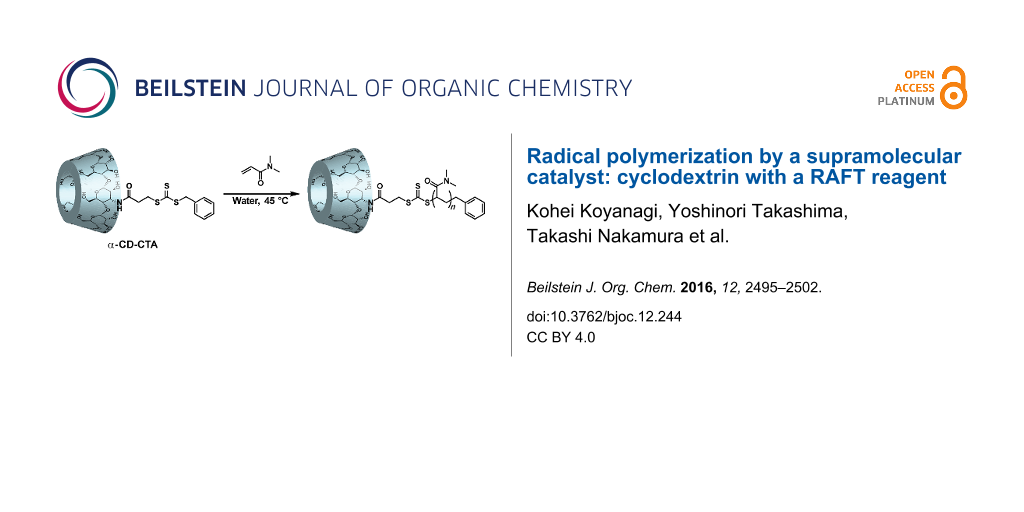
Abstract
Supramolecular catalysts have received a great deal of attention because they improve the selectivity and efficiency of reactions. Catalysts with host molecules exhibit specific reaction properties and recognize substrates via host–guest interactions. Here, we examined radical polymerization reactions with a chain transfer agent (CTA) that has α-cyclodextrin (α-CD) as a host molecule (α-CD-CTA). Prior to the polymerization of N,N-dimethylacrylamide (DMA), we investigated the complex formation of α-CD with DMA. Single X-ray analysis demonstrated that α-CD includes DMA inside its cavity. When DMA was polymerized in the presence of α-CD-CTA using 2,2'-azobis[2-(2-imidazolin-2-yl)propane dihydrochloride (VA-044) as an initiator in an aqueous solution, poly(DMA) was obtained in good yield and with narrow molecular weight distribution. In contrast, the polymerization of DMA without α-CD-CTA produced more widely distributed polymers. In the presence of 1,6-hexanediol (C6 diol) which works as a competitive molecule by being included in the α-CD cavity, the reaction yield was lower than that without C6 diol.
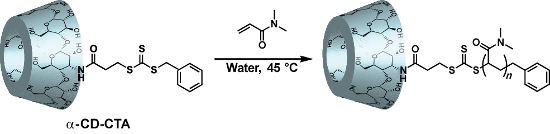
Graphical Abstract
Introduction
The folding of proteins in biological systems, the replication of DNA, and specific substrate recognition by enzymes play important roles in forming supramolecular structures, achieving functions, and maintaining life [1-6]. The crystal structures of RNA polymerase, DNA polymerase, and λ-exonuclease demonstrate that the cylindrical cavities of enzymes can effectively recognize substrates to produce biological polymers [1-6]. Cyclodextrins (CDs) have been widely used as substrate-recognition moieties in artificial enzymes [7-15], which have been used in the hydrolysis of activated esters [16-19] and as phase-transfer catalysts [20-28]. Moreover, via complex formation, modern supramolecular catalysts [29-33] have been used to achieve various highly efficient and selective reactions, including hydrolysis reactions [10-15], C–H bond activation [34-36], olefin epoxidation [37-39], Diels–Alder reactions [40-42], 1,3-dipole cycloadditions [43,44], and polymerizations [45-47], among others. Selective substrate recognition and activation are essential functions of supramolecular catalysts.
CD derivatives are widely used in radical polymerization to dissolve hydrophobic monomers in aqueous solutions [48-54] and to control the aggregation of polymers [55-58]. Although supramolecular catalysts with CDs as monomer recognition sites and catalytic active sites have been designed for polymerization reactions, relatively few reports have described a catalytic design in which the catalytic active site does not leave the CD monomer recognition site during the growing step. In a previous design of radical initiators with CDs, the radical-initiating end group leaves the CD monomer recognition site [59,60]. With this molecular design, an included monomer is distant from the radical species and cannot be involved in the direct polymerization. Here, we will observe the effect of monomer recognition of CD on polymerization if a supramolecular polymerization catalyst capable of inserting the monomer between the active and binding sites can be designed. Based on this concept, we have reported that CDs can include and activate lactones to yield a polymer with a single CD at the end of the polymer chain [61-64]. Subsequently, we reported ring-opening metathesis polymerization involving the use of a Ru complex with a CD-derived monophosphine ligand [47]. In the design of the supramolecular polymerization catalysts, monomers are inserted between the initiating end group and the growing polymer chain.
In this study, the monomer recognition site is introduced to a reversible addition–fragmentation chain transfer (RAFT) polymerization system [65-69]. We have synthesized a chain transfer agent (CTA) bearing the CD moiety (CD-CTA) and have investigated this agent’s polymerization behavior. The polymerization rate constant decreased with the addition of competitive molecules, indicating that complexation between CD-CTA and the monomer plays an important role in determining polymerization rate.
Results and Discussion
Preparation of α-CD-CTA
We designed a CTA reagent with α-CD or β-CD. Figure 1 illustrates the preparation of α-CD-CTA. Mercaptopropionic acid was reacted with benzyl bromide, K3PO4, and carbon bisulfide (CS2) in acetone to afford a trithiocarbonyl derivative, a CTA with a carboxylic acid (CTA-COOH). α-CD-CTA was prepared in 50% yield by the reaction of CTA-COOH and 3-NH2-α-CD with N,N'-dicyclohexylcarbodiimide (DCC)/1-hydroxybenzotriazole (HOBt) in DMF. The α-CD-CTA was purified using reverse-phase chromatography. β-CD-CTA was prepared using the same method as α-CD-CTA in 45% yield (see Supporting Information File 1). α-CD-CTA and β-CD-CTA can be dissolved in water. However, the solubility of β-CD-CTA in water was significantly low, leading to the formation of precipitates. β-CD-CTA forms a self-inclusion complex or a supramolecular dimer complex, which was characterized using 2D ROESY NMR (Supporting Information File 1, Figure S4). We focused on the polymerization activity of α-CD-CTA because the β-CD cavity of β-CD-CTA was capped by the CTA unit, inhibiting the molecular recognition property.
![[1860-5397-12-244-1]](/bjoc/content/figures/1860-5397-12-244-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Preparation scheme of α-CD-CTA.
Figure 1: Preparation scheme of α-CD-CTA.
Crystal structure of the α-CD-DMA and β-CD-DMA complexes
We chose N,N-dimethylacrylamide (DMA), acrylic acid (AA), and acrylamide (AAm) as water-soluble vinyl monomers for radical polymerization. Prior to studying the polymerization of vinyl monomers, we investigated the complex formation of CDs with vinyl monomers. When mixing α-CD and DMA, we obtained single crystals suitable for X-ray crystallography analysis. The X-ray crystallography analysis is important to understand the complex in the condensed phase. Figure 2a shows the crystal structure of α-CD with DMA. α-CD formed a head-to-tail channel structure in the crystal. Figure 2b shows the schematic diagram of the crystal structure of α-CD/DMA. The stoichiometry of α-CD and encapsulated DMA was 1:1.
The N,N-dimethylamino group was located at the wider rim (secondary hydroxy group) of the α-CD cavity and the vinyl group was pointed toward the opposite direction. Based on the crystal structure, it is speculated that the modification of the wider rim of α-CD with the CTA group would bring the vinyl group of DMA and reactive radical species close together during the course of RAFT polymerization.
Figure S5a (Supporting Information File 1) shows the crystal structure of β-CD with DMA. β-CD formed a head-to-head dimeric structure in the crystal. Figure S5b shows a schematic diagram of the crystal structure of β-CD/DMA. The stoichiometry of β-CD and encapsulated DMA was 2:3. For two of the DMA molecules, the N,N-dimethylamino and the vinyl groups were located at the wider and the narrower rims of the β-CD cavity, respectively. The other one of the DMA molecules was packed between the two β-CD molecules in an equatorial plane.
When mixing α-CD and AA in water, we obtained a powder precipitate of a complex between α-CD and AA, although the crystal structure of α-CD and AA was not solved. The formation of precipitate implied the formation of a host-guest complex between them. A mixture of CDs (α-CD, β-CD and γ-CD) and AAm did not result in a precipitate, indicating that the affinities of the CDs for AAm were low.
![[1860-5397-12-244-2]](/bjoc/content/figures/1860-5397-12-244-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Crystal structure of α-CD with N,N-dimethylacrylamide (DMA). (a) The structure of an 1:1 inclusion complex between α-CD and DMA. One of the disordered pattern of DMA is shown. DMA molecules outside the α-CD, hydrogen atoms, and water molecules are omitted for clarity. Colors of the atoms are based on CPK coloring. DMA, space-filling model; CD, stick model. (b) A schematic diagram of the inclusion complex of α-CD/DMA.
Figure 2: Crystal structure of α-CD with N,N-dimethylacrylamide (DMA). (a) The structure of an 1:1 inclusion ...
Polymerization of vinyl monomers mediated by α-CD-CTA
α-CD-CTA-mediated polymerizations of water-soluble vinyl monomers were performed in aqueous media, where the strong molecular recognition property of α-CD is expected due to the hydrophobic effect. As a water-soluble radical initiator, we selected 2,2'-azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (VA-044). Two hundred equivalents (equiv) of monomers, 1 equiv of α-CD-CTA, and 0.4 equiv of VA-044 were mixed in water (monomer: 1 mol/kg). After the reactants were mixed and degassed, the sample tube was heated at 45 °C in an Ar atmosphere. After 24 hours, the polymer solution was lyophilized. The resulting polymers were dissolved in 8 mL MeOH or chloroform, then reprecipitated into 80 mL diethyl ether. This cycle repeated twice to remove monomer. The structures of the obtained polymers were characterized by 1H NMR spectroscopy (Figure S6, Supporting Information File 1). The 1H NMR signals of the benzyl group and those of the CD moiety of α-CD-CTA were both observed in the obtained polymer, which indicates that both groups did not leave the polymer main chain after the radical polymerization and that α-CD-CTA functioned as a good chain transfer agent. Some end groups are formed from the VA 044 initiator.
Table 1 shows results for VA-044-initiated polymerizations of DMA, AA and AAm. Polymers’ molecular weights and distributions were determined by gel permeation chromatography (GPC). The polymerization of DMA and AA mediated by α-CD-CTA gave polymers with similar molecular weights (12.8 and 14.8 kDa) and narrow distributions in good yields (85–88%) (Table 1, entries 2 and 5). In contrast, in the absence of α-CD-CTA, free radical polymerization with VA-044 gave polymers with higher molecular weights and wider distributions (Table 1, entries 1 and 4). In order to investigate the effect of encapsulation of monomer by α-CD moiety, the reactions in the presence of 1,6-hexanediol (C6 diol) as a competitive guest molecule were also investigated. C6 diol was selected as a non-ionic molecule with a high association constant to α-CD (Ka = 134 M−1 ) [70]. In the presence of C6 diol, the reaction yields dropped to 63–72%, whereas the molecular weights of the polymers increased (22.9 and 19.4 kDa in entries 3 and 6, respectively). It is considered that by inhibiting the molecular recognition of α-CD, the inclusion complexation ratio between the monomer and α-CD-CTA was decreased, which lead to lower yields and higher molecular weight of the resulting polymers due to preceding free radical polymerization. In the reaction of AAm monomer, which has a low affinity with α-CD, the properties of the obtained polymers were similar regardless of the presence of the α-CD-CTA or the competitive C6 diol guest. The polymerization of AAm mediated by α-CD-CTA gave polymers with slightly narrower distributions. From the series of experiments, it was demonstrated that the molecular recognition property of α-CD-CTA effectively affected the chain transfer step.
Table 1: Polymerizations of water-soluble vinyl monomers, mediated by α-CD-CTAa.
| Entry | CTAb | Monomerb | Competitorc | Mn / 103 d | Mw / Mnd | Yield% |
|---|---|---|---|---|---|---|
| 1 | – | DMA | – | 9679 | >10 | 96 |
| 2 | α-CD-CTA | DMA | – | 12.8 | 1.2 | 85 |
| 3 | α-CD-CTA | DMA | C6 diol | 22.9 | 1.3 | 63 |
| 4 | – | AA | – | 45.9 | >10 | 98 |
| 5 | α-CD-CTA | AA | – | 14.8 | 1.2 | 88 |
| 6 | α-CD-CTA | AA | C6 diol | 19.4 | 1.2 | 72 |
| 7e | – | AAm | – | 3841 | >10 | 98 |
| 8e | α-CD-CTA | AAm | – | 10.4 | 1.3 | 98 |
| 9e | α-CD-CTA | AAm | C6 diol | 9.9 | 1.4 | 99 |
aPolymerization was performed at 45 °C for 24 h, with VA-044 as an initiator. b[Monomer]0/[CTA]0/[I]0 = 200/1/0.4 ([Monomer]0 = 1 M). c[Competitor]0/[CTA]0 = 50/1. dMn and Mw/Mn were determined by GPC using polystyrene sulfonate sodium salt (PSSNa) or polyacrylamide (PAAm) as references. ePolymerization of AAm was performed in acetate buffer [71,72].
Rate and mechanism of α-CD-CTA-mediated polymerization
We investigated the polymerization rate of DMA mediated by α-CD-CTA and the effect of the competitive molecule in water. A mixture of DMA (200 equiv), α-CD-CTA (1 equiv), and VA-044 (0.4 equiv) in D2O (0.25 mol/kg) was degassed and heated at 45 °C in an Ar atmosphere. When a concentration of 1 mol/kg was used, the polymerization rate was too fast to trace during the initial stages of polymerization. Therefore, we chose to utilize a concentration of 0.25 mol/kg because this concentration allowed the polymerization rate to be easily followed. At regular time intervals, a 0.6 mL sample was collected and analyzed using 1H NMR spectroscopy. The conversions were calculated using the ratio between the integral values for the polymer main chain and the vinyl group of the DMA monomer.
Figure 3a, b and c shows time-conversion curves, kinetic plots, and Mn-conversion for the polymerization of DMA mediated by α-CD-CTA. The time-conversion plots indicate the induction period in the presence and absence of C6 diol. Kinetic rates were determined using the least-square method after the induction period. The presence of C6 diol made a clear difference in the polymerization rate by α-CD-CTA. The kinetic rate was faster for α-CD-CTA (k = 2.2 h−1) than for α-CD-CTA/C6 diol (k = 1.7 h−1). This finding supports that C6 diol inhibits monomer recognition by the α-CD cavity, which was also indicated by the series of experiments in Table 1.
![[1860-5397-12-244-3]](/bjoc/content/figures/1860-5397-12-244-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Time-conversion curves (a), kinetic plots (b) and plots of number-average molecular weight (Mn) versus conversion (c) for the α-CD-CTA-mediated polymerization of DMA. The blue rhombic plots correspond to the α-CD-CTA-mediated polymerization of DMA. The red square plots correspond to the α-CD-CTA/C6 diol-mediated polymerization of DMA.
Figure 3: Time-conversion curves (a), kinetic plots (b) and plots of number-average molecular weight (Mn) ver...
Figure 4 shows the proposed polymerization mechanism of a water-soluble vinyl monomer with α-CD-CTA. The radical (I•) derived from VA-044 attacks the monomer to produce a polymeric radical (Pn•). Pn• reacts with α-CD-CTA to generate the intermediate α-CD-CTA-Pn•. α-CD-CTA-Pn• is held in an equilibrium state with the macro-CTA, α-CD-CTA-Pn, and the benzyl radical (Bn•) with re-initiation activity. As demonstrated by the inhibition experiments, the yields and molecular weights of the polymers were affected by the molecular recognition property of α-CD-CTA. Based on these results, Pn• and Bn• preferentially attack the included monomer to afford the growing polymer in high yield. Therefore, the kinetic rate is faster for α-CD-CTA than for α-CD-CTA/C6 diol, which produced polymers with narrow distribution. In the polymerization reaction, some of the polymer chains (Pn•) form dead chains through termination or irreversible chain transfer. However, the Mw/Mn of the polymer mediated by α-CD-CTA agents that the termination reaction is suppressed by α-CD-CTA compared to free radical polymerization.
![[1860-5397-12-244-4]](/bjoc/content/figures/1860-5397-12-244-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Proposed polymerization mechanism for a water-soluble vinyl monomer with α-CD-CTA as a chain transfer reagent. 2,2'-Azobis[2-(2-imidazolin-2-yl)propane] dihydrochloride (VA-044) was used as a water-soluble radical initiator.
Figure 4: Proposed polymerization mechanism for a water-soluble vinyl monomer with α-CD-CTA as a chain transf...
Conclusion
We studied the radical polymerization of water-soluble vinyl monomers using CD-CTA with molecular recognition property. α-CD was found to include a DMA monomer in a 1:1 manner, which was characterized using single X-ray crystallography analysis. The polymerization of DMA with α-CD-CTA resulted in poly(DMA) with a narrow distribution in high yield, whereas a low yield was obtained for polymerization in the presence of C6 diol, a competitive guest molecule for α-CD. In contrast, the polymerization of AAm, which had a lower affinity for α-CD, was not affected by α-CD-CTA in either the presence or absence of C6 diol. These results indicate that the chain transfer reagent modified with a host molecule provided the site for a reaction between the end group of the growing polymer and monomers. Currently, we are investigating the preparation of supramolecular catalysts with a chain transfer reagent with two CDs to recognize monomers and the growing polymer chain, and exploring its function mimicking a biological molecular clamp.
Supporting Information
| Supporting Information File 1: The preparation of α-CD-CTA and β-CD-CTA and typical polymerization methods. | ||
| Format: PDF | Size: 779.3 KB | Download |
| Supporting Information File 2: Crystallographic information file for α-CD-DMA. | ||
| Format: CIF | Size: 429.9 KB | Download |
| Supporting Information File 3: Crystallographic information file for β-CD-DMA. | ||
| Format: CIF | Size: 163.3 KB | Download |
Acknowledgements
This work was financially supported by the ImPACT Program of Council for Science, Technology and Innovation (Cabinet Office, Government of Japan), a Grant-in-Aid for Scientific Research (B) (no. 26288062) from MEXT of Japan, and the Research Grant Program of the Asahi Glass Foundation.
References
-
Mooney, R. A.; Landick, R. Cell 1999, 98, 687–690. doi:10.1016/S0092-8674(00)81483-X
Return to citation in text: [1] [2] -
Kovall, R.; Matthews, B. W. Science 1997, 277, 1824–1827. doi:10.1126/science.277.5333.1824
Return to citation in text: [1] [2] -
Trakselis, M. A.; Alley, S. C.; Abel-Santos, E.; Benkovic, S. J. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 8368–8375. doi:10.1073/pnas.111006698
Return to citation in text: [1] [2] -
Benkovic, S. J.; Valentine, A. M.; Salinas, F. Annu. Rev. Biochem. 2001, 70, 181–208. doi:10.1146/annurev.biochem.70.1.181
Return to citation in text: [1] [2] -
Breyer, W. A.; Matthews, B. W. Protein Sci. 2001, 10, 1699–1711. doi:10.1110/ps.10301
Return to citation in text: [1] [2] -
Kool, E. T. Acc. Chem. Res. 1998, 31, 502–510. doi:10.1021/ar9602462
Return to citation in text: [1] [2] -
Mallick, I. M.; D'Souza, V. T.; Yamaguchi, M.; Lee, J.; Chalabi, P.; Gadwood, R. C.; Bender, M. L. J. Am. Chem. Soc. 1984, 106, 7252–7254. doi:10.1021/ja00335a070
Return to citation in text: [1] -
D'Souza, V. T.; Hanabusa, K.; O'Leary, T.; Gadwood, R. C.; Bender, M. L. Biochem. Biophys. Res. Commun. 1985, 129, 727–732. doi:10.1016/0006-291X(85)91952-7
Return to citation in text: [1] -
D'Souza, V. T.; Bender, M. L. Acc. Chem. Res. 1987, 20, 146–152. doi:10.1021/ar00136a004
Return to citation in text: [1] -
Komiyama, M.; Breaux, E. J.; Bender, M. L. Bioorg. Chem. 1977, 6, 127–136. doi:10.1016/0045-2068(77)90015-3
Return to citation in text: [1] [2] -
Czarniecki, M. F.; Breslow, R. J. Am. Chem. Soc. 1978, 100, 7771–7772. doi:10.1021/ja00492a079
Return to citation in text: [1] [2] -
Breslow, R.; Czarniecki, M. F.; Emert, J.; Hamaguchi, H. J. Am. Chem. Soc. 1980, 102, 762–770. doi:10.1021/ja00522a054
Return to citation in text: [1] [2] -
Breslow, R.; Trainor, G. L.; Ueno, A. J. Am. Chem. Soc. 1983, 105, 2739–2744. doi:10.1021/ja00347a037
Return to citation in text: [1] [2] -
Breslow, R. Acc. Chem. Res. 1991, 24, 317–324. doi:10.1021/ar00011a001
Return to citation in text: [1] [2] -
Breslow, R. Artificial Enzymes; Wiley-VCH Verlag GmbH & Co. KGaA, 2006.
Return to citation in text: [1] [2] -
Tanaka, Y.; Sakuraba, H.; Nakanishi, H. J. Chem. Soc., Chem. Commun. 1983, 947–948. doi:10.1039/C39830000947
Return to citation in text: [1] -
Sakuraba, H.; Nakai, T.; Tanaka, Y. J. Inclusion Phenom. 1984, 2, 829–839. doi:10.1007/BF00662252
Return to citation in text: [1] -
Trost, B. M.; Van Vranken, D. L. J. Am. Chem. Soc. 1990, 112, 1261–1263. doi:10.1021/ja00159a065
Return to citation in text: [1] -
Trost, B. M.; Van Vranken, D. L. J. Am. Chem. Soc. 1993, 115, 444–458. doi:10.1021/ja00055a013
Return to citation in text: [1] -
Harada, A.; Hu, Y.; Takahashi, S. Chem. Lett. 1986, 2083–2084. doi:10.1246/cl.1986.2083
Return to citation in text: [1] -
Hu, Y.; Harada, A.; Takahashi, S. Synth. Commun. 1988, 18, 1607–1610. doi:10.1080/00397918808081320
Return to citation in text: [1] -
van Dongen, S. F. M.; Elemans, J. A. A. W.; Rowan, A. E.; Nolte, R. J. M. Angew. Chem., Int. Ed. 2014, 53, 11420–11428. doi:10.1002/anie.201404848
Return to citation in text: [1] -
Hubert, C.; Denicourt-Nowicki, A.; Roucoux, A.; Landy, D.; Leger, B.; Crowyn, G.; Monflier, E. Chem. Commun. 2009, 1228–1230. doi:10.1039/b818786j
Return to citation in text: [1] -
Bricout, H.; Hapiot, F.; Ponchel, A.; Tilloy, S.; Monflier, E. Curr. Org. Chem. 2010, 14, 1296–1307. doi:10.2174/138527210791616920
Return to citation in text: [1] -
Hapiot, F.; Bricout, H.; Tilloy, S.; Monflier, E. Eur. J. Inorg. Chem. 2012, 1571–1578. doi:10.1002/ejic.201101316
Return to citation in text: [1] -
Noël, S.; Léger, B.; Ponchel, A.; Philippot, K.; Denicourt-Nowicki, A.; Roucoux, A.; Monflier, E. Catal. Today 2014, 235, 20–32. doi:10.1016/j.cattod.2014.03.030
Return to citation in text: [1] -
Hapiot, F.; Bricout, H.; Menuel, S.; Tilloy, S.; Monflier, E. Catal. Sci. Technol. 2014, 4, 1899–1908. doi:10.1039/c4cy00005f
Return to citation in text: [1] -
Vanbésien, T.; Monflier, E.; Hapiot, F. ACS Catal. 2015, 5, 4288–4292. doi:10.1021/acscatal.5b00861
Return to citation in text: [1] -
Lehn, J.-M. Supramolecular Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA, 1995.
Return to citation in text: [1] -
van Leeuwen, P. W. N. M. Supramolecular Catalysis; Wiley-VCH Verlag GmbH & Co. KGaA; Vol. 2008.
Return to citation in text: [1] -
Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P. W. N. M. Chem. Soc. Rev. 2014, 43, 1660–1733. doi:10.1039/C3CS60027K
Return to citation in text: [1] -
Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P. W. N. M. Chem. Soc. Rev. 2014, 43, 1734–1787. doi:10.1039/C3CS60037H
Return to citation in text: [1] -
Vriezema, D. M.; Aragonès, M. C.; Elemans, J. A. A. W.; Cornelissen, J. J. L. M.; Rowan, A. E.; Nolte, R. J. M. Chem. Rev. 2005, 105, 1445–1490. doi:10.1021/cr0300688
Return to citation in text: [1] -
Leung, D. H.; Fiedler, D.; Bergman, R. G.; Raymond, K. N. Angew. Chem., Int. Ed. 2004, 43, 963–966. doi:10.1002/anie.200352772
Return to citation in text: [1] -
Das, S.; Incarvito, C. D.; Crabtree, R. H.; Brudvig, G. W. Science 2006, 312, 1941–1943. doi:10.1126/science.1127899
Return to citation in text: [1] -
Leung, D. H.; Bergman, R. G.; Raymond, K. N. J. Am. Chem. Soc. 2006, 128, 9781–9797. doi:10.1021/ja061412w
Return to citation in text: [1] -
Thordarson, P.; Bijsterveld, E. J. A.; Rowan, A. E.; Nolte, R. J. M. Nature 2003, 424, 915–918. doi:10.1038/nature01925
Return to citation in text: [1] -
Coumans, R. G. E.; Elemans, J. A. A. W.; Nolte, R. J. M.; Rowan, A. E. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 19647–19651. doi:10.1073/pnas.0603036103
Return to citation in text: [1] -
Jónsson, S.; Odille, F. G. J.; Norrby, P.-O.; Warnmark, K. Chem. Commun. 2005, 549–551. doi:10.1039/B411978A
Return to citation in text: [1] -
Walter, C. J.; Anderson, H. L.; Sanders, J. K. M. J. Chem. Soc., Chem. Commun. 1993, 458–460. doi:10.1039/C39930000458
Return to citation in text: [1] -
Marty, M.; Clyde-Watson, Z.; Twyman, L. J.; Nakash, M.; Sanders, J. K. M. Chem. Commun. 1998, 2265–2266. doi:10.1039/a806070c
Return to citation in text: [1] -
Yoshizawa, M.; Tamura, M.; Fujita, M. Science 2006, 312, 251–254. doi:10.1126/science.1124985
Return to citation in text: [1] -
Mock, W. L.; Irra, T. A.; Wepsiec, J. P.; Adhya, M. J. Org. Chem. 1989, 54, 5302–5308. doi:10.1021/jo00283a024
Return to citation in text: [1] -
Chen, J.; Rebek, J., Jr. Org. Lett. 2002, 4, 327–329. doi:10.1021/ol0168115
Return to citation in text: [1] -
Armspach, D.; Matt, D.; Peruch, F.; Lutz, P. Eur. J. Inorg. Chem. 2003, 805–809. doi:10.1002/ejic.200390109
Return to citation in text: [1] -
Jouffroy, M.; Armspach, D.; Matt, D.; Osakada, K.; Takeuchi, D. Angew. Chem., Int. Ed. 2016, 55, 8367–8370. doi:10.1002/anie.201603191
Return to citation in text: [1] -
Takashima, Y.; Uramatsu, K.; Jomori, D.; Harima, A.; Otsubo, M.; Yamaguchi, H.; Harada, A. ACS Macro Lett. 2013, 2, 384–387. doi:10.1021/mz4001942
Return to citation in text: [1] [2] -
Köllisch, H. S.; Barner-Kowollik, C.; Ritter, H. Chem. Commun. 2009, 1097–1099. doi:10.1039/b818897a
Return to citation in text: [1] -
Storsberg, J.; Ritter, H. Macromol. Rapid Commun. 2000, 21, 236–241. doi:10.1002/(SICI)1521-3927(20000301)21:5<236::AID-MARC236>3.0.CO;2-K
Return to citation in text: [1] -
Glöckner, P.; Metz, N.; Ritter, H. Macromolecules 2000, 33, 4288–4290. doi:10.1021/ma992012e
Return to citation in text: [1] -
Storsberg, J.; Hartenstein, M.; Müller, A. H. E.; Ritter, H. Macromol. Rapid Commun. 2000, 21, 1342–1346. doi:10.1002/1521-3927(20001201)21:18<1342::AID-MARC1342>3.0.CO;2-Z
Return to citation in text: [1] -
Hetzer, M.; Schmidt, B. V. K. J.; Barner-Kowollik, C.; Ritter, H. J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 2504–2517. doi:10.1002/pola.26644
Return to citation in text: [1] -
Köllisch, H.; Barner-Kowollik, C.; Ritter, H. Macromol. Rapid Commun. 2006, 27, 848–853. doi:10.1002/marc.200600067
Return to citation in text: [1] -
Choi, S. W.; Kretschmann, O.; Ritter, H.; Ragnoli, M.; Galli, G. Macromol. Chem. Phys. 2003, 204, 1475–1479. doi:10.1002/macp.200350010
Return to citation in text: [1] -
Yao, F.; Xu, L.; Fu, G.-D.; Lin, B. Macromolecules 2010, 43, 9761–9770. doi:10.1021/ma102039n
Return to citation in text: [1] -
Liu, Y.-Y.; Zhong, Y.-B.; Nan, J.-K.; Tian, W. Macromolecules 2010, 43, 10221–10230. doi:10.1021/ma1019973
Return to citation in text: [1] -
Chechik, V.; Ionita, G. Org. Biomol. Chem. 2006, 4, 3505–3510. doi:10.1039/b607676a
Return to citation in text: [1] -
Ionita, G.; Chechik, V. Org. Biomol. Chem. 2005, 3, 3096–3098. doi:10.1039/b508256k
Return to citation in text: [1] -
Kakuchi, T.; Narumi, A.; Miura, Y.; Matsuya, S.; Sugimoto, N.; Satoh, T.; Kaga, H. Macromolecules 2003, 36, 3909–3913. doi:10.1021/ma021295z
Return to citation in text: [1] -
Kakuchi, T.; Narumi, A.; Matsuda, T.; Miura, Y.; Sugimoto, N.; Satoh, T.; Kaga, H. Macromolecules 2003, 36, 3914–3920. doi:10.1021/ma021296r
Return to citation in text: [1] -
Takashima, Y.; Osaki, M.; Harada, A. J. Am. Chem. Soc. 2004, 126, 13588–13589. doi:10.1021/ja047171e
Return to citation in text: [1] -
Osaki, M.; Takashima, Y.; Yamaguchi, H.; Harada, A. J. Am. Chem. Soc. 2007, 129, 14452–14457. doi:10.1021/ja075140o
Return to citation in text: [1] -
Harada, A.; Osaki, M.; Takashima, Y.; Yamaguchi, H. Acc. Chem. Res. 2008, 41, 1143–1152. doi:10.1021/ar800079v
Return to citation in text: [1] -
Takashima, Y.; Osaki, M.; Ishimaru, Y.; Yamaguchi, H.; Harada, A. Angew. Chem., Int. Ed. 2011, 50, 7524–7528. doi:10.1002/anie.201102834
Return to citation in text: [1] -
Chiefari, J.; Chong, Y. K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T. P. T.; Mayadunne, R. T. A.; Meijs, G. F.; Moad, C. L.; Moad, G.; Rizzardo, E.; Thang, S. H. Macromolecules 1998, 31, 5559–5562. doi:10.1021/ma9804951
Return to citation in text: [1] -
McCormick, C. L.; Lowe, A. B. Acc. Chem. Res. 2004, 37, 312–325. doi:10.1021/ar0302484
Return to citation in text: [1] -
Moad, G.; Chong, Y. K.; Postma, A.; Rizzardo, E.; Thang, S. H. Polymer 2005, 46, 8458–8468. doi:10.1016/j.polymer.2004.12.061
Return to citation in text: [1] -
Semsarilar, M.; Perrier, S. Nat. Chem. 2010, 2, 811–820. doi:10.1038/nchem.853
Return to citation in text: [1] -
Barner-Kowollik, C., Ed. Handbook of RAFT Polymerization; Wiley-VCH Verlag GmbH & Co. KGaA, 2008. doi:10.1002/9783527622757
Return to citation in text: [1] -
Bastos, M.; Briggner, L.-E.; Shehatta, I.; Wadsö, I. J. Chem. Thermodyn. 1990, 22, 1181–1190. doi:10.1016/0021-9614(90)90111-3
Return to citation in text: [1] -
Thomas, D. B.; Sumerlin, B. S.; Lowe, A. B.; McCormick, C. L. Macromolecules 2003, 36, 1436–1439. doi:10.1021/ma025960f
Return to citation in text: [1] -
Thomas, D. B.; Convertine, A. J.; Myrick, L. J.; Scales, C. W.; Smith, A. E.; Lowe, A. B.; Vasilieva, Y. A.; Ayres, N.; McCormick, C. L. Macromolecules 2004, 37, 8941–8950. doi:10.1021/ma048199d
Return to citation in text: [1]
| 70. | Bastos, M.; Briggner, L.-E.; Shehatta, I.; Wadsö, I. J. Chem. Thermodyn. 1990, 22, 1181–1190. doi:10.1016/0021-9614(90)90111-3 |
| 47. | Takashima, Y.; Uramatsu, K.; Jomori, D.; Harima, A.; Otsubo, M.; Yamaguchi, H.; Harada, A. ACS Macro Lett. 2013, 2, 384–387. doi:10.1021/mz4001942 |
| 65. | Chiefari, J.; Chong, Y. K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T. P. T.; Mayadunne, R. T. A.; Meijs, G. F.; Moad, C. L.; Moad, G.; Rizzardo, E.; Thang, S. H. Macromolecules 1998, 31, 5559–5562. doi:10.1021/ma9804951 |
| 66. | McCormick, C. L.; Lowe, A. B. Acc. Chem. Res. 2004, 37, 312–325. doi:10.1021/ar0302484 |
| 67. | Moad, G.; Chong, Y. K.; Postma, A.; Rizzardo, E.; Thang, S. H. Polymer 2005, 46, 8458–8468. doi:10.1016/j.polymer.2004.12.061 |
| 68. | Semsarilar, M.; Perrier, S. Nat. Chem. 2010, 2, 811–820. doi:10.1038/nchem.853 |
| 69. | Barner-Kowollik, C., Ed. Handbook of RAFT Polymerization; Wiley-VCH Verlag GmbH & Co. KGaA, 2008. doi:10.1002/9783527622757 |
| 1. | Mooney, R. A.; Landick, R. Cell 1999, 98, 687–690. doi:10.1016/S0092-8674(00)81483-X |
| 2. | Kovall, R.; Matthews, B. W. Science 1997, 277, 1824–1827. doi:10.1126/science.277.5333.1824 |
| 3. | Trakselis, M. A.; Alley, S. C.; Abel-Santos, E.; Benkovic, S. J. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 8368–8375. doi:10.1073/pnas.111006698 |
| 4. | Benkovic, S. J.; Valentine, A. M.; Salinas, F. Annu. Rev. Biochem. 2001, 70, 181–208. doi:10.1146/annurev.biochem.70.1.181 |
| 5. | Breyer, W. A.; Matthews, B. W. Protein Sci. 2001, 10, 1699–1711. doi:10.1110/ps.10301 |
| 6. | Kool, E. T. Acc. Chem. Res. 1998, 31, 502–510. doi:10.1021/ar9602462 |
| 20. | Harada, A.; Hu, Y.; Takahashi, S. Chem. Lett. 1986, 2083–2084. doi:10.1246/cl.1986.2083 |
| 21. | Hu, Y.; Harada, A.; Takahashi, S. Synth. Commun. 1988, 18, 1607–1610. doi:10.1080/00397918808081320 |
| 22. | van Dongen, S. F. M.; Elemans, J. A. A. W.; Rowan, A. E.; Nolte, R. J. M. Angew. Chem., Int. Ed. 2014, 53, 11420–11428. doi:10.1002/anie.201404848 |
| 23. | Hubert, C.; Denicourt-Nowicki, A.; Roucoux, A.; Landy, D.; Leger, B.; Crowyn, G.; Monflier, E. Chem. Commun. 2009, 1228–1230. doi:10.1039/b818786j |
| 24. | Bricout, H.; Hapiot, F.; Ponchel, A.; Tilloy, S.; Monflier, E. Curr. Org. Chem. 2010, 14, 1296–1307. doi:10.2174/138527210791616920 |
| 25. | Hapiot, F.; Bricout, H.; Tilloy, S.; Monflier, E. Eur. J. Inorg. Chem. 2012, 1571–1578. doi:10.1002/ejic.201101316 |
| 26. | Noël, S.; Léger, B.; Ponchel, A.; Philippot, K.; Denicourt-Nowicki, A.; Roucoux, A.; Monflier, E. Catal. Today 2014, 235, 20–32. doi:10.1016/j.cattod.2014.03.030 |
| 27. | Hapiot, F.; Bricout, H.; Menuel, S.; Tilloy, S.; Monflier, E. Catal. Sci. Technol. 2014, 4, 1899–1908. doi:10.1039/c4cy00005f |
| 28. | Vanbésien, T.; Monflier, E.; Hapiot, F. ACS Catal. 2015, 5, 4288–4292. doi:10.1021/acscatal.5b00861 |
| 59. | Kakuchi, T.; Narumi, A.; Miura, Y.; Matsuya, S.; Sugimoto, N.; Satoh, T.; Kaga, H. Macromolecules 2003, 36, 3909–3913. doi:10.1021/ma021295z |
| 60. | Kakuchi, T.; Narumi, A.; Matsuda, T.; Miura, Y.; Sugimoto, N.; Satoh, T.; Kaga, H. Macromolecules 2003, 36, 3914–3920. doi:10.1021/ma021296r |
| 16. | Tanaka, Y.; Sakuraba, H.; Nakanishi, H. J. Chem. Soc., Chem. Commun. 1983, 947–948. doi:10.1039/C39830000947 |
| 17. | Sakuraba, H.; Nakai, T.; Tanaka, Y. J. Inclusion Phenom. 1984, 2, 829–839. doi:10.1007/BF00662252 |
| 18. | Trost, B. M.; Van Vranken, D. L. J. Am. Chem. Soc. 1990, 112, 1261–1263. doi:10.1021/ja00159a065 |
| 19. | Trost, B. M.; Van Vranken, D. L. J. Am. Chem. Soc. 1993, 115, 444–458. doi:10.1021/ja00055a013 |
| 61. | Takashima, Y.; Osaki, M.; Harada, A. J. Am. Chem. Soc. 2004, 126, 13588–13589. doi:10.1021/ja047171e |
| 62. | Osaki, M.; Takashima, Y.; Yamaguchi, H.; Harada, A. J. Am. Chem. Soc. 2007, 129, 14452–14457. doi:10.1021/ja075140o |
| 63. | Harada, A.; Osaki, M.; Takashima, Y.; Yamaguchi, H. Acc. Chem. Res. 2008, 41, 1143–1152. doi:10.1021/ar800079v |
| 64. | Takashima, Y.; Osaki, M.; Ishimaru, Y.; Yamaguchi, H.; Harada, A. Angew. Chem., Int. Ed. 2011, 50, 7524–7528. doi:10.1002/anie.201102834 |
| 7. | Mallick, I. M.; D'Souza, V. T.; Yamaguchi, M.; Lee, J.; Chalabi, P.; Gadwood, R. C.; Bender, M. L. J. Am. Chem. Soc. 1984, 106, 7252–7254. doi:10.1021/ja00335a070 |
| 8. | D'Souza, V. T.; Hanabusa, K.; O'Leary, T.; Gadwood, R. C.; Bender, M. L. Biochem. Biophys. Res. Commun. 1985, 129, 727–732. doi:10.1016/0006-291X(85)91952-7 |
| 9. | D'Souza, V. T.; Bender, M. L. Acc. Chem. Res. 1987, 20, 146–152. doi:10.1021/ar00136a004 |
| 10. | Komiyama, M.; Breaux, E. J.; Bender, M. L. Bioorg. Chem. 1977, 6, 127–136. doi:10.1016/0045-2068(77)90015-3 |
| 11. | Czarniecki, M. F.; Breslow, R. J. Am. Chem. Soc. 1978, 100, 7771–7772. doi:10.1021/ja00492a079 |
| 12. | Breslow, R.; Czarniecki, M. F.; Emert, J.; Hamaguchi, H. J. Am. Chem. Soc. 1980, 102, 762–770. doi:10.1021/ja00522a054 |
| 13. | Breslow, R.; Trainor, G. L.; Ueno, A. J. Am. Chem. Soc. 1983, 105, 2739–2744. doi:10.1021/ja00347a037 |
| 14. | Breslow, R. Acc. Chem. Res. 1991, 24, 317–324. doi:10.1021/ar00011a001 |
| 15. | Breslow, R. Artificial Enzymes; Wiley-VCH Verlag GmbH & Co. KGaA, 2006. |
| 48. | Köllisch, H. S.; Barner-Kowollik, C.; Ritter, H. Chem. Commun. 2009, 1097–1099. doi:10.1039/b818897a |
| 49. | Storsberg, J.; Ritter, H. Macromol. Rapid Commun. 2000, 21, 236–241. doi:10.1002/(SICI)1521-3927(20000301)21:5<236::AID-MARC236>3.0.CO;2-K |
| 50. | Glöckner, P.; Metz, N.; Ritter, H. Macromolecules 2000, 33, 4288–4290. doi:10.1021/ma992012e |
| 51. | Storsberg, J.; Hartenstein, M.; Müller, A. H. E.; Ritter, H. Macromol. Rapid Commun. 2000, 21, 1342–1346. doi:10.1002/1521-3927(20001201)21:18<1342::AID-MARC1342>3.0.CO;2-Z |
| 52. | Hetzer, M.; Schmidt, B. V. K. J.; Barner-Kowollik, C.; Ritter, H. J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 2504–2517. doi:10.1002/pola.26644 |
| 53. | Köllisch, H.; Barner-Kowollik, C.; Ritter, H. Macromol. Rapid Commun. 2006, 27, 848–853. doi:10.1002/marc.200600067 |
| 54. | Choi, S. W.; Kretschmann, O.; Ritter, H.; Ragnoli, M.; Galli, G. Macromol. Chem. Phys. 2003, 204, 1475–1479. doi:10.1002/macp.200350010 |
| 1. | Mooney, R. A.; Landick, R. Cell 1999, 98, 687–690. doi:10.1016/S0092-8674(00)81483-X |
| 2. | Kovall, R.; Matthews, B. W. Science 1997, 277, 1824–1827. doi:10.1126/science.277.5333.1824 |
| 3. | Trakselis, M. A.; Alley, S. C.; Abel-Santos, E.; Benkovic, S. J. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 8368–8375. doi:10.1073/pnas.111006698 |
| 4. | Benkovic, S. J.; Valentine, A. M.; Salinas, F. Annu. Rev. Biochem. 2001, 70, 181–208. doi:10.1146/annurev.biochem.70.1.181 |
| 5. | Breyer, W. A.; Matthews, B. W. Protein Sci. 2001, 10, 1699–1711. doi:10.1110/ps.10301 |
| 6. | Kool, E. T. Acc. Chem. Res. 1998, 31, 502–510. doi:10.1021/ar9602462 |
| 55. | Yao, F.; Xu, L.; Fu, G.-D.; Lin, B. Macromolecules 2010, 43, 9761–9770. doi:10.1021/ma102039n |
| 56. | Liu, Y.-Y.; Zhong, Y.-B.; Nan, J.-K.; Tian, W. Macromolecules 2010, 43, 10221–10230. doi:10.1021/ma1019973 |
| 57. | Chechik, V.; Ionita, G. Org. Biomol. Chem. 2006, 4, 3505–3510. doi:10.1039/b607676a |
| 58. | Ionita, G.; Chechik, V. Org. Biomol. Chem. 2005, 3, 3096–3098. doi:10.1039/b508256k |
| 37. | Thordarson, P.; Bijsterveld, E. J. A.; Rowan, A. E.; Nolte, R. J. M. Nature 2003, 424, 915–918. doi:10.1038/nature01925 |
| 38. | Coumans, R. G. E.; Elemans, J. A. A. W.; Nolte, R. J. M.; Rowan, A. E. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 19647–19651. doi:10.1073/pnas.0603036103 |
| 39. | Jónsson, S.; Odille, F. G. J.; Norrby, P.-O.; Warnmark, K. Chem. Commun. 2005, 549–551. doi:10.1039/B411978A |
| 43. | Mock, W. L.; Irra, T. A.; Wepsiec, J. P.; Adhya, M. J. Org. Chem. 1989, 54, 5302–5308. doi:10.1021/jo00283a024 |
| 44. | Chen, J.; Rebek, J., Jr. Org. Lett. 2002, 4, 327–329. doi:10.1021/ol0168115 |
| 34. | Leung, D. H.; Fiedler, D.; Bergman, R. G.; Raymond, K. N. Angew. Chem., Int. Ed. 2004, 43, 963–966. doi:10.1002/anie.200352772 |
| 35. | Das, S.; Incarvito, C. D.; Crabtree, R. H.; Brudvig, G. W. Science 2006, 312, 1941–1943. doi:10.1126/science.1127899 |
| 36. | Leung, D. H.; Bergman, R. G.; Raymond, K. N. J. Am. Chem. Soc. 2006, 128, 9781–9797. doi:10.1021/ja061412w |
| 45. | Armspach, D.; Matt, D.; Peruch, F.; Lutz, P. Eur. J. Inorg. Chem. 2003, 805–809. doi:10.1002/ejic.200390109 |
| 46. | Jouffroy, M.; Armspach, D.; Matt, D.; Osakada, K.; Takeuchi, D. Angew. Chem., Int. Ed. 2016, 55, 8367–8370. doi:10.1002/anie.201603191 |
| 47. | Takashima, Y.; Uramatsu, K.; Jomori, D.; Harima, A.; Otsubo, M.; Yamaguchi, H.; Harada, A. ACS Macro Lett. 2013, 2, 384–387. doi:10.1021/mz4001942 |
| 10. | Komiyama, M.; Breaux, E. J.; Bender, M. L. Bioorg. Chem. 1977, 6, 127–136. doi:10.1016/0045-2068(77)90015-3 |
| 11. | Czarniecki, M. F.; Breslow, R. J. Am. Chem. Soc. 1978, 100, 7771–7772. doi:10.1021/ja00492a079 |
| 12. | Breslow, R.; Czarniecki, M. F.; Emert, J.; Hamaguchi, H. J. Am. Chem. Soc. 1980, 102, 762–770. doi:10.1021/ja00522a054 |
| 13. | Breslow, R.; Trainor, G. L.; Ueno, A. J. Am. Chem. Soc. 1983, 105, 2739–2744. doi:10.1021/ja00347a037 |
| 14. | Breslow, R. Acc. Chem. Res. 1991, 24, 317–324. doi:10.1021/ar00011a001 |
| 15. | Breslow, R. Artificial Enzymes; Wiley-VCH Verlag GmbH & Co. KGaA, 2006. |
| 71. | Thomas, D. B.; Sumerlin, B. S.; Lowe, A. B.; McCormick, C. L. Macromolecules 2003, 36, 1436–1439. doi:10.1021/ma025960f |
| 72. | Thomas, D. B.; Convertine, A. J.; Myrick, L. J.; Scales, C. W.; Smith, A. E.; Lowe, A. B.; Vasilieva, Y. A.; Ayres, N.; McCormick, C. L. Macromolecules 2004, 37, 8941–8950. doi:10.1021/ma048199d |
| 29. | Lehn, J.-M. Supramolecular Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA, 1995. |
| 30. | van Leeuwen, P. W. N. M. Supramolecular Catalysis; Wiley-VCH Verlag GmbH & Co. KGaA; Vol. 2008. |
| 31. | Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P. W. N. M. Chem. Soc. Rev. 2014, 43, 1660–1733. doi:10.1039/C3CS60027K |
| 32. | Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuwen, P. W. N. M. Chem. Soc. Rev. 2014, 43, 1734–1787. doi:10.1039/C3CS60037H |
| 33. | Vriezema, D. M.; Aragonès, M. C.; Elemans, J. A. A. W.; Cornelissen, J. J. L. M.; Rowan, A. E.; Nolte, R. J. M. Chem. Rev. 2005, 105, 1445–1490. doi:10.1021/cr0300688 |
| 40. | Walter, C. J.; Anderson, H. L.; Sanders, J. K. M. J. Chem. Soc., Chem. Commun. 1993, 458–460. doi:10.1039/C39930000458 |
| 41. | Marty, M.; Clyde-Watson, Z.; Twyman, L. J.; Nakash, M.; Sanders, J. K. M. Chem. Commun. 1998, 2265–2266. doi:10.1039/a806070c |
| 42. | Yoshizawa, M.; Tamura, M.; Fujita, M. Science 2006, 312, 251–254. doi:10.1126/science.1124985 |
© 2016 Koyanagi et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)