [1] -
Burés, J.; Isart, C.; Vilarrasa, J. Org. Lett. 2007, 9, 4635–4638. doi:10.1021/ol702212n
Return to citation in text: [1] -
Wang, H.; Huang, D.; Cheng, D.; Li, L.; Shi, Y. Org. Lett. 2011, 13, 1650–1653. doi:10.1021/ol200127n
Return to citation in text: [1] -
Lin, A.; Fang, L.; Zhu, X.; Zhu, C.; Cheng, Y. Adv. Synth. Catal. 2011, 353, 545–549. doi:10.1002/adsc.201000679
Return to citation in text: [1] -
Li, X.; Liu, C.; Xue, X.-S.; Cheng, J.-P. Org. Lett. 2012, 14, 4374–4377. doi:10.1021/ol301833f
Return to citation in text: [1] -
Cai, Y.; Li, J.; Chen, W.; Xie, M.; Liu, X.; Lin, L.; Feng, X. Org. Lett. 2012, 14, 2726–2729. doi:10.1021/ol3009446
Return to citation in text: [1] -
Shirakawa, S.; Tokuda, T.; Kasai, A.; Maruoka, K. Org. Lett. 2013, 15, 3350–3353. doi:10.1021/ol4013926
Return to citation in text: [1] -
Denmark, S. E.; Chi, H. M. J. Am. Chem. Soc. 2014, 136, 8915–8918. doi:10.1021/ja5046296
Return to citation in text: [1] -
Xu, M.; Qiao, B.; Duan, S.; Liu, H.; Jiang, Z. Tetrahedron 2014, 70, 8696–8702. doi:10.1016/j.tet.2014.09.037
Return to citation in text: [1] -
Hostier, T.; Ferey, V.; Ricci, G.; Gomez Pardo, D.; Cossy, J. Org. Lett. 2015, 17, 3898–3901. doi:10.1021/acs.orglett.5b01889
Return to citation in text: [1] -
Hostier, T.; Ferey, V.; Ricci, G.; Pardo, D. G.; Cossy, J. Chem. Commun. 2015, 51, 13898–13901. doi:10.1039/c5cc05421d
Return to citation in text: [1] -
You, Y.; Wu, Z.-J.; Wang, Z.-H.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. J. Org. Chem. 2015, 80, 8470–8477. doi:10.1021/acs.joc.5b01491
Return to citation in text: [1] -
Liao, K.; Zhou, F.; Yu, J.-S.; Gao, W.-M.; Zhou, J. Chem. Commun. 2015, 51, 16255–16258. doi:10.1039/c5cc07010d
Return to citation in text: [1] -
Nalbandian, C. J.; Miller, E. M.; Toenjes, S. T.; Gustafson, J. L. Chem. Commun. 2017, 53, 1494–1497. doi:10.1039/c6cc09998j
Return to citation in text: [1] -
Roy, S. J. S.; Mukherjee, S. Org. Biomol. Chem. 2017, 15, 6921–6925. doi:10.1039/c7ob01714f
Return to citation in text: [1] -
Denmark, S. E.; Kornfilt, D. J. P. J. Org. Chem. 2017, 82, 3192–3222. doi:10.1021/acs.joc.7b00295
Return to citation in text: [1] -
Chen, Z.-C.; Chen, P.; Chen, Z.; Ouyang, Q.; Liang, H.-P.; Du, W.; Chen, Y.-C. Org. Lett. 2018, 20, 6279–6283. doi:10.1021/acs.orglett.8b02764
Return to citation in text: [1] -
Wang, J.-J.; Yang, H.; Gou, B.-B.; Zhou, L.; Chen, J. J. Org. Chem. 2018, 83, 4730–4738. doi:10.1021/acs.joc.8b00487
Return to citation in text: [1] -
Chaitanya, M.; Anbarasan, P. Org. Lett. 2018, 20, 1183–1186. doi:10.1021/acs.orglett.8b00065
Return to citation in text: [1] -
Nalbandian, C. J.; Brown, Z. E.; Alvarez, E.; Gustafson, J. L. Org. Lett. 2018, 20, 3211–3214. doi:10.1021/acs.orglett.8b01066
Return to citation in text: [1] -
Panger, J. L.; Denmark, S. E. Org. Lett. 2020, 22, 2501–2505. doi:10.1021/acs.orglett.9b04347
Return to citation in text: [1] -
Nagao, Y.; Hiroya, K. Synlett 2020, 31, 813–817. doi:10.1055/s-0039-1690836
Return to citation in text: [1] -
Jiang, Q.; Li, H.; Zhao, X. Org. Lett. 2021, 23, 8777–8782. doi:10.1021/acs.orglett.1c03270
Return to citation in text: [1] -
Liang, Y.; Jiao, H.; Zhang, H.; Wang, Y.-Q.; Zhao, X. Org. Lett. 2022, 24, 7210–7215. doi:10.1021/acs.orglett.2c03009
Return to citation in text: [1] -
Tan, Q.; Chen, Q.; Zhu, Z.; Liu, X. Chem. Commun. 2022, 58, 9686–9689. doi:10.1039/d2cc03443c
Return to citation in text: [1] -
Kanikarapu, S.; Gogoi, M. P.; Dutta, S.; Sahoo, A. K. Org. Lett. 2022, 24, 8289–8294. doi:10.1021/acs.orglett.2c03225
Return to citation in text: [1] -
Xue, Y.-N.; Feng, K.; Tian, J.; Zhang, J.; Chang, H.-H.; Gao, W.-C. Org. Chem. Front. 2023, 10, 2070–2074. doi:10.1039/d3qo00159h
Return to citation in text: [1] -
Yu, J.; Gao, C.; Song, Z.; Yang, H.; Fu, H. Org. Biomol. Chem. 2015, 13, 4846–4850. doi:10.1039/c5ob00252d
Return to citation in text: [1] -
Tian, H.; Yang, H.; Zhu, C.; Fu, H. Adv. Synth. Catal. 2015, 357, 481–488. doi:10.1002/adsc.201400929
Return to citation in text: [1] -
Tian, H.; Yu, J.; Yang, H.; Zhu, C.; Fu, H. Adv. Synth. Catal. 2016, 358, 1794–1800. doi:10.1002/adsc.201501181
Return to citation in text: [1] -
Song, E.; Kim, H. Y.; Oh, K. Org. Biomol. Chem. 2017, 15, 1776–1779. doi:10.1039/c6ob02772e
Return to citation in text: [1] -
Li, X.; Guo, Y.; Shen, Z. J. Org. Chem. 2018, 83, 2818–2829. doi:10.1021/acs.joc.7b03263
Return to citation in text: [1] -
Yakan, H.; Kütük, H. Monatsh. Chem. 2018, 149, 2047–2057. doi:10.1007/s00706-018-2261-4
Return to citation in text: [1] -
Zhang, X.; Zheng, Y.; Qiu, L.; Xu, X. Org. Biomol. Chem. 2018, 16, 70–76. doi:10.1039/c7ob02488f
Return to citation in text: [1] -
Lei, X.; Wang, Y.; Fan, E.; Sun, Z. Org. Lett. 2019, 21, 1484–1487. doi:10.1021/acs.orglett.9b00275
Return to citation in text: [1] -
Han, J.; Zhang, Y.; Wu, X.-Y.; Wong, H. N. C. Chem. Commun. 2019, 55, 397–400. doi:10.1039/c8cc09049a
Return to citation in text: [1] -
Huang, X.; Li, J.; Li, X.; Wang, J.; Peng, Y.; Song, G. RSC Adv. 2019, 9, 26419–26424. doi:10.1039/c9ra05708k
Return to citation in text: [1] -
Mondal, M.; Saha, A. Tetrahedron Lett. 2019, 60, 150965. doi:10.1016/j.tetlet.2019.150965
Return to citation in text: [1]
| 55. | Graßl, S.; Hamze, C.; Koller, T. J.; Knochel, P. Chem. – Eur. J. 2019, 25, 3752–3755. doi:10.1002/chem.201806261 |
| 56. | Lin, Y.; Guanghui, L. Ü.; Liu, Y.; Zheng, Y.; Nie, R.; Guo, L.; Wu, Y. Catal. Commun. 2018, 112, 68–73. doi:10.1016/j.catcom.2018.04.021 |
| 100. | Yakan, H.; Kütük, H. Monatsh. Chem. 2018, 149, 2047–2057. doi:10.1007/s00706-018-2261-4 |
| 57. | Gao, W.-C.; Cheng, Y.-F.; Chang, H.-H.; Li, X.; Wei, W.-L.; Yang, P. J. Org. Chem. 2019, 84, 4312–4317. doi:10.1021/acs.joc.9b00256 |
| 101. | Zhang, X.; Zheng, Y.; Qiu, L.; Xu, X. Org. Biomol. Chem. 2018, 16, 70–76. doi:10.1039/c7ob02488f |
| 98. | Song, E.; Kim, H. Y.; Oh, K. Org. Biomol. Chem. 2017, 15, 1776–1779. doi:10.1039/c6ob02772e |
| 99. | Li, X.; Guo, Y.; Shen, Z. J. Org. Chem. 2018, 83, 2818–2829. doi:10.1021/acs.joc.7b03263 |
| 97. | Tian, H.; Yu, J.; Yang, H.; Zhu, C.; Fu, H. Adv. Synth. Catal. 2016, 358, 1794–1800. doi:10.1002/adsc.201501181 |
| 64. | Kumari, A. H.; Kumar, J. J.; Krishna, G. R.; Reddy, R. J. Synthesis 2021, 53, 2850–2864. doi:10.1055/a-1482-2486 |
| 65. | Li, Y.-F.; Wei, Y.-F.; Tian, J.; Zhang, J.; Chang, H.-H.; Gao, W.-C. Org. Lett. 2022, 24, 5736–5740. doi:10.1021/acs.orglett.2c02160 |
| 62. | Ghorai, J.; Kesavan, A.; Anbarasan, P. Chem. Commun. 2021, 57, 10544–10547. doi:10.1039/d1cc03760a |
| 63. | Dodds, A. C.; Sutherland, A. J. Org. Chem. 2021, 86, 5922–5932. doi:10.1021/acs.joc.1c00448 |
| 60. | Yu, X.; Shang, Y.-Z.; Cheng, Y.-F.; Tian, J.; Niu, Y.; Gao, W.-C. Org. Biomol. Chem. 2020, 18, 1806–1811. doi:10.1039/d0ob00050g |
| 104. | Huang, X.; Li, J.; Li, X.; Wang, J.; Peng, Y.; Song, G. RSC Adv. 2019, 9, 26419–26424. doi:10.1039/c9ra05708k |
| 61. | Tian, J.; Yuan, K.-N.; Liu, W.; Chang, H.-H.; Li, X.; Gao, W.-C. Chem. Commun. 2021, 57, 1943–1946. doi:10.1039/d0cc07988j |
| 105. | Mondal, M.; Saha, A. Tetrahedron Lett. 2019, 60, 150965. doi:10.1016/j.tetlet.2019.150965 |
| 58. | Augustin, A. U.; Jones, P. G.; Werz, D. B. Chem. – Eur. J. 2019, 25, 11620–11624. doi:10.1002/chem.201902160 |
| 102. | Lei, X.; Wang, Y.; Fan, E.; Sun, Z. Org. Lett. 2019, 21, 1484–1487. doi:10.1021/acs.orglett.9b00275 |
| 59. | Yu, W.; Yang, S.; Wang, P.-L.; Li, P.; Li, H. Org. Biomol. Chem. 2020, 18, 7165–7173. doi:10.1039/d0ob01388a |
| 103. | Han, J.; Zhang, Y.; Wu, X.-Y.; Wong, H. N. C. Chem. Commun. 2019, 55, 397–400. doi:10.1039/c8cc09049a |
| 66. | Tian, J.; Feng, K.; Yuan, K.-N.; Li, X.; Chang, H.-H.; Gao, W.-C. J. Org. Chem. 2022, 87, 2402–2409. doi:10.1021/acs.joc.1c02269 |
| 67. | Wang, W.; Li, H.; Wang, J.; Liao, L. Tetrahedron Lett. 2004, 45, 8229–8231. doi:10.1016/j.tetlet.2004.09.021 |
| 68. | Tudge, M.; Tamiya, M.; Savarin, C.; Humphrey, G. R. Org. Lett. 2006, 8, 565–568. doi:10.1021/ol052615c |
| 75. | Denmark, S. E.; Chi, H. M. J. Am. Chem. Soc. 2014, 136, 8915–8918. doi:10.1021/ja5046296 |
| 76. | Xu, M.; Qiao, B.; Duan, S.; Liu, H.; Jiang, Z. Tetrahedron 2014, 70, 8696–8702. doi:10.1016/j.tet.2014.09.037 |
| 73. | Cai, Y.; Li, J.; Chen, W.; Xie, M.; Liu, X.; Lin, L.; Feng, X. Org. Lett. 2012, 14, 2726–2729. doi:10.1021/ol3009446 |
| 74. | Shirakawa, S.; Tokuda, T.; Kasai, A.; Maruoka, K. Org. Lett. 2013, 15, 3350–3353. doi:10.1021/ol4013926 |
| 71. | Lin, A.; Fang, L.; Zhu, X.; Zhu, C.; Cheng, Y. Adv. Synth. Catal. 2011, 353, 545–549. doi:10.1002/adsc.201000679 |
| 72. | Li, X.; Liu, C.; Xue, X.-S.; Cheng, J.-P. Org. Lett. 2012, 14, 4374–4377. doi:10.1021/ol301833f |
| 69. | Burés, J.; Isart, C.; Vilarrasa, J. Org. Lett. 2007, 9, 4635–4638. doi:10.1021/ol702212n |
| 70. | Wang, H.; Huang, D.; Cheng, D.; Li, L.; Shi, Y. Org. Lett. 2011, 13, 1650–1653. doi:10.1021/ol200127n |
| 78. | Hostier, T.; Ferey, V.; Ricci, G.; Pardo, D. G.; Cossy, J. Chem. Commun. 2015, 51, 13898–13901. doi:10.1039/c5cc05421d |
| 79. | You, Y.; Wu, Z.-J.; Wang, Z.-H.; Xu, X.-Y.; Zhang, X.-M.; Yuan, W.-C. J. Org. Chem. 2015, 80, 8470–8477. doi:10.1021/acs.joc.5b01491 |
| 77. | Hostier, T.; Ferey, V.; Ricci, G.; Gomez Pardo, D.; Cossy, J. Org. Lett. 2015, 17, 3898–3901. doi:10.1021/acs.orglett.5b01889 |
| 1. | Beletskaya, I. P.; Ananikov, V. P. Chem. Rev. 2011, 111, 1596–1636. doi:10.1021/cr100347k |
| 2. | Liu, L.; Stelmach, J. E.; Natarajan, S. R.; Chen, M.-H.; Singh, S. B.; Schwartz, C. D.; Fitzgerald, C. E.; O'Keefe, S. J.; Zaller, D. M.; Schmatz, D. M.; Doherty, J. B. Bioorg. Med. Chem. Lett. 2003, 13, 3979–3982. doi:10.1016/j.bmcl.2003.08.059 |
| 3. | Fontecave, M.; Ollagnier-de-Choudens, S.; Mulliez, E. Chem. Rev. 2003, 103, 2149–2166. doi:10.1021/cr020427j |
| 4. | El-Aasr, M.; Fujiwara, Y.; Takeya, M.; Ikeda, T.; Tsukamoto, S.; Ono, M.; Nakano, D.; Okawa, M.; Kinjo, J.; Yoshimitsu, H.; Nohara, T. J. Nat. Prod. 2010, 73, 1306–1308. doi:10.1021/np100105u |
| 5. | Scott, K. A.; Njardarson, J. T. Analysis of US FDA-Approved Drugs Containing Sulfur Atoms. In Sulfur Chemistry; Jiang, X., Ed.; Topics in Current Chemistry Collections; Springer: Cham, 2019; pp 1–34. doi:10.1007/978-3-030-25598-5_1 |
| 14. | Wang, Y.; Chackalamannil, S.; Hu, Z.; Clader, J. W.; Greenlee, W.; Billard, W.; Binch, H., III; Crosby, G.; Ruperto, V.; Duffy, R. A.; McQuade, R.; Lachowicz, J. E. Bioorg. Med. Chem. Lett. 2000, 10, 2247–2250. doi:10.1016/s0960-894x(00)00457-1 |
| 15. | Nielsen, S. F.; Nielsen, E. Ø.; Olsen, G. M.; Liljefors, T.; Peters, D. J. Med. Chem. 2000, 43, 2217–2226. doi:10.1021/jm990973d |
| 39. | Gillis, H. M.; Greene, L.; Thompson, A. Synlett 2009, 112–116. doi:10.1055/s-0028-1087486 |
| 40. | Merricks, D.; Sammes, P. G.; Walker, E. R. H.; Henrick, K.; McPartlin, M. M. J. Chem. Soc., Perkin Trans. 1 1991, 2169–2176. doi:10.1039/p19910002169 |
| 86. | Chaitanya, M.; Anbarasan, P. Org. Lett. 2018, 20, 1183–1186. doi:10.1021/acs.orglett.8b00065 |
| 12. | Stantchev, T. S.; Paciga, M.; Lankford, C. R.; Schwartzkopff, F.; Broder, C. C.; Clouse, K. A. Retrovirology 2012, 9, 97. doi:10.1186/1742-4690-9-97 |
| 13. | Markovic, I.; Stantchev, T. S.; Fields, K. H.; Tiffany, L. J.; Tomiç, M.; Weiss, C. D.; Broder, C. C.; Strebel, K.; Clouse, K. A. Blood 2004, 103, 1586–1594. doi:10.1182/blood-2003-05-1390 |
| 41. | Mampuys, P.; McElroy, C. R.; Clark, J. H.; Orru, R. V. A.; Maes, B. U. W. Adv. Synth. Catal. 2020, 362, 3–64. doi:10.1002/adsc.201900864 |
| 42. | Wang, X.; Meng, J.; Zhao, D.; Tang, S.; Sun, K. Chin. Chem. Lett. 2023, 34, 107736. doi:10.1016/j.cclet.2022.08.016 |
| 43. | Wei, Y.-F.; Gao, W.-C.; Chang, H.-H.; Jiang, X. Org. Chem. Front. 2022, 9, 6684–6707. doi:10.1039/d2qo01447e |
| 9. | Liu, G.; Huth, J. R.; Olejniczak, E. T.; Mendoza, R.; DeVries, P.; Leitza, S.; Reilly, E. B.; Okasinski, G. F.; Fesik, S. W.; von Geldern, T. W. J. Med. Chem. 2001, 44, 1202–1210. doi:10.1021/jm000503f |
| 10. | Altiparmak, I. H.; Erkus, M. E.; Sezen, H.; Demirbag, R.; Kaya, Z.; Sezen, Y.; Gunebakmaz, O.; Asoglu, R.; Besli, F.; Neselioglu, S.; Erel, O. Coron. Artery Dis. 2016, 27, 295–301. doi:10.1097/mca.0000000000000362 |
| 11. | Hoffman, S.; Nolin, J.; McMillan, D.; Wouters, E.; Janssen‐Heininger, Y.; Reynaert, N. J. Cell. Biochem. 2015, 116, 884–892. doi:10.1002/jcb.25017 |
| 34. | Iwasaki, M.; Nishihara, Y. Dalton Trans. 2016, 45, 15278–15284. doi:10.1039/c6dt02167k |
| 35. | Yan, S.-Y.; Liu, Y.-J.; Liu, B.; Liu, Y.-H.; Shi, B.-F. Chem. Commun. 2015, 51, 4069–4072. doi:10.1039/c4cc10446c |
| 36. | Li, B.; Chen, Z.; Cao, H.; Zhao, H. Org. Lett. 2018, 20, 3291–3295. doi:10.1021/acs.orglett.8b01168 |
| 37. | Kibriya, G.; Mondal, S.; Hajra, A. Org. Lett. 2018, 20, 7740–7743. doi:10.1021/acs.orglett.8b03549 |
| 84. | Chen, Z.-C.; Chen, P.; Chen, Z.; Ouyang, Q.; Liang, H.-P.; Du, W.; Chen, Y.-C. Org. Lett. 2018, 20, 6279–6283. doi:10.1021/acs.orglett.8b02764 |
| 6. | De Martino, G.; La Regina, G.; Coluccia, A.; Edler, M. C.; Barbera, M. C.; Brancale, A.; Wilcox, E.; Hamel, E.; Artico, M.; Silvestri, R. J. Med. Chem. 2004, 47, 6120–6123. doi:10.1021/jm049360d |
| 7. | Eryilmaz, M. A.; Kozanhan, B.; Solak, I.; Çetinkaya, Ç. D.; Neselioglu, S.; Erel, Ö. J. Cancer Res. Ther. (Mumbai, India) 2019, 15, 1062–1066. doi:10.4103/jcrt.jcrt_553_17 |
| 8. | Dirican, N.; Dirican, A.; Sen, O.; Aynali, A.; Atalay, S.; Bircan, H. A.; Oztürk, O.; Erdogan, S.; Cakir, M.; Akkaya, A. Redox Rep. 2016, 21, 197–203. doi:10.1179/1351000215y.0000000027 |
| 38. | Vásquez-Céspedes, S.; Ferry, A.; Candish, L.; Glorius, F. Angew. Chem., Int. Ed. 2015, 54, 5772–5776. doi:10.1002/anie.201411997 |
| 85. | Wang, J.-J.; Yang, H.; Gou, B.-B.; Zhou, L.; Chen, J. J. Org. Chem. 2018, 83, 4730–4738. doi:10.1021/acs.joc.8b00487 |
| 26. | Denmark, S. E.; Kornfilt, D. J. P.; Vogler, T. J. Am. Chem. Soc. 2011, 133, 15308–15311. doi:10.1021/ja2064395 |
| 29. | Capozzi, G.; Menichetti, S.; Nicastro, M.; Taddei, M. Heterocycles 1989, 29, 1703–1708. doi:10.3987/com-89-5010 |
| 30. | Capozzi, G.; Ottana', R.; Romeo, G. Heterocycles 1987, 26, 39–42. doi:10.3987/r-1987-01-0039 |
| 82. | Roy, S. J. S.; Mukherjee, S. Org. Biomol. Chem. 2017, 15, 6921–6925. doi:10.1039/c7ob01714f |
| 23. | Preuss, R.; Schmidt, R. R. Synthesis 1988, 694–697. doi:10.1055/s-1988-27673 |
| 24. | Nicolaou, K. C.; Seitz, S. P.; Sipio, W. J.; Blount, J. F. J. Am. Chem. Soc. 1979, 101, 3884–3893. doi:10.1021/ja00508a028 |
| 25. | Young, R. N.; Coombs, W.; Guindon, Y.; Rokach, J.; Ethier, D.; Hall, R. Tetrahedron Lett. 1981, 22, 4933–4936. doi:10.1016/s0040-4039(01)92385-8 |
| 31. | Eichman, C. C.; Stambuli, J. P. Molecules 2011, 16, 590–608. doi:10.3390/molecules16010590 |
| 32. | Sinha, A. K.; Equbal, D.; Rastogi, S. K.; Kumar, S.; Kumar, R. Asian J. Org. Chem. 2022, 11, e202100744. doi:10.1002/ajoc.202100744 |
| 33. | Abedinifar, F.; Bahadorikhalili, S.; Larijani, B.; Mahdavi, M.; Verpoort, F. Appl. Organomet. Chem. 2022, 36, e6482. doi:10.1002/aoc.6482 |
| 83. | Denmark, S. E.; Kornfilt, D. J. P. J. Org. Chem. 2017, 82, 3192–3222. doi:10.1021/acs.joc.7b00295 |
| 19. | Taniguchi, N. J. Org. Chem. 2006, 71, 7874–7876. doi:10.1021/jo060834l |
| 20. | Movassagh, B.; Navidi, M. Tetrahedron Lett. 2008, 49, 6712–6714. doi:10.1016/j.tetlet.2008.09.071 |
| 21. | Trost, B. M.; Ochiai, M.; McDougal, P. G. J. Am. Chem. Soc. 1978, 100, 7103–7106. doi:10.1021/ja00490a072 |
| 22. | Yan, S.-Y.; Liu, Y.-J.; Liu, B.; Liu, Y.-H.; Zhang, Z.-Z.; Shi, B.-F. Chem. Commun. 2015, 51, 7341–7344. doi:10.1039/c5cc01436k |
| 80. | Liao, K.; Zhou, F.; Yu, J.-S.; Gao, W.-M.; Zhou, J. Chem. Commun. 2015, 51, 16255–16258. doi:10.1039/c5cc07010d |
| 16. | Lee, C.-F.; Liu, Y.-C.; Badsara, S. S. Chem. – Asian J. 2014, 9, 706–722. doi:10.1002/asia.201301500 |
| 17. | Gensch, T.; Klauck, F. J. R.; Glorius, F. Angew. Chem., Int. Ed. 2016, 55, 11287–11291. doi:10.1002/anie.201605193 |
| 18. | Kianmehr, E.; Doraghi, F.; Foroumadi, A. Synthesis 2022, 54, 2464–2472. doi:10.1055/s-0041-1737337 |
| 27. | Ramesh, S.; Franck, R. W. J. Chem. Soc., Chem. Commun. 1989, 960–962. doi:10.1039/c39890000960 |
| 28. | Furukawa, N.; Morishita, T.; Akasaka, T.; Oae, S. Tetrahedron Lett. 1979, 20, 3973–3976. doi:10.1016/s0040-4039(01)86480-7 |
| 81. | Nalbandian, C. J.; Miller, E. M.; Toenjes, S. T.; Gustafson, J. L. Chem. Commun. 2017, 53, 1494–1497. doi:10.1039/c6cc09998j |
| 46. | Tian, H.; Zhu, C.; Yang, H.; Fu, H. Chem. Commun. 2014, 50, 8875–8877. doi:10.1039/c4cc03600j |
| 44. | Liang, G.; Chen, J.; Chen, J.; Li, W.; Chen, J.; Wu, H. Tetrahedron Lett. 2012, 53, 6768–6770. doi:10.1016/j.tetlet.2012.09.132 |
| 45. | Saravanan, P.; Anbarasan, P. Org. Lett. 2014, 16, 848–851. doi:10.1021/ol4036209 |
| 90. | Jiang, Q.; Li, H.; Zhao, X. Org. Lett. 2021, 23, 8777–8782. doi:10.1021/acs.orglett.1c03270 |
| 87. | Nalbandian, C. J.; Brown, Z. E.; Alvarez, E.; Gustafson, J. L. Org. Lett. 2018, 20, 3211–3214. doi:10.1021/acs.orglett.8b01066 |
| 88. | Panger, J. L.; Denmark, S. E. Org. Lett. 2020, 22, 2501–2505. doi:10.1021/acs.orglett.9b04347 |
| 53. | Chaitanya, M.; Anbarasan, P. Org. Lett. 2018, 20, 3362–3366. doi:10.1021/acs.orglett.8b01281 |
| 54. | Lv, L.; Li, Z. J. Org. Chem. 2018, 83, 10985–10994. doi:10.1021/acs.joc.8b01621 |
| 51. | Gao, W.-C.; Liu, T.; Cheng, Y.-F.; Chang, H.-H.; Li, X.; Zhou, R.; Wei, W.-L.; Qiao, Y. J. Org. Chem. 2017, 82, 13459–13467. doi:10.1021/acs.joc.7b02498 |
| 95. | Yu, J.; Gao, C.; Song, Z.; Yang, H.; Fu, H. Org. Biomol. Chem. 2015, 13, 4846–4850. doi:10.1039/c5ob00252d |
| 52. | Liu, T.; Tian, J.; Gao, W.-C.; Chang, H.-H.; Liu, Q.; Li, X.; Wei, W.-L. Org. Biomol. Chem. 2017, 15, 5983–5992. doi:10.1039/c7ob01225j |
| 96. | Tian, H.; Yang, H.; Zhu, C.; Fu, H. Adv. Synth. Catal. 2015, 357, 481–488. doi:10.1002/adsc.201400929 |
| 49. | Zhao, J.-Q.; Luo, S.-W.; Zhang, X.-M.; Xu, X.-Y.; Zhou, M.-Q.; Yuan, W.-C. Tetrahedron 2017, 73, 5444–5450. doi:10.1016/j.tet.2017.07.053 |
| 93. | Kanikarapu, S.; Gogoi, M. P.; Dutta, S.; Sahoo, A. K. Org. Lett. 2022, 24, 8289–8294. doi:10.1021/acs.orglett.2c03225 |
| 50. | Ramesh, E.; Guntreddi, T.; Sahoo, A. K. Eur. J. Org. Chem. 2017, 4405–4413. doi:10.1002/ejoc.201700607 |
| 94. | Xue, Y.-N.; Feng, K.; Tian, J.; Zhang, J.; Chang, H.-H.; Gao, W.-C. Org. Chem. Front. 2023, 10, 2070–2074. doi:10.1039/d3qo00159h |
| 47. | Yu, J.; Jiang, M.; Song, Z.; He, T.; Yang, H.; Fu, H. Adv. Synth. Catal. 2016, 358, 2806–2810. doi:10.1002/adsc.201600133 |
| 91. | Liang, Y.; Jiao, H.; Zhang, H.; Wang, Y.-Q.; Zhao, X. Org. Lett. 2022, 24, 7210–7215. doi:10.1021/acs.orglett.2c03009 |
| 48. | Gao, W.-C.; Liu, T.; Zhang, B.; Li, X.; Wei, W.-L.; Liu, Q.; Tian, J.; Chang, H.-H. J. Org. Chem. 2016, 81, 11297–11304. doi:10.1021/acs.joc.6b02271 |
| 92. | Tan, Q.; Chen, Q.; Zhu, Z.; Liu, X. Chem. Commun. 2022, 58, 9686–9689. doi:10.1039/d2cc03443c |
© 2023 Doraghi et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.
