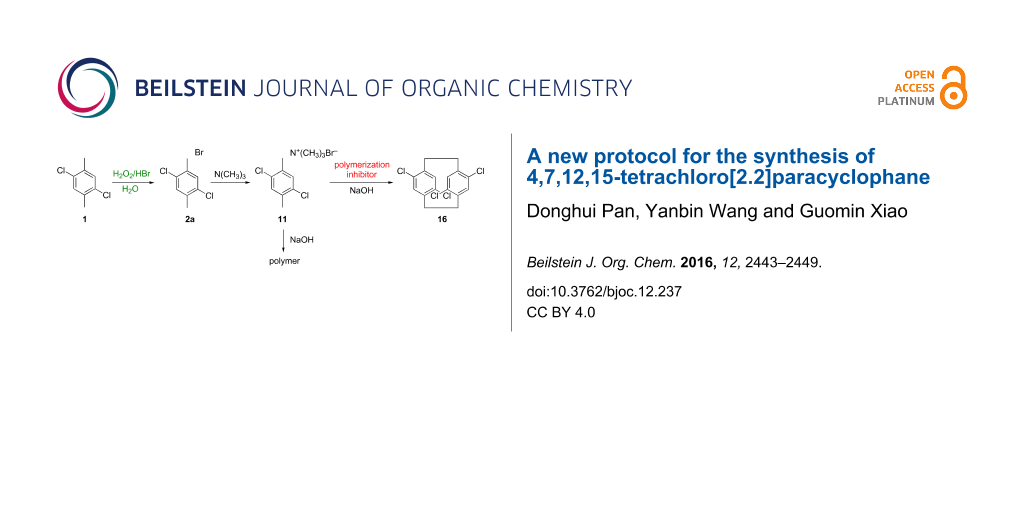
Abstract
We report a green and convenient protocol to prepare 4,7,12,15-tetrachloro[2.2]paracyclophane, the precursor of parylene D, from 2,5-dichloro-p-xylene. In the first bromination step, with H2O2–HBr as a bromide source, this procedure becomes organic-waste-free and organic-solvent-free and can appropriately replace the existing bromination methods. The Winberg elimination–dimerization step, using aqueous sodium hydroxide solution instead of silver oxide for anion exchange, results in a significant improvement in product yield. Furthermore, four substituted [2.2]paracyclophanes were also prepared in this convenient way.

Graphical Abstract
Introduction
Parylene films (Figure 1) are desired uniform coating materials that are widely used in microelectronic engineering, automotive and medical industries, owing to their low dielectricity, high thermal and oxidative stability, and chemical inertness [1-4]. Parylene N was firstly commercialized, and its precursor [2.2]paracyclophane (Figure 2) was typically produced by Hofmann elimination [5,6]. As reported, the uniform coating properties of parylene films were improved by introducing halogen atoms to the structure of the parent [2.2]paracyclophane [7]. Therefore, the two chloride atoms on the benzene ring make parylene D superior to parylene N and parylene C. There are some creative strategies for the synthesis of 4,7,12,15-tetrachloro[2.2]paracyclophane (Figure 2), the precursor of parylene D [8]. Theoretically, direct chlorination of [2.2]paracyclophane is an ideal route to prepare tetrachloroparacyclophane, but a pure polysubstituted product is difficult to obtain by electrophilic substitution without repeated crystallization or chromatographic purification [9]. Thus, we report an improved synthesis method using the Winberg dimerization of 2,5-dichloro-(4-methylbenzyl)trimethylammonium hydroxide without tedious purification.
![[1860-5397-12-237-1]](/bjoc/content/figures/1860-5397-12-237-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chemical structures of parylene N, parylene C, and parylene D.
Figure 1: Chemical structures of parylene N, parylene C, and parylene D.
![[1860-5397-12-237-2]](/bjoc/content/figures/1860-5397-12-237-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Chemical structures of [2.2]paracyclophane and 4,7,12,15-tetrachloro[2.2]paracyclophane.
Figure 2: Chemical structures of [2.2]paracyclophane and 4,7,12,15-tetrachloro[2.2]paracyclophane.
The important chemical 1-(bromomethyl)-2,5-dichloro-4-methylbenzene is an intermediate in the preparation of 2,5-dichloro-(4-methylbenzyl)trimethylammonium hydroxide. During our investigation of the synthesis of 4,7,12,15-tetrachloro[2.2]paracyclophane, we also adopted an improved bromination process to prepare 1-(bromomethyl)-2,5-dichloro-4-methylbenzene. Traditionally, there are several disadvantages when molecular bromine is used as a brominating reagent, such as toxicity, inconvenient handling and high reactivity, which lead to unsatisfactory results in the bromination process [10-12]. In addition, the release of corrosive HBr as a byproduct and the use of organic solvents make this protocol less environmentally friendly [13]. The use of other brominating agents, such as N-bromosuccinimide (NBS) and pyridinium tribromides, also has the drawbacks such as low atom efficiency and the requirement of reagent residue elimination [14]. In contrast to traditional brominating reagents, the H2O2–HBr system, which generates active bromine in situ, is a convenient and green brominating agent [15]. Furthermore, the use of the H2O2–HBr couple improves the selectivity and allows for the complete utilization of bromine atoms, thus increasing the atom economy [16]. These advantages prompted us to develop a novel method to prepare 1-(bromomethyl)-2,5-dichloro-4-methylbenzene and 4,7,12,15-tetrachloro[2.2]paracyclophane in a convenient and green way.
Results and Discussion
We initially planned to optimize the reaction conditions for the bromination of the benzylic position of 2,5-dichloro-p-xylene (1) by using the H2O2–HBr system, and investigated various factors, including the activation mode, the reagent stoichiometry, the solvent, and the reaction temperature (Table 1).
Table 1: Bromination of 2,5-dichloro-p-xylene (1) with H2O2–HBr.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-237-i2.svg?max-width=637&scale=1.0)
|
|||||||
| Entry | 1/H2O2/HBr | Mode of initiationa | Solvent | Methodb | Temp. (°C) | Yieldc (%) | |
|---|---|---|---|---|---|---|---|
| 2a | 2b | ||||||
| 1 | 1:1:1 | dark | CCl4 | A | 25 | 22.8 | – |
| 2 | 1:1:1 | dark | CCl4 | A | 75 | 62.9 | 4.2 |
| 3 | 1:1:1 | 3% DBP | CCl4 | A | 75 | 65.8 | 8.1 |
| 4 | 1:1:1 | 3% AMPA | CCl4 | A | 75 | 62.3 | 7.8 |
| 5 | 1:1:1 | incandescent light | CCl4 | A | 25 | 70.2 | 3.5 |
| 6 | 1:1:1 | incandescent light | H2O | A | 25 | 68.8 | 2.5 |
| 7 | 1:1.5:1 | incandescent light | H2O | A | 25 | 73.1 | 4.6 |
| 8 | 1:2:1 | incandescent light | H2O | A | 25 | 80.4 | 4.2 |
| 9 | 1:2:1.1 | incandescent light | H2O | A | 25 | 85.1 | 3.5 |
| 10 | 1:2:1.5 | incandescent light | H2O | A | 25 | 82.7 | 10.1 |
| 11 | 1:2:1.1 | incandescent light | H2O | B | 25 | 89.9 | 1.2 |
| 12 | 1:2:1.1 | incandescent light | H2O | B | 80 | 65.1 | 28.2 |
aRadical initiators: DBP (dibenzoyl peroxide), AMPA (2,2’-azobis(2-methylpropionamidine) dihydrochloride), 40 W incandescent light bulb. bMethod A: H2O2 and HBr were added in one portion; Method B: H2O2 was added gradually (1 equiv per 2.5 h). cYields were determined by 1H NMR spectroscopy and were based on starting compound 1.
The bromination reaction activated by heating in the dark produced a 62.9% yield of the monobrominated product 1-(bromomethyl)-2,5-dichloro-4-methylbenzene (2a) accompanied by a small amount of 1,4-bis(bromomethyl)-2,5-dichlorobenzene (2b) (Table 1, entry 2). Next, a radical reaction was induced by adding 3 mol % of radical initiator (DBP or AMPA) and proceeded at 75 °C for 4 h (Table 1, entries 3 and 4). Though the yields in both processes increased, the selectivity of 2a decreased due to the formation of some excessive brominated byproducts. Then, we tried visible light as activator of the racial process. Interestingly, the yield and the selectivity of 2a increased when a 40 W incandescent light bulb was used at 25 °C for 6 h (Table 1, entry 5) compared to other activation modes.
To make the chemical process green, we designed a bromination process with water as the reaction medium rather than organic solvents. Despite the low solubility of the organic substrates, the yields of 2a were improved without significant formation of byproducts (Table 1, entries 5 and 6). Furthermore, it was convenient to separate the organic product from the reaction mixtures. In small-scale experiments, a simple extraction with an appropriate organic solvent was efficient to obtain the product. However, in large-scale bromination processes, a clear phase separation occurred, so the product could be obtained by drying the organic phase after separation from the aqueous phase.
Considering the H2O2 decomposition in the presence of HBr and Br2 in the reaction, the effect of the amount of H2O2 was investigated. Actually, the yields of 2a increased to 73.1% and 80.4% when 1.5 and 2.0 equiv of H2O2 were used (Table 1, entries 7 and 8), respectively, in the bromination process. Similarly, when the amount of HBr increased to 1.1 equiv, the yield of 2a was maximized (Table 1, entry 9). However, a large amount of 2b was found when excessive HBr (1.5 equiv) was used, which decreased the selectivity of this bromination protocol (Table 1, entry 10).
The effect of reagent addition modes on the bromination yields was also studied. The results showed that gradual addition of H2O2 (method B) improved the yield of the main product 2a in contrast to a one-time addition of H2O2 (method A). This may be due to a significant decrease of H2O2 decomposition during the slow addition process. In addition, the Br2 generated in situ was reduced by stepwise addition of H2O2, which would improve the selectivity of 2a by preventing the side reactions.
Next, the bromination of other para-xylene derivatives under optimized conditions (see Table 1, entry 11) were investigated to examine the versatility of the protocol. As can be seen in Table 2, para-xylene (3), 2-chloro-1,4-dimethylbenzene (5) and 2-bromo-1,4-dimethylbenzene (7) were converted to the corresponding benzyl bromides in high yields with a small amount of dibrominated byproducts. However, in the case of 1-nitro-2,5-dimethylbenzene (9), a lower yield of benzyl brominated product was obtained. This could be explained by the deactivating effect of the nitro group [16]. Therefore, a 100 W high pressure mercury lamp (‘solar’ light) was used to increase the formation of bromide radical in the repeated bromination experiment of 9. On this occasion the yield of the monobrominated product 10a was high, and this was in agreement with the literature [16].
Table 2: Visible-light induced free-radical bromination of substituted p-xylenes with H2O2–HBr.
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-237-i3.svg?max-width=637&scale=1.0)
|
||
| Substrate | Time(h) | Yielda (%) |
|---|---|---|
| 3: R = H | 16 | 4a: 89.2, 4b: 3.2 |
| 5: R = Cl | 22 | 6a: 85.3, 6b: 2.5 |
| 7: R = Br | 25 | 8a: 82.7, 8b: 4.2 |
| 9: R = NO2b | 60 | 10a: 78.5, 10b: 2.3 |
aYields were determined by 1H NMR spectroscopy and were based on starting compounds. bThe reaction mixture was irradiated with a 100 W high pressure mercury lamp.
Five brominated products were obtained through the above bromination protocol, and were used to synthesize substituted (4-methylbenzyl)trimethylammonium bromides in diethyl ether at 0 °C with quantitative yields [17] (Scheme 1).
![[1860-5397-12-237-i1]](/bjoc/content/inline/1860-5397-12-237-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of substituted (4-methylbenzyl)trimethylammonium bromides from substituted (4-methylbenzyl)bromides.
Scheme 1: Synthesis of substituted (4-methylbenzyl)trimethylammonium bromides from substituted (4-methylbenzy...
Then, we used 2,5-dichloro-(4-methylbenzyl)trimethylammonium bromide (11) as starting material to prepare tetrachloro[2.2]paracyclophane in an aqueous sodium hydroxide solution according to Winberg’s method [18,19]. The intermediate 2,5-dichloro-(4-methylbenzyl)trimethylammonium hydroxide was formed and then decomposed in boiling toluene, resulting in a small amount of a dimer product 16 and a quantity of polymer byproduct (Table 3, entry 1). After the reaction, the polymer byproduct was removed by filtration, and the dimer product was obtained by concentrating the filtrate under reduced pressure. Thus, a chromatographic purification was not necessary in the improved dimerization protocol.
Table 3: Synthesis of 4,7,12,15-tetrachloro[2.2]paracyclophane 16 from 11.
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-237-i4.svg?max-width=637&scale=1.0)
|
||
| Entry | Polymerization inhibitor | Yielda (%) |
|---|---|---|
| 1 | – | 12 |
| 2 | phenothiazine | 25 |
| 3 | 2-chlorophenothiazine | 35 |
aYields of products were based on compound 11.
To suppress the polymerization and to improve the yield of the dimer product, we attempted the addition of a polymerization inhibitor. As expected, the addition of 3 mol % phenothiazine significantly improved the yield of 16 to 25% (Table 3, entry 2). The addition of 2-chlorophenothiazine increased the yield to 35% (Table 3, entry 3), which was about three times than that without any inhibitor. In addition, the 35% yield of dimer product was about two times the yield (20%) when the protocol with silver oxide for anion exchange was used [17], and it was comparable to the commercial synthetic protocol with 36.5% yield [20]. Although two isomers from the dimerization reaction could be formed, only the 4,7,12,15-tetrachloro isomer was obtained. The structure of the product was confirmed by 1H and 13C NMR spectral analysis, and the data matched well with the reported results [17]. Furthermore, the 1H NMR spectra of the CH2CH2 bridge in the paracyclophane structure was consistent with the data reported in the literature, which also identified the 4,7,12,15-tetrachloro isomer [21].
Then, four substituted [2.2]paracyclophanes were synthesized from substituted (4-methylbenzyl)trimethylammonium bromides in aqueous sodium hydroxide solution in the presence of a polymerization inhibitor (Table 4). It was found that the yields of dimer products were improved dramatically compared to the results obtained with silver oxide used for anion exchange reported by Chow [17]. We speculated that the replacement of silver oxide by aqueous sodium hydroxide solution might promote the formation of substituted (4-methylbenzyl)trimethylammonium hydroxide, but we are unable to provide any conclusive evidence at presence. For the dimerization of 12, the [2.2]paracyclophane (17) was obtained in 33% yield, and its structure was confirmed by NMR spectroscopy and elemental analysis. Similarly, dimerization of 13, 14, and 15 resulted in regiospecific 4,16-disubstituted [2.2]paracyclophanes 18, 19, and 20, respectively, in about 35% yield (Table 4, entries 2, 3 and 4). The structures of the synthesized 4,16-disubstituted [2.2]paracyclophanes were also consistent with their NMR spectral data.
Table 4: Synthesis of substituted [2.2]paracyclophanes from substituted (4-methylbenzyl)trimethylammonium bromides.
| Entry | Starting material | Product | Yielda (%) |
|---|---|---|---|
| 1 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-237-i5.svg?max-width=637&scale=1.0)
12 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-12-237-i6.svg?max-width=637&scale=1.0)
17 |
33 (23) |
| 2 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-12-237-i7.svg?max-width=637&scale=1.0)
13 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-12-237-i8.svg?max-width=637&scale=1.0)
18 |
36 (24) |
| 3 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-12-237-i9.svg?max-width=637&scale=1.0)
14 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-12-237-i10.svg?max-width=637&scale=1.0)
19 |
33 (19) |
| 4 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-12-237-i11.svg?max-width=637&scale=1.0)
15 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-12-237-i12.svg?max-width=637&scale=1.0)
20 |
32 (18) |
aIn the presence of 2-chlorophenothiazine. The numbers in parenthesis are the yields in the presence of phenothiazine.
Conclusion
A convenient protocol was reported to synthesize 4,7,12,15-tetrachloro[2.2]paracyclophane. In the first bromination step, 1-(bromomethyl)-2,5-dichloro-4-methylbenzene was synthesized with high yield and selectivity from 2,5-dichloro-p-xylene by using a H2O2–HBr couple in water. The use of H2O2–HBr as a bromide source made this procedure organic-waste-free, organic-solvent-free and an appropriate replacement of the existing bromination methods. In the Winberg elimination–dimerization step, 35% yield of 4,7,12,15-tetrachloro[2.2]paracyclophane was obtained from 2,5-dichloro-(4-methylbenzyl)trimethylammonium bromide and aqueous sodium hydroxide solution in the presence of a polymerization inhibitor, which was about two folds than that used silver oxide as anion exchange. Moreover, four substituted [2.2]paracyclophanes were prepared in this convenient way.
Experimental
General
2,5-Dichloro-p-xylene, para-xylene, 2-chloro-1,4-dimethylbenzene, 2-bromo-1,4-dimethylbenzene and 1-nitro-2,5-dimethylbenzene were purchased from commercial suppliers. All chemicals were used as received without further purification. 1H NMR spectra were recorded in CDCl3 using an AVANCE III 400WB spectrometer. IR spectra were recorded on a Nicolet AVATAR 5700 FTIR spectrophotometer in the range of 4000–400 cm−1 using KBr pellets. Melting points were determined using a Beijing TaiKe X-4 melting point apparatus and were uncorrected. Mass spectra were obtained using an Agilent 1260-6224 spectrometer with electron impact ionization (EI, 70 eV). Elemental analyses were recorded on an Elementar vario MICRO cube.
Typical reaction procedure for visible-light induced bromination with the H2O2–HBr system
Analogous as described in [16], substituted p-xylene (1.0 mmol) was added to 2.0 mL solution (CCl4 or water) of 2.0 mmol of H2O2 (0.23 g, 30% H2O2 aqueous) and 1.1 mmol of HBr (0.22 g, 30% HBr aqueous). The mixture was stirred at 300 rpm at appropriate temperature under irradiation from a 40 W incandescent light bulb. At the end of the bromination reaction (6–20 h), the mixture was transferred into a separating funnel and 4 mL of 0.005 M NaHSO3 was added. The crude product was extracted using 3 × 5 mL CH2Cl2 and the combined organic phase was dried over MgSO4. Then the solvent was evaporated under reduced pressure and the crude mixture was analyzed by 1H NMR spectroscopy. Lastly the products were separated by column chromatography (SiO2, hexane/EtOAc) and identified by comparison with literature data.
2a: colorless oil. 1H NMR (CDCl3) δ 2.24 (s, 3H, ArCH3), 5.12 (s, 2H, ArCH2), 7.29 (s, 1H, ArH), 7.33 (s, 1H, ArH); EIMS m/z: 254, 175, 173, 102.
4a: colorless oil. 1H NMR (CDCl3) δ 2.19 (s, 3H, ArCH3), 4.66 (s, 2H, ArCH2), 7.07–7.11 (m, 2H, ArH), 7.25–7.31 (m, 1H, ArH); anal. calcd for C8H9Br (185.06): C, 51.92; H, 4.90; found: C, 51.81; H, 4.96.
6a: colorless oil. 1H NMR (CDCl3) δ 2.31 (s, 3H, ArCH3), 4.95 (s, 2H, ArCH2), 6.96–6.98 (m, 1H, ArH), 6.99–7.24 (m, 1H, ArH), 7.26–7.37 (m, 1H, ArH); anal. calcd for C8H8BrCl (219.51): C, 43.77; H, 3.67; Cl, 16.15; found: C, 43.68; H, 3.72; Cl, 16.06.
8a: mp 53–55 °C; 1H NMR (CDCl3) δ 2.31 (s, 3H, ArCH3), 4.93 (s, 2H, ArCH2), 7.02–7.54 (m, 3H, ArH); anal. calcd for C8H8BrCl (219.51): C, 43.77; H, 3.67; Cl, 16.15; found: C, 43.68; H, 3.72; Cl, 16.06.
10a: mp 72–74 °C; 1H NMR (CDCl3) δ 2.41 (s, 3H, ArCH3), 4.95 (s, 2H, ArCH2), 6.96–7.37 (m, 3H, ArH); anal. calcd for C8H8BrNO2 (230.06): C, 41.77; H, 3.51; N, 6.09; found: C, 41.68; H, 3.57; N, 6.12.
Typical reaction procedure for the preparation of substituted (4-methylbenzyl)trimethylammonium bromides
Substituted 4-methylbenzyl bromide (5.0 mmol) was added to 50.0 mL Et2O solution in a 100 mL three-necked flask. The mixture was cooled at 0 °C and was stirred at 300 rpm. Me3N was generated by heating an aqueous Me3N solution (40% w/w, 15 mL) and passed into the flask for 4 h. The product was precipitated as a white solid. Then the mixture was stirred at room temperature overnight and the quaternary ammonium salt was obtained on a Büchner funnel and dried in a vacuum oven at 80 °C for 24 h.
11: highly hygroscopic solid. IR (KBr) ν/cm−1: 3004, 1635, 1617, 1477, 1375, 1190, 980.
12: highly hygroscopic solid. IR (KBr) v/cm−1: 2989, 1521, 1483, 1382, 1125, 910, 805, 722.
13: highly hygroscopic solid. IR (KBr) v/cm−1: 2968, 2935, 1632, 1452, 1371, 1154, 725, 672.
14: highly hygroscopic solid. IR (KBr) v/cm−1: 3009, 2946, 1642, 1458, 1381, 1205, 653.
15: highly hygroscopic solid. IR (KBr) v/cm−1: 2979, 1621, 1550, 1508, 1472, 1376, 1345, 1135, 663.
Typical reaction procedure for the synthesis of substituted tetrachloro[2.2]paracyclophanes
In a 100 mL three-necked flask equipped with a stirrer and a Dean–Stark water separator attached to a reflux condenser was placed 15 mL aqueous sodium hydroxide solution (40% w/w) and 45 mL toluene. With vigorous stirring, a solution of benzyltrimethylammonium bromides (50 mmol), dissolved in 5 mL water, was added dropwise in 30 min. The inhibitor (0.15 mmol) was then added to the solution and the mixture was heated under reflux for 4 h. After all water had been separated, a pale yellow solid polymer began to precipitate. When the evolution of Me3N was finished, the reaction system was heated and stirred for another 1 h. The mixture was cooled and the solid was filtrated and washed with toluene (5 mL × 3). The filtrates were combined and evaporated under vacuum to give a solid product which was further washed with hexane (5 mL × 3).
16: white solid, mp >280 °C (dec); 1H NMR (CDCl3) δ 2.91 (m, 2H, ArCH2), 3.26 (m, 2H, ArCH2), 6.95 (s, 2H, ArH); 13C NMR (CDCl3) δ 30.8, 77.0, 131.8, 133.9, 138.6; anal. calcd for C16H12Cl4 (346.07): C, 55.53; H, 3.50; Cl, 40.97; found: C, 55.47; H, 3.62; Cl, 40.89.
17: white solid, mp 281–283 °C; 1H NMR (CDCl3) δ 3.09 (s, 8H, ArCH2), 6.50 (s, 8H, ArH); anal. calcd for C16H16 (208.30): C, 92.26; H, 7.74; found: C, 92.15; H, 7.82.
18: white solid, mp 163–165 °C; 1H NMR (CDCl3) δ 2.85–2.97 (m, 4H, ArCH2), 3.03–3.37 (m, 4H, ArCH2), 6.92–7.54 (m, 6H, ArH). anal. calcd for C16H14Cl2 (277.19): C, 69.33; H, 5.09; Cl, 25.58; found: C, 69.27; H, 5.05; Cl, 25.65.
19: white solid, mp 238–240 °C; 1H NMR (CDCl3) δ 2.86–3.12 (m, 4H, ArCH2), 3.15–3.34 (4H, m, ArCH2), 6.43–7.15 (m, 6H, ArH); anal. calcd for C16H14Br2 (366.10): C, 52.49; H, 3.85; found: C, 52.38; H, 3.82.
20: 1H NMR (CDCl3) δ 2.81–3.07 (m, 4H, ArCH2), 3.27–3.35 (m, 4H, ArCH2), 7.25–8.23 (m, 6H, ArH); anal. calcd for C16H14N2O4 (298.30): C, 64.42; H, 4.73; N, 9.39; found: C, 64.32; H, 4.75; N, 9.45.
Supporting Information
| Supporting Information File 1: Copies of MS, 1H and 13C NMR spectra of the synthesized compounds. | ||
| Format: PDF | Size: 581.8 KB | Download |
References
-
Dolbier, W. R., Jr.; Duan, J.-X.; Roche, A. J. Org. Lett. 2000, 2, 1867–1869. doi:10.1021/ol005943f
Return to citation in text: [1] -
Dolbier, W. R., Jr.; Xie, P.; Zhang, L.; Xu, W.; Chang, Y.; Abboud, K. A. J. Org. Chem. 2008, 73, 2469–2472. doi:10.1021/jo7026849
Return to citation in text: [1] -
Rossen, K.; Pye, P. J.; Maliakal, A.; Volante, R. P. J. Org. Chem. 1997, 62, 6462–6463. doi:10.1021/jo971300a
Return to citation in text: [1] -
Hicks, C.; Duffy, B.; Hargaden, G. C. Org. Chem. Front. 2014, 1, 716–725. doi:10.1039/c4qo00110a
Return to citation in text: [1] -
Morphy, J. R.; Rankovic, Z.; Rees, D. C. Tetrahedron Lett. 1996, 37, 3209–3212. doi:10.1016/0040-4039(96)00497-2
Return to citation in text: [1] -
Seuron, P.; Solladie, G. J. Org. Chem. 1980, 45, 715–719. doi:10.1021/jo01292a033
Return to citation in text: [1] -
Amii, H.; Hayashi, R.; Seo, M.; Katahira, Y.; Kobayashi, A.; Uneyama, K. J. Fluorine Chem. 2013, 152, 90–93. doi:10.1016/j.jfluchem.2013.04.001
Return to citation in text: [1] -
Paradies, J. Synthesis 2011, 3749–3766. doi:10.1055/s-0031-1289296
Return to citation in text: [1] -
Bartholomew, G. P.; Bazan, G. C. J. Am. Chem. Soc. 2002, 124, 5183–5196. doi:10.1021/ja0121383
Return to citation in text: [1] -
Pravst, I.; Zupan, M.; Stavber, S. Green Chem. 2006, 8, 1001–1005. doi:10.1039/B608446J
Return to citation in text: [1] -
Heropoulos, G. A.; Cravotto, G.; Screttas, C. G.; Steele, B. R. Tetrahedron Lett. 2007, 48, 3247–3250. doi:10.1016/j.tetlet.2007.03.023
Return to citation in text: [1] -
Pravst, I.; Zupan, M.; Stavber, S. Tetrahedron 2008, 64, 5191–5199. doi:10.1016/j.tet.2008.03.048
Return to citation in text: [1] -
Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Green Chem. 2007, 9, 1212–1218. doi:10.1039/b707065a
Return to citation in text: [1] -
Guha, S. K.; Wu, B.; Kim, B. S.; Baik, W.; Koo, S. Tetrahedron Lett. 2006, 47, 291–293. doi:10.1016/j.tetlet.2005.11.023
Return to citation in text: [1] -
Podgoršek, A.; Stavber, S.; Zupan, M. Tetrahedron 2009, 65, 4429–4439. doi:10.1016/j.tet.2009.03.034
Return to citation in text: [1] -
Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Tetrahedron Lett. 2006, 47, 7245–7247. doi:10.1016/j.tetlet.2006.07.109
Return to citation in text: [1] [2] [3] [4] -
Chow, H.-F.; Low, K.-H.; Wong, K. Y. Synlett 2005, 2130–2134. doi:10.1055/s-2005-872270
Return to citation in text: [1] [2] [3] [4] -
Winberg, H. E.; Fawcett, F. S.; Mochel, W. E.; Theobald, C. W. J. Am. Chem. Soc. 1960, 82, 1428–1435. doi:10.1021/ja01491a037
Return to citation in text: [1] -
Winberg, H. E.; Fawcett, F. S. Org. Synth., Coll. Vol. V; John Wiley and Sons, Ltd.: New York, 1973; pp 883–886.
Return to citation in text: [1] -
Galley, R. A.; Landon, R. S.; Senior, K. C. [2.2]paracyclophane and derivatives thereof. U.S. Patent 5302767, April 12, 1994.
Return to citation in text: [1] -
Dix, I.; Hopf, H.; Satyanarayana, T. B. N.; Ernst, L. Beilstein J. Org. Chem. 2010, 6, 932–937. doi:10.3762/bjoc.6.104
Return to citation in text: [1]
| 17. | Chow, H.-F.; Low, K.-H.; Wong, K. Y. Synlett 2005, 2130–2134. doi:10.1055/s-2005-872270 |
| 17. | Chow, H.-F.; Low, K.-H.; Wong, K. Y. Synlett 2005, 2130–2134. doi:10.1055/s-2005-872270 |
| 21. | Dix, I.; Hopf, H.; Satyanarayana, T. B. N.; Ernst, L. Beilstein J. Org. Chem. 2010, 6, 932–937. doi:10.3762/bjoc.6.104 |
| 1. | Dolbier, W. R., Jr.; Duan, J.-X.; Roche, A. J. Org. Lett. 2000, 2, 1867–1869. doi:10.1021/ol005943f |
| 2. | Dolbier, W. R., Jr.; Xie, P.; Zhang, L.; Xu, W.; Chang, Y.; Abboud, K. A. J. Org. Chem. 2008, 73, 2469–2472. doi:10.1021/jo7026849 |
| 3. | Rossen, K.; Pye, P. J.; Maliakal, A.; Volante, R. P. J. Org. Chem. 1997, 62, 6462–6463. doi:10.1021/jo971300a |
| 4. | Hicks, C.; Duffy, B.; Hargaden, G. C. Org. Chem. Front. 2014, 1, 716–725. doi:10.1039/c4qo00110a |
| 9. | Bartholomew, G. P.; Bazan, G. C. J. Am. Chem. Soc. 2002, 124, 5183–5196. doi:10.1021/ja0121383 |
| 17. | Chow, H.-F.; Low, K.-H.; Wong, K. Y. Synlett 2005, 2130–2134. doi:10.1055/s-2005-872270 |
| 20. | Galley, R. A.; Landon, R. S.; Senior, K. C. [2.2]paracyclophane and derivatives thereof. U.S. Patent 5302767, April 12, 1994. |
| 7. | Amii, H.; Hayashi, R.; Seo, M.; Katahira, Y.; Kobayashi, A.; Uneyama, K. J. Fluorine Chem. 2013, 152, 90–93. doi:10.1016/j.jfluchem.2013.04.001 |
| 17. | Chow, H.-F.; Low, K.-H.; Wong, K. Y. Synlett 2005, 2130–2134. doi:10.1055/s-2005-872270 |
| 5. | Morphy, J. R.; Rankovic, Z.; Rees, D. C. Tetrahedron Lett. 1996, 37, 3209–3212. doi:10.1016/0040-4039(96)00497-2 |
| 6. | Seuron, P.; Solladie, G. J. Org. Chem. 1980, 45, 715–719. doi:10.1021/jo01292a033 |
| 18. | Winberg, H. E.; Fawcett, F. S.; Mochel, W. E.; Theobald, C. W. J. Am. Chem. Soc. 1960, 82, 1428–1435. doi:10.1021/ja01491a037 |
| 19. | Winberg, H. E.; Fawcett, F. S. Org. Synth., Coll. Vol. V; John Wiley and Sons, Ltd.: New York, 1973; pp 883–886. |
| 15. | Podgoršek, A.; Stavber, S.; Zupan, M. Tetrahedron 2009, 65, 4429–4439. doi:10.1016/j.tet.2009.03.034 |
| 16. | Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Tetrahedron Lett. 2006, 47, 7245–7247. doi:10.1016/j.tetlet.2006.07.109 |
| 14. | Guha, S. K.; Wu, B.; Kim, B. S.; Baik, W.; Koo, S. Tetrahedron Lett. 2006, 47, 291–293. doi:10.1016/j.tetlet.2005.11.023 |
| 16. | Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Tetrahedron Lett. 2006, 47, 7245–7247. doi:10.1016/j.tetlet.2006.07.109 |
| 13. | Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Green Chem. 2007, 9, 1212–1218. doi:10.1039/b707065a |
| 16. | Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Tetrahedron Lett. 2006, 47, 7245–7247. doi:10.1016/j.tetlet.2006.07.109 |
| 10. | Pravst, I.; Zupan, M.; Stavber, S. Green Chem. 2006, 8, 1001–1005. doi:10.1039/B608446J |
| 11. | Heropoulos, G. A.; Cravotto, G.; Screttas, C. G.; Steele, B. R. Tetrahedron Lett. 2007, 48, 3247–3250. doi:10.1016/j.tetlet.2007.03.023 |
| 12. | Pravst, I.; Zupan, M.; Stavber, S. Tetrahedron 2008, 64, 5191–5199. doi:10.1016/j.tet.2008.03.048 |
| 16. | Podgoršek, A.; Stavber, S.; Zupan, M.; Iskra, J. Tetrahedron Lett. 2006, 47, 7245–7247. doi:10.1016/j.tetlet.2006.07.109 |
© 2016 Pan et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)