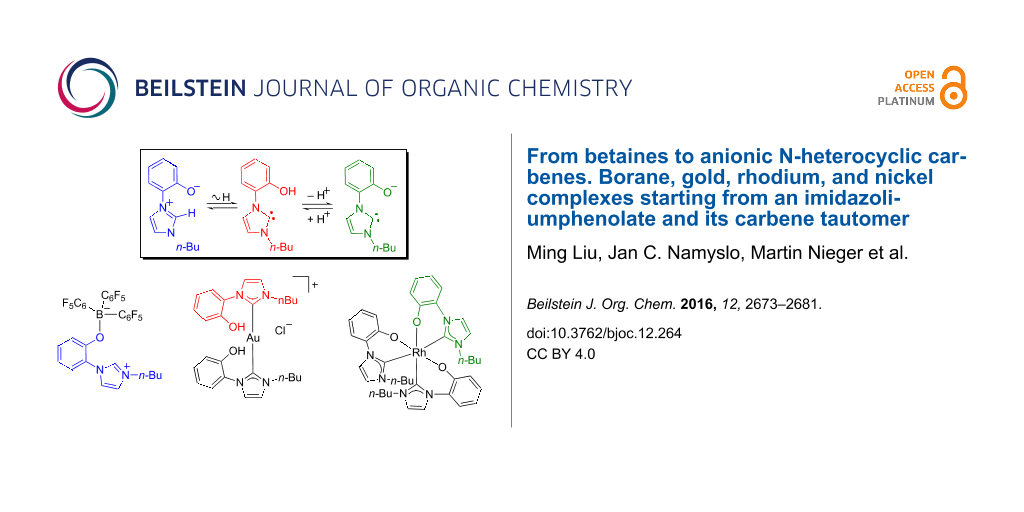
Abstract
The mesomeric betaine imidazolium-1-ylphenolate forms a borane adduct with tris(pentafluorophenyl)borane by coordination with the phenolate oxygen, whereas its NHC tautomer 1-(2-phenol)imidazol-2-ylidene reacts with (triphenylphosphine)gold(I) chloride to give the cationic NHC complex [Au(NHC)2][Cl] by coordination with the carbene carbon atom. The anionic N-heterocyclic carbene 1-(2-phenolate)imidazol-2-ylidene gives the complexes [K][Au(NHC−)2], [Rh(NHC−)3] and [Ni(NHC−)2], respectively. Results of four single crystal analyses are presented.

Graphical Abstract
Introduction
Since the first isolation of a stable N-heterocyclic carbene (NHC) [1] in 1991 this compound class has provided numerous highly efficient ligands of NHC-metal catalysts for cross-coupling reactions [2-7], versatile organocatalysts [8-10], and starting materials for heterocycle syntheses [11-13]. Consequently books [14-16] and reviews cover the range from NHC structures in the light of their early history [17], syntheses [6,18], coordination chemistry [19,20], and catalysis [21,22], to biological activities of NHC complexes [23,24]. In the last decade, attention has also been directed to anionic N-heterocyclic carbenes [25]. In this context, mesomeric betaines are interesting from two viewpoints. They are not only able to undergo tautomerisations to neutral NHCs and thus provide a safe storage form of otherwise unstable species [26,27], but proved also to be suitable starting materials for the generation of anionic NHCs by deprotonation. As examples of the latter mentioned species, sydnone anions 1 [28], imidazole-2-ylidene-4-olate 2 [29] as well as its 4-aminide derivatives [30] have been prepared from mesoionic compounds (Figure 1). The carbenes 3 [31] and 4 [32,33] originate from a conjugated ylide and a cross-conjugated mesomeric betaine, respectively. A review elucidates the interconversions of mesomeric betaines to different types of N-heterocyclic carbenes (NHC, aNHC, rNHC, MICs) and vice versa [34].
![[1860-5397-12-264-1]](/bjoc/content/figures/1860-5397-12-264-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of anionic N-heterocyclic carbenes.
Figure 1: Examples of anionic N-heterocyclic carbenes.
Herein, conjugated mesomeric betaine 2-(1-butyl-1H-imidazolium-3-yl)phenolate, its tautomeric NHC and its anionic NHC are shown to serve as monodentate and bidentate ligands, respectively.
Results and Discussion
Salt 5 is available via a three-step procedure [35]. Deprotonation with potassium carbonate resulted in the formation of the tautomeric equilibrium of the mesomeric betaine 6A and its NHC 6B (Scheme 1). The NMR spectra measured in a variety of solvents (DMSO-d6, MeCN-d3, MeOD-d4, D2O, THF-d8) show that the polar mesomeric betaine 6A is the only detectable tautomer under these conditions; unfortunately the addition of less polar solvents induces a precipitation from solution so that detection of the NHC tautomer by NMR spectroscopy is not possible under these conditions. Base screening revealed that the anionic N-heterocyclic carbene 7 can be generated in quantitative yield at rt with lithium bis(trimethylsilyl)amide in THF/pyridine due to its superior solubility. As evidenced by 1H NMR and 13C NMR spectroscopy, the carbene 7 proved to be stable in pyridine-d5 solution up to 50 °C. The resonance frequency of the carbene carbon atom can be detected at δ = 203 ppm. Minute traces of water protonate the anionic NHC 7 spontaneously to give 6A/B without any traces of decomposition products.
![[1860-5397-12-264-i1]](/bjoc/content/inline/1860-5397-12-264-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: A postulated mesomeric betaine – NHC equilibrium (6A/6B) and formation of an anionic NHC 7. Formation of borane adduct 8 of tautomer 6A, a cationic gold complex 9 of tautomer 6B, and an anionic gold complex 10 of anionic NHC 7. a) K2CO3, MeOH, reflux, 4 h, 98%. b) LiN(TMS)2, pyridine, rt, 100%. c) B(C6F5)3, dioxane, reflux, 43%. d) (PPh3)AuCl, THF, reflux, 60%; X = Cl/Br. e) K2CO3, MeOH, 100%.
Scheme 1: A postulated mesomeric betaine – NHC equilibrium (6A/6B) and formation of an anionic NHC 7. Formati...
The betaine 6A reacted with tris(pentafluorophenyl)borane in dioxane at reflux temperature to give the borane adduct 8. The proton attached to C-2 of the imidazolium ring (crystallographic numbering, cf. Figure 2: C9) is clearly detectable at δ = 9.22 ppm in DMSO-d6, while the boron and fluorine atoms give resonance frequencies at δ = −3.45 ppm, and δ = −133.91, −159.97, −165.18 ppm in the 11B NMR and 19F NMR spectra, respectively. A single crystal X-ray analysis of the borane adduct displays two different conformers in the unit cell which differ inter alia in the torsion angles around the Nimidazole–Cphenol bonds [−53.32(16)° vs 114.12(13)°] and the Cphenyl–O bonds [49.43(17)° vs −15.29(18)°] (Figure 2).
![[1860-5397-12-264-2]](/bjoc/content/figures/1860-5397-12-264-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular drawing of one of the two crystallographic independent molecules of borane adduct 8 (displacement parameters are drawn at 50% probability level; crystallographic numbering).
Figure 2: Molecular drawing of one of the two crystallographic independent molecules of borane adduct 8 (disp...
A gold complex of the N-heterocyclic carbene tautomer 6B can be obtained on reaction with (triphenylphosphine)gold(I) chloride in boiling anhydrous THF under a nitrogen atmosphere, under which conditions the colorless gold complex [Au(6B)2][Cl] (9) is formed in 60% yield (Scheme 1). The protons of the OH resonate at δ = 10.34 ppm in DMSO-d6, and the 13C NMR chemical shift of the carbene atom appears at δ = 183.9 ppm. Single crystals of the gold complex 9 were obtained by slow evaporation of a concentrated solution of 9 in MeOH and a molecular drawing is shown in Figure 3. The X-ray structure revealed a mixed occupation of the same position with chloride and bromide anions (4:1). The bond length between the gold atom and the carbene carbon atom (crystallographic numbering: Au1–C2) was found to be 202.11(15) pm and this value corresponds to the distances in other NHC gold complexes [36,37].
![[1860-5397-12-264-3]](/bjoc/content/figures/1860-5397-12-264-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Molecular drawing of the cation of the gold complex 9 (displacement parameters are drawn at 50% probability level; crystallographic numbering).
Figure 3: Molecular drawing of the cation of the gold complex 9 (displacement parameters are drawn at 50% pro...
The coordination about the Au(I) center is almost linear, as a C2–Au(1)–C2A bond angle of 177.35(8)° was found. However, in contrast to other gold complexes, the two imidazole rings are not coplanar [38]. In addition, the imidazole and the phenol rings are twisted about −119.84(16)° (C5–N1–C6–C7) relative to the imidazole ring. Two Au complexes are connected via one halide anion which is involved in hydrogen bonds between two OH groups. On treatment with methanolic potassium carbonate, the two OH groups of gold complex 9 can be deprotonated to give the anionic complex [K][Au(7)2] 10 of the anionic N-heterocyclic carbene 7 in quantitative yield (Scheme 1). ESI mass spectra taken in the anion detection mode show the peak of 10 as base peak at m/z = 627.
The anionic N-heterocyclic carbene 7 also forms a rhodium and a nickel complex (Scheme 2). The colorless rhodium complex [Rh(7)3] 11 was prepared on reaction of the tautomeric mixture 6A/B with either chlorido(1,5-cyclooctadiene)rhodium(I) dimer, or with bis(triphenylphosphine)rhodium(I) carbonyl chloride in anhydrous toluene at reflux temperature, respectively. During these reactions, the water of crystallization of the starting material is – at least partially – removed by azeotropic distillation and Rh(I) is obviously oxidized to Rh(III). In order to balance the chemical equation, the formation of elemental hydrogen and of one equivalent of HCl can be postulated. Neither hydrogen nor reduced species such as cyclooctane or cyclooctene, however, have been detected. In the ESI mass spectra the base peak was detected at m/z = 749.2 which corresponds to the molecular peak of a [Rh(7)3 + H]+ complex. In the 13C NMR spectra, the Ccarbene resonance frequencies were detected at 173.5 ppm, 171.0 ppm, and 164.4 ppm and have been identified by their 1JRhCcarbene couplings of 35.6 Hz, 35.6 Hz, and 48.5 Hz, respectively. These chemical shifts are in a more upfield region and the coupling constants are smaller than in other complexes such as the neutral N-heterocyclic oxocarbene (NHOC) rhodium complex ([Rh(NHOC)Cl(COD)] of 2 (R = Mes; δCcarbene = 229.7 ppm; 1JRhCcarbene = 51.5 Hz) as well as its enol ethers (δCcarbene = 171–177 ppm) [29,39], and other Rh complexes [40,41]. Correspondingly the phenol proton signals of the three geometrically non-equivalent ligands appear at different resonance frequencies. Thus, the three unsubstituted ortho positions of the phenolate moieties are detectable at 6.93–6.97 ppm (overlapped), 6.72 ppm, and 5.81 ppm, respectively.
![[1860-5397-12-264-i2]](/bjoc/content/inline/1860-5397-12-264-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The anionic NHC 7 forms a rhodium complex and a nickel complex. d) [RhCl(COD)]2, toluene, rt to reflux (50%). e) [RhCl(PPh3)2(CO)], toluene, reflux, 57%. f) NiCl2(PPh3)2, THF, rt to reflux, 45%.
Scheme 2: The anionic NHC 7 forms a rhodium complex and a nickel complex. d) [RhCl(COD)]2, toluene, rt to ref...
Single crystals of [Rh(7)3] (11) were obtained by slow evaporation of a concentrated solution of 11 in a mixture of EtOAc and MeOH. The single crystal X-ray analysis proved that three anionic N-heterocyclic carbenes 7 serve as bidentate ligands, respectively (Figure 4). The n-butyl group connected to N25 is disordered. The bond lengths between the rhodium atom and the carbene atoms (C1, C21 and C41) were determined to be 196.6(2), 204.7(2) and 204.4(2) pm. In the literature-known complexes [Rh(ICy3)(CO)][PF6], [Rh(IiPr2Me2)3(CO)][PF6], and [Rh(ICy)(IiPr2Me2)2(CO)][PF6] bond lengths between 206.38(11) and 214.90(13) pm were found [40], whereas the two independent molecules of [Rh(IBioxMe4)3][BArF4] in the elemental cell have one Rh–Ccarbene bond of 194.1(2) pm and 193.4(2) pm, respectively, trans to a free coordination site of this naked low-coordinate rhodium complex [41]. Correspondingly the Rh–O15 (202.49(14) pm) and Rh–O35 bond lengths cis to the shortened Rh–Ccarbene bond in 11 are identical (2.0244(14) pm), whereas the Rh–O55 bond trans to this bond is longer (209.98(15) pm). The phenolate rings are twisted in relation to the imidazole rings and the determined dihedral angles for C1–N2–C10–C15, C21–N22–C30–C35, and C41–N42–C50–C55 are 26.371(5)°, 18.939(5)°, and −33.008(5)°, respectively. The carbenes are twisted by approximately 28.1° to 37.0° in relation to the Rh–Ccarbene bonds.
![[1860-5397-12-264-4]](/bjoc/content/figures/1860-5397-12-264-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Molecular drawing of rhodium complex 11 (minor disorder parts omitted for clarity, displacement parameters are drawn at 50% probability level; crystallographic numberings).
Figure 4: Molecular drawing of rhodium complex 11 (minor disorder parts omitted for clarity, displacement par...
The reaction of 6A/6B with bis(triphenylphosphine)nickel(II) chloride at reflux temperature resulted in the formation of the nickel complex [Ni(7)2] (12, Scheme 2). Single crystals were obtained by slow evaporation of 12 from EtOAc/MeOH. The X-ray analysis shows two independent nickel complexes which are connected via hydrogen bonds to two water molecules (Figure 5). The nickel atom is in the center of an essentially square planar environment, as the sums of the bond angles are 363.81° and 364.76°, respectively. The ligand adopts a cis arrangement around the Ni atom. The Ni–O bond lengths were determined to be 187.60(13) pm and 190.11(13) pm, whereas the Ni–Ccarbene bonds have lengths between 184.06(19) pm and 184.82(19) pm, respectively. These values correspond to literature-known bond lengths of cis arranged bidentate ligands around Ni [42-44], but, as expected, they differ from those of [(PEt3)(Ph)Ni(imidazo[1,5-a]quinolin-9-olate-1-ylidene)] [45] as well as Ni complexes with tridentate ligands [46,47].
![[1860-5397-12-264-5]](/bjoc/content/figures/1860-5397-12-264-5.png?scale=1.8&max-width=1024&background=FFFFFF)
Figure 5: Molecular drawing of the dimeric nickel complex 12 (displacement parameters are drawn at 50% probability level; crystallographic numberings).
Figure 5: Molecular drawing of the dimeric nickel complex 12 (displacement parameters are drawn at 50% probab...
Conclusion
In summary, this publication presents four molecules which differ only in their number and positions of protons. Starting from cation 5, the neutral compound 6A (betaine) was formed and detected spectroscopically, whereas its tautomeric carbene 6B could not be characterized in non-complexed form. Deprotonation of 6A/B gave the anionic NHC 7. The two tautomers 6A/6B and the anionic NHC 7 could be trapped as complexes. These results supplement our knowledge about the interesting area of overlap between mesomeric betaines and N-heterocyclic carbenes [34] and of structurally related zwitterionic motifs known in the literature [48-52].
Experimental
General considerations. Silica gel 60 (0.040–0.063 mm) was used for flash-chromatographic separations. The NMR spectra were obtained with a Bruker Avance 400 and Bruker Avance III 600 MHz spectrometer, and 1H NMR spectra were recorded at 400 MHz or 600 MHz. 13C NMR spectra were recorded at 100 MHz or 150 MHz. The solvent peak or tetramethylsilane was used as the internal reference. The multiplicities are described here by using the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, and m = multiplet, and the signal orientations in DEPT experiments were described as follows: o = no signal; + = up (CH, CH3); − = down (CH2). For NMR peak assignments we numbered the compounds not necessarily in accordance with IUPAC rules to allow comparisons. Thus, “C-2” always refers to C-2 of the imidazolium and imidazol-2-ylidene ring, respectively. The spectroscopic numbering, which differ from the crystallographic numbering, is presented on the NMR spectra in Supporting Information File 1. All FTIR spectra were measured with a Bruker Alpha T spectrometer in the range of 400 to 4000 cm−1. The mass spectra were obtained with a Varian 320 MS Triple Quad GC/MS/MS instrument with a Varian 450-GC. All electrospray ionisation mass spectra (ESIMS) were measured with a Hewlett-Packard/Agilent LCMSD series HP 1100 spectrometer with APIES. The compound samples were sprayed from MeOH at 4000 V capillary voltage and fragmentor voltages of 30 V, unless otherwise noted. Melting points are uncorrected and were determined in an apparatus according to Dr. Tottoli (Büchi). All HRMS spectra were obtained with a Bruker Daltonik Tesla-Fourier transform–ion cyclotron resonance mass spectrometer with electrospray ionisation.
The single-crystal X-ray diffraction studies were carried out on a Bruker D8 Venture diffractometer with Photon100 detector at 123(2) K using Cu Kα (8) and Mo Kα radiation (9, 11, 12) (λ = 1.54178 Å and 0.71073 Å). Dual space methods (8, SHELXT) [53] and direct methods (9, 11, 12, SHELXS-97) [54] were used for structure solution and refinement was carried out using SHELXL-2014 (full-matrix least-squares on F2). Hydrogen atoms were localized by difference electron density determination and refined using a riding model (H(O) free). Semi-empirical absorption corrections were applied. For 8, 9, and 11 an extinction correction was applied. In 9 the halogen counter anion (occupancy Cl/Br approx. 81:19) and in 11 one n-butyl group are disordered.
8: colorless crystals, C31H16BF15N2O, Mr = 728.27, crystal size 0.35 × 0.25 × 0.15 mm, monoclinic, space group P21/n (No. 14), a = 21.9006(7) Å, b = 13.1620(4) Å, c = 22.0149(7) Å, β = 117.982(1)°, V = 5650.1(3) Å3, Z = 8, ρ = 1.712 Mg/m−3, µ(Cu Kα) = 1.552 mm−1, F(000) = 2912, 2Θmax = 144.4°, 56172 reflections, of which 11099 were independent (Rint = 0.024), 902 parameters, R1 = 0.030 (for 10343 I > 2σ(I)), wR2 = 0.074 (all data), S = 1.04, largest diff. peak/hole = 0.359/−0.191 e Å−3. There are 2 different conformers in the asymmetric unit.
9: colorless crystals, C26H32AuN4O2∙0.19(Br)∙0.81(Cl), Mr = 673.42, crystal size 0.24 × 0.12 × 0.06 mm, monoclinic, space group C2/c (No. 15), a = 15.0275(9) Å, b = 13.1392(7) Å, c = 13.0205(7) Å, β = 104.161(2)°, V = 2492.8(2) Å3, Z = 4, ρ = 1.794 Mg/m−3, µ(Mo Kα) = 6.324 mm−1, F(000) = 1326, 2Θmax = 55.2°, 46420 reflections, of which 2867 were independent (Rint = 0.026), 162 parameters, R1 = 0.011 (for 2788 I > 2σ(I)), wR2 = 0.026 (all data), S = 1.10, largest diff. peak/hole = 0.404/−0.489 e Å−3, disorder of the halogen counter anion (occupancy Cl/Br 0.813(2):0.187(2)).
11: yellow crystals, C39H45N6O3Rh, Mr = 748.72, crystal size 0.38 × 0.32 × 0.16 mm, monoclinic, space group P21/n (No. 14), a = 11.7692(5) Å, b = 18.8111(7) Å, c = 16.2459(7) Å, β = 104.615(2)°, V = 3480.3(2) Å3, Z = 4, ρ = 1.429 Mg/m−3, µ(Mo Kα) = 0.538 mm−1, F(000) = 1560, 2Θmax = 55.2°, 90530 reflections, of which 8005 were independent (Rint = 0.046), 436 parameters, 146 restraints, R1 = 0.032 (for 6902 I > 2σ(I)), wR2 = 0.071 (all data), S = 1.05, largest diff. peak/hole = 0.645/−0.996 e Å−3, disorder of the n-butyl group.
12: yellow crystals, C26H30N4NiO2∙H2O, Mr = 507.26, crystal size 0.22 × 0.12 × 0.04 mm, monoclinic, space group P21/n (No. 14), a = 11.2267(5) Å, b = 22.8310(9) Å, c = 19.1274(8) Å, β = 100.099(2)°, V = 4826.7(4) Å3, Z = 8, ρ = 1.396 Mg/m−3, µ(Mo Kα) = 0.839 mm−1, F(000) = 2144, 2Θmax = 55.2°, 89393 reflections, of which 11117 were independent (Rint = 0.070), 625 parameters, 6 restraints, R1 = 0.038 (for 8729 I > 2σ(I)), wR2 = 0.095 (all data), S = 1.06, largest diff. peak/hole = 1.063/−0.370 e Å−3, dimer.
CCDC 1489189 (8), 1489190 (9), 1489191 (11), and 1489192 (12) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Preparation of lithium 2-(3-butyl-1H-imidazol-2-ylidene-1-yl)phenolate (7): A solution of 0.02 g (0.09 mmol) of 2-(3-butyl-1H-imidazolium-1-yl)phenolate [35] and 0.10 mL of lithium bis(trimethylsilyl)amide (1.0 M solution in THF) in 0.7 mL of pyridine was stirred for 30 minutes at rt. The product was characterized in solution, as traces of moisture reconstituted the starting material. Concentration of the solution in vacuo, filtration and crystallization of the mother liquor in the presence of one drop of water gave 6A/B in quantitative yield. Yield of 7: 0.02 g (100%). 1H NMR (600 MHz, pyridine-d5) δ 7.44 (d, J = 0.8 Hz, 1H, H-5), 7.40 (dd, J1 = 1.0 Hz, J2 = 7.6 Hz, 1H, H-11), 7.16–7.12 (m, 2H, H-9/8), 7.05 (d, J = 0.8 Hz, 1H, H-4), 6.59 (ddd, J1 = 0.9 Hz, J2 = 7.3 Hz, J3 = 7.6 Hz, 1H, H-10), 3.94 (t, J = 7.0 Hz, 2H, H-12), 1.68–1.62 (m, 2H, H-13), 1.19–1.15 (m, 2H, H-14), 0.75 (t, J = 7.3 Hz, 3H, H-15) ppm; 13C NMR (150 MHz, pyridine-d5) δ 203.1 (o, C-2), 162.2 (o, C-7), 131.0 (o, C-6), 127.2 (+, C-9), 123.0 (+, C-8), 122.7 (+, C-11), 119.5 (+, C-5), 118.5 (+, C-4), 111. 2 (+, C-10), 50.7 (−, C-12), 33.5 (−, C-13), 19.8 (−, C-14), 13.5 (+, C-15) ppm; ESIMS (50 V) m/z (%): 215.1 (100) [M − Li]−.
Preparation of (2-(3-butyl-1H-imidazolium-1-yl)phenoxy)- tris(perfluorophenyl)borate (8): Under an inert atmosphere a solution of 0.108 g (0.50 mmol) of 2-(3-butyl-1H-imidazolium-1-yl)phenolate and 0.152 mg (1.0 mmol) of tris(pentafluorophenyl)borane in 10 mL of dry dioxane was stirred at reflux for 4 h in a bomb tube. Then, the solvent was distilled off in vacuo. The product was separated by column chromatography (silica gel, EtOAc). Yield: 0.155 g (43%) of a colorless solid, mp 230 °C; 1H NMR (600 MHz, DMSO-d6) δ 9.22 (dd, J1 = 1.4 Hz, J2 = 1.5 Hz, 1H, H-2), 7.90 (dd, J1 = 1.4 Hz, J2 = 1.5 Hz, 1H, H-4), 7.82 (dd, J1 = 1.4 Hz, J2 = 1.5 Hz, 1H, H-5), 7.43 (dd, J1 = 2.3 Hz, J2 = 9.8 Hz, 1H, H-11), 7.17 (ddd, J1 = 2.3 Hz, J2 = 7.4 Hz, J3 = 8.4 Hz, 1H, H-9), 6.81 (ddd, J1 = 1.0 Hz, J2 = 7.4 Hz, J3 = 9.8 Hz, 1H, H-10), 6.60 (dd, J1 = 1.0 Hz, J2 = 8.4 Hz, 1H, H-8), 4.18 (t, J = 10.8 Hz, 2H, H-12), 1.72–1.64 (overlapped, 2H, H-13), 1.22–1.13 (overlapped, 2H, H-14), 0.84 (t, J = 11.0 Hz, 3H, H-15) ppm; 13C NMR (150 MHz, DMSO-d6) δ 152.5 (o, C-7), 147.2 (o, d, 1JC,F = 242.1 Hz, C-20), 138.2 (o, d, 1JC,F = 230.3 Hz, C-21), 136.3 (+, C-2), 135.8 (o, d, 1JC,F = 235.5 Hz, C-19), 130.2 (+, C-9), 125.4 (o, C-6), 125.0 (+, C-11), 123.5 (+, C-5), 121.8 (o, C-4), 122.2–121.3 (+/o, C-18), 117.7 (+, C-10), 117.4 (+, C-8), 48.7 (−,C-12), 31.4 (−, C-13), 18.6 (−, C-14), 13.0 (+, C-15) ppm; 11B NMR (DMSO-d6, 128 MHz, external reference) δ −3.45 ppm; 19F NMR (DMSO-d6, 565 MHz, Cl3CF) δ −133.91 (d, 3JF,F = 21.5 Hz, 6F, F-19/19'), −159.97 (t, 3JF,F = 21.5 Hz, 3F, F-21), −165.18 (dd, 3JF,F = 21.5 Hz, 6F, F-20/20', partially overlapped) ppm; IR (ATR) ![[Graphic 1]](/bjoc/content/inline/1860-5397-12-264-i3.svg?max-width=637&scale=1.18182) : 1511, 1498, 1456, 1304, 1277, 1262, 1079, 1038, 974, 965, 945, 936, 802, 768, 762, 755, 733, 691, 673, 667, 654, 649 cm−1; ESIMS (50 V) m/z (%): 727.0 (100) [M − H]−. HRESIMS: calcd for C31H15N2OF15B−, 727.1038; found, 727.1038.
: 1511, 1498, 1456, 1304, 1277, 1262, 1079, 1038, 974, 965, 945, 936, 802, 768, 762, 755, 733, 691, 673, 667, 654, 649 cm−1; ESIMS (50 V) m/z (%): 727.0 (100) [M − H]−. HRESIMS: calcd for C31H15N2OF15B−, 727.1038; found, 727.1038.
Preparation of bis(3-butyl-1-(2-hydroxyphenyl)-1H-imidazolium-2-yl)gold monochloride (9): A solution of 0.43 g (0.20 mmol) of 2-(3-butyl-1H-imidazolium-1-yl)phenolate in 5 mL of anhydrous THF was treated with 0.05 g (0.10 mmol) of (triphenylphosphine)gold(I) chloride and stirred under an inert atmosphere overnight at reflux temperature. The resulting solid was filtered off, washed with THF, and dried in vacuo. Yield: 0.40 g (60%) of a yellow solid, mp 242 °C; 1H NMR (600 MHz, CD3OD) δ 7.39 (d, J = 2.0 Hz, 2H, H-4/4'), 7.37 (dd, J1 = 1.6 Hz, J2 = 7.7 Hz, 2H, H-11/11'), 7.38–7.33 (overlapped signals, 4H, H-5/5'/9/9'), 7.01 (dd, J1 = 1.3 Hz, J2 = 8.2 Hz, 2H, H-8/8'), 6.93 (ddd, J1 = 1.3 Hz, J2 = 7.5 Hz, J3 = 7.7 Hz, 2H, H-10/10´), 4.01 (t, J = 7.1 Hz, 4H, H-12/12'), 1.71–1.66 (m, 4H, H-13/13'), 1.22–1.15 (m, 4H, H-14/14'), 0.88 (t, J = 7.3 Hz, 6H, H-15/15') ppm; 1H NMR (600 MHz, DMSO-d6) δ 10.34 (s broad, 2H, OH), 7.62 (d, J = 1.9 Hz, 2H, H-2/2'), 7.57 (d, J = 1.9 Hz, 2H, H-3/3'), 7.38 (dd, J1 = 1.7 Hz, J2 = 7.8 Hz, 2H, H-11/11'), 7.34 (ddd, J1 = 1.7 Hz, J2 = 7.6 Hz, J3 = 8.2 Hz, 2H, H-9/9'), 7.06 (dd, J1 = 1.3 Hz, J2 = 8.2 Hz, 2H, H-8/8'), 6.93 (ddd, J1 = 1.3 Hz, J2 = 7.6 Hz, J3 = 7.8 Hz, 2H, H-10/10'), 3.96 (t, J = 7.0 Hz, 4H, H-12/12'), 1.59–1.64 (m, 4H, H-13/13'), 1.05–1.11 (m, 4H, H-14/14'), 0.81 (t, J = 7.3 Hz, 6H, H-15/15') ppm; 13C NMR (150 MHz, CD3OD) δ183.9 (o, C-2/2'), 152.5 (o, C-7/7'), 130.3 (+, C-9/9'), 128.1 (+, C-11/11'), 126.8 (o, C-6/6'), 123.8 (+, C-5/5'), 120.7 (+, C-4/4'), 119.1 (+, C-10/10'), 116.5 (+, C-8/8'), 50.4 (−, C-12/12'), 32.9 (−, C-13/13'), 19.2 (−, C-14/14'), 13.6 (+, C-15/15') ppm; IR (ATR) ![[Graphic 2]](/bjoc/content/inline/1860-5397-12-264-i4.svg?max-width=637&scale=1.18182) : 2956, 1598, 1509, 1463, 1455, 1285, 1241, 829, 766, 752, 734, 687 cm−1; MS (ESI 5 V) m/z (%) = 629.1 (100) M+. HRESIMS: calcd for C26H32N4O2Au+, 629.2192; found, 629.2191.
: 2956, 1598, 1509, 1463, 1455, 1285, 1241, 829, 766, 752, 734, 687 cm−1; MS (ESI 5 V) m/z (%) = 629.1 (100) M+. HRESIMS: calcd for C26H32N4O2Au+, 629.2192; found, 629.2191.
Preparation of potassium bis(3-butyl-1-(2-phenolate)-1H-imidazolium-2-yl)gold (10): A solution of 0.066 g (0.10 mmol) of bis(3-butyl-1-(2-hydroxyphenyl)-1H-imidazolium-2-yl)gold chloride (9) and 0.014 g (0.10 mmol) of K2CO3 in 5 mL of methanol was stirred for 30 minutes under ultrasound irradiation. After concentrating the solution in vacuo, the resulting colorless solid was filtered off. Yield: 0.066 g (100%), mp 240 °C; 1H NMR (600 MHz, CD3OD) δ 7.34 (d, J = 1.8 Hz, 2H, H-4/4'), 7.21–7.23 (overlapped signals, 4H, H-4/4' and H-11/11'), 7.08 (ddd, J1 = 1.86 Hz, J2 = 7.3 Hz, J3 = 8.3 Hz, 2H, H-9/9'), 6.80 (dd, J1 = 1.2 Hz, J2 = 8.3 Hz, 2H, H-8/8'), 6.43 (ddd, J1 = 1.2 Hz, J2 = 7.3 Hz, J3 = 7.5 Hz, 2H, H-10/10'), 4.00 (t, J = 7.1 Hz, 4H, H-12/12'), 1.70–1.65 (m, 4H, H-13/13'), 1.26–1.19 (m, 4H, H-14/14'), 0.88 (t, J = 7.3 Hz, 6H, H15/15') ppm; 13C NMR (150 MHz, CD3OD) δ 183.5 (o, C-2/2'), 162.5 (o, C-7), 160.1(o, C-7'), 129.5 (+, C-9/9'), 129.2 (o, C-6/6'), 127.4 (+, C-11/11'), 124.1 (+, C-5/5'), 121.2 (+, C-8/8'), 119.7 (+, C-4/4'), 111.9 (+, C-10/10'), 50.2 (−, C-12/12'), 33.1 (−, C-13/13'), 19.3 (−, C-14/14'), 12.7 (+, C-15/15') ppm; IR (ATR) ![[Graphic 3]](/bjoc/content/inline/1860-5397-12-264-i5.svg?max-width=637&scale=1.18182) : 3157, 2958, 1637, 1591, 1483, 1447, 1311, 1246, 1076, 1061, 844, 748, 701, 566 cm−1; ESIMS (50 V) m/z (%): 627.2 (100) [M − K]−.
: 3157, 2958, 1637, 1591, 1483, 1447, 1311, 1246, 1076, 1061, 844, 748, 701, 566 cm−1; ESIMS (50 V) m/z (%): 627.2 (100) [M − K]−.
Preparation of tris(3-butyl-1-(2-oxidophenyl)-1H-imidazolium-2-yl)rhodium (11): Method A: A solution of 0.16 g (0.72 mmol) of 2-(3-butyl-1H-imidazolium-1-yl)phenolate in 5 mL of anhydrous toluene was treated with 0.06 g (0.12 mmol) of chloro(1,5-cyclooctadiene)rhodium(I) dimer and stirred under an inert atmosphere overnight at reflux temperature. The resulting colorless solid was filtered off, washed with THF and dried in vacuo. Yield: 0.09g (50%) of a yellow solid, mp: 280 °C. Method B: A solution of 0.16 g (0.72 mmol) of 2-(3-butyl-1H-imidazolium-1-yl)phenolate in 5 mL of anhydrous toluene was treated with 0.17 g (0.24 mmol) of bis(triphenylphosphine)rhodium(I) carbonyl chloride and stirred under an inert atmosphere overnight at reflux temperature. The resulting colorless solid was filtered off, washed with THF and dried in vacuo. Yield: 0.10g (57%) of a yellow solid, mp 280 °C; 1H NMR (600 MHz, CD3OD) δ 8.00 (d, J = 2.2 Hz, 1H, H-5), 7.82 (d, J = 2.2 Hz, 1H, H-5'), 7.52 dd, J1 = 1.5 Hz, J2 = 8.2 Hz, 1H, H-11), 7.44 (d, J = 2.2 Hz, 1H, H-4), 7.39 (d, J = 2.0 Hz, 1H, H-4'), 7.29 (d, J = 2.2 Hz, 1H, H-5''), 7.12 (d, J = 2.0 Hz, 1H, H-5'), 7.11 (dd, J1 = 1.5 Hz, J2 = 8.2 Hz, 1H, H-11'), 7.05 (dd, J1 = 1.4 Hz, J2 = 8.1 Hz, 1H, H-11''), 6.97–6.93 (m, 2H, H-8/9), 6.80 (ddd, J1 = 1.5 Hz, J2 = 7.0 Hz, J3 = 8.2 Hz, 1H, H-9'), 6.72 (dd, J1 = 1.5 Hz, J2 = 8.3 Hz, 1H, H-8'), 6.64–6.60 (overlapped signals, 2H, H-9''/10), 6.57 (ddd, J1 = 1.5 Hz, J2 = 7.0 Hz, J3 = 8.3 Hz, 1H, H-10'), 6.41 (ddd, J1 = 1.4 Hz, J2 = 7.3 Hz, J3 = 8.1 Hz, 1H , H-10''), 5.81 (dd, J1 = 1.3 Hz, J2 = 8.1 Hz, 1H, H-8''), 4.33 (ddd, J1 = 4.8 Hz, J2 = 11.8 Hz, J3 = 16.7 Hz, 1H, H-12), 4.49 (ddd, J1 = 4.8 Hz, J2 = 11.8 Hz, J3 = 16.7 Hz, 1H, H12), 3.78 (t, J = 8.5 Hz, 2H, H-12'), 3.78–3.64 (m, 2H, H-12''), 1.70–1.64 (m, 1H, H-13), 1.62–1.56 (m, 1H, H-13), 1.53–1.45 (m, 1H, H-13'), 1.37–1.30 (m, 1H, H-13'), 1.27–1.18 (m, 1H, H-14), 1.08–0.89 (m, 8H, 13''/14/ 2 × H-14'/14''/15), 0.88–0.82 (m, 1H, H-14'), 0.79–0.72 (m, 1H, H-13''), 0.68 (t, J = 7.3 Hz, 3H, H-15'), 0.63 (t, J = 7.4 Hz, 3H, H-15'') ppm; 13C NMR (150 MHz, CD3OD) δ 173.5 (o, d, 1JC,Rh = 35.6 Hz, C-2), 171.0 (o, d, 1JC,Rh = 35.6 Hz, C-2'), 164.4 (o, d, 1JC,Rh = 48.5 Hz, C-2''), 160.2 (o, C-7), 159.8(o, C-7'), 157.3 (o, C-7''), 130.5 (o, C-6), 129.8 (o, C-6'), 128.2 (o, C-6''), 126.6 (+, C-9), 126.1 (+, C-9'), 125.9 (+, C-9''), 123.4 (+, C-8), 122.9 (+, C-4), 122.7 (+, C-8'), 122.2 (+, C-4'), 121.3 (+, C-8''), 120.8 (+, C-4''), 120.7 (+, C-11), 119.6 (+, C-11'), 118.7 (+, C-11''), 118.4 (+, C-5), 118.4 (+, C-5'), 117.8 (+, C-5''), 115.1 (+, C-10), 114.1 (+, C-10'), 113.6 (+, C-10''), 48.7/48.3/48.2 (−, C-12/12'/12''), 32.5/32.3/32.0 (−, C-13/13'/13''), 20.0/19.7/19.7 (−, C-14/14'/14''), 12.9/12.4/12.4 (+, C-15/15'/15'') ppm; IR (ATR) ![[Graphic 4]](/bjoc/content/inline/1860-5397-12-264-i6.svg?max-width=637&scale=1.18182) : 2955, 1590, 1419, 1374, 1302, 1268, 1122, 950, 850, 742, 716, 698, 690, 683, 657 cm−1; ESIMS (30 V) m/z (%): 749.2 (100) [M+H]+; HRESIMS: calcd for C39H46N6O3Rh+, 749.2686; found, 749.2685.
: 2955, 1590, 1419, 1374, 1302, 1268, 1122, 950, 850, 742, 716, 698, 690, 683, 657 cm−1; ESIMS (30 V) m/z (%): 749.2 (100) [M+H]+; HRESIMS: calcd for C39H46N6O3Rh+, 749.2686; found, 749.2685.
Preparation of bis(3-butyl-1-(2-oxidophenyl)-1H-imidazolium-2-yl)nickel (12): A solution of 0.15 g (0.70 mmol) of 2-(3-butyl-1H-imidazolium-1-yl)phenolate in 5 mL of anhydrous toluene was treated with 0.23 g (0.35 mmol) of bis(triphenylphosphine)nickel(II) dichloride and stirred under an inert atmosphere overnight at reflux temperature. The resulting white solid was filtered off, washed with THF and dried in vacuo. Yield: 0.09 g (45%) of a yellow solid, mp: 153 °C; 1H NMR (600 MHz, CD3OD) δ 7.67 (d, J = 2.0 Hz, 2H, H-4/4'), 7.44 (dd, J1 = 1.3 Hz, J2 = 7.5 Hz, 2H, H-11/11'), 7.36 (d, J = 2.0 Hz, 2H, H-5/5'), 7.11 (dd, J1 = 1.9 Hz, J2 = 8.2 Hz, 2H, H-8/8'), 7.08 (ddd, J1 = 1.3 Hz, J2 = 8.2 Hz, J3 = 8.9 Hz, 2H, H-9/9'), 6.72 (ddd, J1 = 1.9 Hz, J2 = 7.5 Hz, J3 = 8.9 Hz, 2H, H-10/10'), 3.68–3.63 (m, 2H, H-12/12'), 3.06–3.01 (m, 2H, H-12/12'), 2.42–2.35 (m, 2H, H-13/13'), 1.84–1.76 (m, 2H, H-13/13'), 1.27–1.21 (m, 4H, H-14/14'), 0.77 (t, J1 = 7.4 Hz, 6H, H-15/15') ppm; 13C NMR (600 MHz, CD3OD) δ 156.8 (o, C-7/7'), 156.2 (o, C-2/2'), 128.8 (o, C-6/6'), 127.4 (+, C-9/9'), 124.2 (+, C-5/5'), 120.9 (+, C-8/8'), 118.4 (+, C-11/11'), 118.3 (+, C-4/4'), 115.1 (+, C-10/10'), 49.8 (+, C-12/12'), 33.4 (+, C-13/13'), 19.5 (+, C-14/14'), 12.3 (+, C-15/15') ppm; IR (ATR) ![[Graphic 5]](/bjoc/content/inline/1860-5397-12-264-i7.svg?max-width=637&scale=1.18182) : 2958, 2929, 2872, 1593, 1487, 1457, 1417, 1395, 1300, 1273, 1235, 1154, 952, 840, 742, 724, 681 cm−1; ESIMS (5 V) m/z (%): 511.0 (100) [M + Na]+; HRESIMS: calcd for C26H31N4O2Ni+; 489.1800; found, 489.1800.
: 2958, 2929, 2872, 1593, 1487, 1457, 1417, 1395, 1300, 1273, 1235, 1154, 952, 840, 742, 724, 681 cm−1; ESIMS (5 V) m/z (%): 511.0 (100) [M + Na]+; HRESIMS: calcd for C26H31N4O2Ni+; 489.1800; found, 489.1800.
Supporting Information
| Supporting Information File 1: NMR spectra and molecular drawings. | ||
| Format: PDF | Size: 1.3 MB | Download |
References
-
Arduengo, A. J., III; Harlow, R. L.; Kline, M. J. Am. Chem. Soc. 1991, 113, 361–363. doi:10.1021/ja00001a054
Return to citation in text: [1] -
Hopkinson, M. N.; Richter, C.; Schedler, M.; Glorius, F. Nature 2014, 510, 485–496. doi:10.1038/nature13384
Return to citation in text: [1] -
Zhao, Q.; Curran, D. P.; Malacria, M.; Fensterbank, L.; Goddard, J.-P.; Lacôte, E. Chem. – Eur. J. 2011, 17, 9911–9914. doi:10.1002/chem.201101822
Return to citation in text: [1] -
Mata, J. A.; Poyatos, M. Curr. Org. Chem. 2011, 15, 3309–3324. doi:10.2174/138527211797247969
Return to citation in text: [1] -
Schmidt, A.; Rahimi, A. Chem. Commun. 2010, 46, 2995–2997. doi:10.1039/c001362e
Return to citation in text: [1] -
Hahn, F. E.; Jahnke, M. C. Angew. Chem. 2008, 120, 3166–3216. doi:10.1002/ange.200703883
Angew. Chem., Int. Ed. 2008, 47, 3122–3172. doi:10.1002/anie.200703883
Return to citation in text: [1] [2] -
Crabtree, R. H. J. Organomet. Chem. 2005, 690, 5451–5457. doi:10.1016/j.jorganchem.2005.07.099
Return to citation in text: [1] -
Chen, J.; Meng, S.; Wang, L.; Tang, H.; Huang, Y. Chem. Sci. 2015, 6, 4184–4189. doi:10.1039/C5SC00878F
Return to citation in text: [1] -
Cheng, J.; Huang, Z.; Chi, Y. R. Angew. Chem. 2013, 125, 8754–8758. doi:10.1002/ange.201303247
Angew. Chem., Int. Ed. 2013, 52, 8592–8596. doi:10.1002/anie.201303247
Return to citation in text: [1] -
Grossmann, A.; Enders, D. Angew. Chem. 2012, 124, 320–332. doi:10.1002/ange.201105415
Angew. Chem., Int. Ed. 2012, 51, 314–325. doi:10.1002/anie.201105415
Return to citation in text: [1] -
Guan, Z.; Hillrichs, K.; Ünlü, C.; Rissanen, K.; Nieger, M.; Schmidt, A. Tetrahedron 2015, 71, 276–282. doi:10.1016/j.tet.2014.11.054
Return to citation in text: [1] -
Tskhovrebov, A. G.; Naested, L. C. E.; Solari, E.; Scopelliti, R.; Severin, K. Angew. Chem. 2015, 127, 1305–1308. doi:10.1002/ange.201410067
Angew. Chem., Int. Ed. 2015, 54, 1289–1292. doi:10.1002/anie.201410067
Return to citation in text: [1] -
Guan, Z.; Namyslo, J. C.; Drafz, M. H. H.; Nieger, M.; Schmidt, A. Beilstein J. Org. Chem. 2014, 10, 832–840. doi:10.3762/bjoc.10.79
Return to citation in text: [1] -
Nolan, S. P. N-Heterocyclic Carbenes. Effective Tools for Organometallic Synthesis; Wiley-VCH: Hoboken, 2014.
Return to citation in text: [1] -
Moss, R. A.; Doyle, M. P. Contemporary Carbene Chemistry; Wiley: Hoboken, New Jersey, 2014. doi:10.1002/9781118730379
Return to citation in text: [1] -
Díez-González, S., Ed. N-Heterocyclic Carbenes. Form Laboratory Curiosities to Efficient Synthetic Tools; RSC Catalysis Series No. 6; RSC Publishing: Cambridge, 2011.
Return to citation in text: [1] -
Schmidt, A.; Wiechmann, S.; Otto, C. F. Adv. Heterocycl. Chem. 2016, 119, 143–172. doi:10.1016/bs.aihch.2016.02.002
Return to citation in text: [1] -
Merceron-Saffon, N.; Baceiredo, A.; Gornitzka, H.; Bertrand, G. Science 2003, 301, 1223–1225. doi:10.1126/science.1086860
Return to citation in text: [1] -
Ezugwu, C. I.; Kabir, N. A.; Yusubov, M.; Verpoort, F. Coord. Chem. Rev. 2016, 307, 188–210. doi:10.1016/j.ccr.2015.06.012
Return to citation in text: [1] -
Hille, C.; Kühn, F. E. Dalton Trans. 2016, 45, 15–31. doi:10.1039/C5DT03641K
Return to citation in text: [1] -
Ohki, Y.; Seino, H. Dalton Trans. 2016, 45, 874–880. doi:10.1039/C5DT04298D
Return to citation in text: [1] -
Ritleng, V.; Henrion, M.; Chetcuti, M. J. ACS Catal. 2016, 6, 890–906. doi:10.1021/acscatal.5b02021
Return to citation in text: [1] -
Sandtorv, A. H.; Leitch, C.; Bedringaas, S. L.; Gjertsen, B. T.; Bjørsvik, H.-R. ChemMedChem 2015, 10, 1522–1527. doi:10.1002/cmdc.201500234
Return to citation in text: [1] -
Budagumpi, S.; Haque, R. A.; Endud, S.; Rehman, G. U.; Salman, A. W. Eur. J. Inorg. Chem. 2013, 4367–4388. doi:10.1002/ejic.201300483
Return to citation in text: [1] -
Nasr, A.; Winkler, A.; Tamm, M. Coord. Chem. Rev. 2016, 316, 68–124. doi:10.1016/j.ccr.2016.02.011
Return to citation in text: [1] -
Liu, M.; Nieger, M.; Schmidt, A. Chem. Commun. 2015, 51, 477–479. doi:10.1039/C4CC08032G
Return to citation in text: [1] -
Färber, C.; Leibold, M.; Bruhn, C.; Maurer, M.; Siemeling, U. Chem. Commun. 2012, 48, 227–229. doi:10.1039/C1CC16460K
Return to citation in text: [1] -
Wiechmann, S.; Freese, T.; Drafz, M. H. H.; Hübner, E. G.; Namyslo, J. C.; Nieger, M.; Schmidt, A. Chem. Commun. 2014, 50, 11822–11824. doi:10.1039/C4CC05461J
Return to citation in text: [1] -
Benhamou, L.; Vujkovic, N.; César, V.; Gornitzka, H.; Lugan, N.; Lavigne, G. Organometallics 2010, 29, 2616–2630. doi:10.1021/om1003607
Return to citation in text: [1] [2] -
Danopoulos, A. A.; Monakhov, K. Yu.; Braunstein, P. Chem. – Eur. J. 2013, 19, 450–455. doi:10.1002/chem.201203488
Return to citation in text: [1] -
Pidlypnyi, N.; Namyslo, J. C.; Drafz, M. H. H.; Nieger, M.; Schmidt, A. J. Org. Chem. 2013, 78, 1070–1079. doi:10.1021/jo302479p
Return to citation in text: [1] -
César, V.; Lugan, N.; Lavigne, G. Chem. – Eur. J. 2010, 16, 11432–11442. doi:10.1002/chem.201000870
Return to citation in text: [1] -
César, V.; Lugan, N.; Lavigne, G. J. Am. Chem. Soc. 2008, 130, 11286–11287. doi:10.1021/ja804296t
Return to citation in text: [1] -
Schmidt, A.; Wiechmann, S.; Freese, T. ARKIVOC 2013, i, 424–469.
Return to citation in text: [1] [2] -
Liu, M.; Nieger, M.; Hübner, E. G.; Schmidt, A. Chem. – Eur. J. 2016, 22, 5416–5424. doi:10.1002/chem.201505042
Return to citation in text: [1] [2] -
Xu, X.; Kim, S. H.; Zhang, X.; Das, A. K.; Hirao, H.; Hong, S. H. Organometallics 2013, 32, 164–171. doi:10.1021/om3009603
Return to citation in text: [1] -
Jothibasu, R.; Huynh, H. V.; Koh, L. L. J. Organomet. Chem. 2008, 693, 374–380. doi:10.1016/j.jorganchem.2007.11.003
Return to citation in text: [1] -
Catalano, V. J.; Etogo, A. O. Inorg. Chem. 2007, 46, 5608–5615. doi:10.1021/ic070260i
Return to citation in text: [1] -
Benhamou, L.; César, V.; Gornitzka, H.; Lugan, N.; Lavigne, G. Chem. Commun. 2009, 4720–4722. doi:10.1039/b907908d
Return to citation in text: [1] -
Burling, S.; Douglas, S.; Mahon, M. F.; Nama, D.; Pregosin, P. S.; Whittlesey, M. K. Organometallics 2006, 25, 2642–2648. doi:10.1021/om060202p
Return to citation in text: [1] [2] -
Chaplin, A. B. Organometallics 2014, 33, 624–626. doi:10.1021/om401223w
Return to citation in text: [1] [2] -
Nirmala, M.; Prakash, G.; Ramachandran, R.; Viswanathamurthi, P.; Malecki, J. G.; Linert, W. J. Mol. Catal. A: Chem. 2015, 397, 56–67. doi:10.1016/j.molcata.2014.10.031
Return to citation in text: [1] -
Kong, Y.; Cheng, M.; Ren, H.; Xu, S.; Song, H.; Yang, M.; Liu, B.; Wang, B. Organometallics 2011, 30, 1677–1681. doi:10.1021/om1011825
Return to citation in text: [1] -
Boydston, A. J.; Rice, J. D.; Sanderson, M. D.; Dykhno, O. L.; Bielawski, C. W. Organometallics 2006, 25, 6087–6098. doi:10.1021/om060494u
Return to citation in text: [1] -
Tao, W.-j.; Nakano, R.; Ito, S.; Nozaki, K. Angew. Chem. 2016, 128, 2885–2889. doi:10.1002/ange.201510077
Angew. Chem., Int. Ed. 2016, 55, 2835–2839. doi:10.1002/anie.201510077
Return to citation in text: [1] -
Borré, E.; Dahm, G.; Aliprandi, A.; Mauro, M.; Dagorne, S.; Bellemin-Laponnaz, S. Organometallics 2014, 33, 4374–4384. doi:10.1021/om5003446
Return to citation in text: [1] -
Matson, E. M.; Espinosa Martinez, G.; Ibrahim, A. D.; Jackson, B. J.; Bertke, J. A.; Fout, A. R. Organometallics 2015, 34, 399–407. doi:10.1021/om5007177
Return to citation in text: [1] -
Arduengo, A. J., III; Gurau, G.; Kelley, S. P.; Marshall, W. J.; Runyon, J. W. Angew. Chem. 2013, 125, 11071–11073. doi:10.1002/ange.201305714
Angew. Chem. ,Int. Ed. 2013, 52, 10871–10873. doi:10.1002/anie.201305714
Return to citation in text: [1] -
Samantaray, M. K.; Shaikh, M. M.; Ghosh, P. Organometallics 2009, 28, 2267–2275. doi:10.1021/om801186f
Return to citation in text: [1] -
Prasad, L.; Taylor, M. R. Acta Crystallogr., Sect. C 1983, 39, 1686–1688. doi:10.1107/S0108270183009750
Return to citation in text: [1] -
Weinberg, D. R.; Hazari, N.; Labinger, J. A.; Bercaw, J. E. Organometallics 2010, 29, 89–100. doi:10.1021/om900803r
Return to citation in text: [1] -
Ledoux, N.; Allaert, B.; Verpoort, F. Eur. J. Inorg. Chem. 2007, 5578–5583. doi:10.1002/ejic.200700665
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/S2053273314026370
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. doi:10.1107/S0108767307043930
Return to citation in text: [1]
| 46. | Borré, E.; Dahm, G.; Aliprandi, A.; Mauro, M.; Dagorne, S.; Bellemin-Laponnaz, S. Organometallics 2014, 33, 4374–4384. doi:10.1021/om5003446 |
| 47. | Matson, E. M.; Espinosa Martinez, G.; Ibrahim, A. D.; Jackson, B. J.; Bertke, J. A.; Fout, A. R. Organometallics 2015, 34, 399–407. doi:10.1021/om5007177 |
| 48. |
Arduengo, A. J., III; Gurau, G.; Kelley, S. P.; Marshall, W. J.; Runyon, J. W. Angew. Chem. 2013, 125, 11071–11073. doi:10.1002/ange.201305714
Angew. Chem. ,Int. Ed. 2013, 52, 10871–10873. doi:10.1002/anie.201305714 |
| 49. | Samantaray, M. K.; Shaikh, M. M.; Ghosh, P. Organometallics 2009, 28, 2267–2275. doi:10.1021/om801186f |
| 50. | Prasad, L.; Taylor, M. R. Acta Crystallogr., Sect. C 1983, 39, 1686–1688. doi:10.1107/S0108270183009750 |
| 51. | Weinberg, D. R.; Hazari, N.; Labinger, J. A.; Bercaw, J. E. Organometallics 2010, 29, 89–100. doi:10.1021/om900803r |
| 52. | Ledoux, N.; Allaert, B.; Verpoort, F. Eur. J. Inorg. Chem. 2007, 5578–5583. doi:10.1002/ejic.200700665 |
| 1. | Arduengo, A. J., III; Harlow, R. L.; Kline, M. J. Am. Chem. Soc. 1991, 113, 361–363. doi:10.1021/ja00001a054 |
| 14. | Nolan, S. P. N-Heterocyclic Carbenes. Effective Tools for Organometallic Synthesis; Wiley-VCH: Hoboken, 2014. |
| 15. | Moss, R. A.; Doyle, M. P. Contemporary Carbene Chemistry; Wiley: Hoboken, New Jersey, 2014. doi:10.1002/9781118730379 |
| 16. | Díez-González, S., Ed. N-Heterocyclic Carbenes. Form Laboratory Curiosities to Efficient Synthetic Tools; RSC Catalysis Series No. 6; RSC Publishing: Cambridge, 2011. |
| 30. | Danopoulos, A. A.; Monakhov, K. Yu.; Braunstein, P. Chem. – Eur. J. 2013, 19, 450–455. doi:10.1002/chem.201203488 |
| 11. | Guan, Z.; Hillrichs, K.; Ünlü, C.; Rissanen, K.; Nieger, M.; Schmidt, A. Tetrahedron 2015, 71, 276–282. doi:10.1016/j.tet.2014.11.054 |
| 12. |
Tskhovrebov, A. G.; Naested, L. C. E.; Solari, E.; Scopelliti, R.; Severin, K. Angew. Chem. 2015, 127, 1305–1308. doi:10.1002/ange.201410067
Angew. Chem., Int. Ed. 2015, 54, 1289–1292. doi:10.1002/anie.201410067 |
| 13. | Guan, Z.; Namyslo, J. C.; Drafz, M. H. H.; Nieger, M.; Schmidt, A. Beilstein J. Org. Chem. 2014, 10, 832–840. doi:10.3762/bjoc.10.79 |
| 31. | Pidlypnyi, N.; Namyslo, J. C.; Drafz, M. H. H.; Nieger, M.; Schmidt, A. J. Org. Chem. 2013, 78, 1070–1079. doi:10.1021/jo302479p |
| 8. | Chen, J.; Meng, S.; Wang, L.; Tang, H.; Huang, Y. Chem. Sci. 2015, 6, 4184–4189. doi:10.1039/C5SC00878F |
| 9. |
Cheng, J.; Huang, Z.; Chi, Y. R. Angew. Chem. 2013, 125, 8754–8758. doi:10.1002/ange.201303247
Angew. Chem., Int. Ed. 2013, 52, 8592–8596. doi:10.1002/anie.201303247 |
| 10. |
Grossmann, A.; Enders, D. Angew. Chem. 2012, 124, 320–332. doi:10.1002/ange.201105415
Angew. Chem., Int. Ed. 2012, 51, 314–325. doi:10.1002/anie.201105415 |
| 28. | Wiechmann, S.; Freese, T.; Drafz, M. H. H.; Hübner, E. G.; Namyslo, J. C.; Nieger, M.; Schmidt, A. Chem. Commun. 2014, 50, 11822–11824. doi:10.1039/C4CC05461J |
| 2. | Hopkinson, M. N.; Richter, C.; Schedler, M.; Glorius, F. Nature 2014, 510, 485–496. doi:10.1038/nature13384 |
| 3. | Zhao, Q.; Curran, D. P.; Malacria, M.; Fensterbank, L.; Goddard, J.-P.; Lacôte, E. Chem. – Eur. J. 2011, 17, 9911–9914. doi:10.1002/chem.201101822 |
| 4. | Mata, J. A.; Poyatos, M. Curr. Org. Chem. 2011, 15, 3309–3324. doi:10.2174/138527211797247969 |
| 5. | Schmidt, A.; Rahimi, A. Chem. Commun. 2010, 46, 2995–2997. doi:10.1039/c001362e |
| 6. |
Hahn, F. E.; Jahnke, M. C. Angew. Chem. 2008, 120, 3166–3216. doi:10.1002/ange.200703883
Angew. Chem., Int. Ed. 2008, 47, 3122–3172. doi:10.1002/anie.200703883 |
| 7. | Crabtree, R. H. J. Organomet. Chem. 2005, 690, 5451–5457. doi:10.1016/j.jorganchem.2005.07.099 |
| 29. | Benhamou, L.; Vujkovic, N.; César, V.; Gornitzka, H.; Lugan, N.; Lavigne, G. Organometallics 2010, 29, 2616–2630. doi:10.1021/om1003607 |
| 21. | Ohki, Y.; Seino, H. Dalton Trans. 2016, 45, 874–880. doi:10.1039/C5DT04298D |
| 22. | Ritleng, V.; Henrion, M.; Chetcuti, M. J. ACS Catal. 2016, 6, 890–906. doi:10.1021/acscatal.5b02021 |
| 25. | Nasr, A.; Winkler, A.; Tamm, M. Coord. Chem. Rev. 2016, 316, 68–124. doi:10.1016/j.ccr.2016.02.011 |
| 35. | Liu, M.; Nieger, M.; Hübner, E. G.; Schmidt, A. Chem. – Eur. J. 2016, 22, 5416–5424. doi:10.1002/chem.201505042 |
| 19. | Ezugwu, C. I.; Kabir, N. A.; Yusubov, M.; Verpoort, F. Coord. Chem. Rev. 2016, 307, 188–210. doi:10.1016/j.ccr.2015.06.012 |
| 20. | Hille, C.; Kühn, F. E. Dalton Trans. 2016, 45, 15–31. doi:10.1039/C5DT03641K |
| 26. | Liu, M.; Nieger, M.; Schmidt, A. Chem. Commun. 2015, 51, 477–479. doi:10.1039/C4CC08032G |
| 27. | Färber, C.; Leibold, M.; Bruhn, C.; Maurer, M.; Siemeling, U. Chem. Commun. 2012, 48, 227–229. doi:10.1039/C1CC16460K |
| 6. |
Hahn, F. E.; Jahnke, M. C. Angew. Chem. 2008, 120, 3166–3216. doi:10.1002/ange.200703883
Angew. Chem., Int. Ed. 2008, 47, 3122–3172. doi:10.1002/anie.200703883 |
| 18. | Merceron-Saffon, N.; Baceiredo, A.; Gornitzka, H.; Bertrand, G. Science 2003, 301, 1223–1225. doi:10.1126/science.1086860 |
| 53. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/S2053273314026370 |
| 17. | Schmidt, A.; Wiechmann, S.; Otto, C. F. Adv. Heterocycl. Chem. 2016, 119, 143–172. doi:10.1016/bs.aihch.2016.02.002 |
| 23. | Sandtorv, A. H.; Leitch, C.; Bedringaas, S. L.; Gjertsen, B. T.; Bjørsvik, H.-R. ChemMedChem 2015, 10, 1522–1527. doi:10.1002/cmdc.201500234 |
| 24. | Budagumpi, S.; Haque, R. A.; Endud, S.; Rehman, G. U.; Salman, A. W. Eur. J. Inorg. Chem. 2013, 4367–4388. doi:10.1002/ejic.201300483 |
| 54. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. doi:10.1107/S0108767307043930 |
| 35. | Liu, M.; Nieger, M.; Hübner, E. G.; Schmidt, A. Chem. – Eur. J. 2016, 22, 5416–5424. doi:10.1002/chem.201505042 |
| 32. | César, V.; Lugan, N.; Lavigne, G. Chem. – Eur. J. 2010, 16, 11432–11442. doi:10.1002/chem.201000870 |
| 33. | César, V.; Lugan, N.; Lavigne, G. J. Am. Chem. Soc. 2008, 130, 11286–11287. doi:10.1021/ja804296t |
| 42. | Nirmala, M.; Prakash, G.; Ramachandran, R.; Viswanathamurthi, P.; Malecki, J. G.; Linert, W. J. Mol. Catal. A: Chem. 2015, 397, 56–67. doi:10.1016/j.molcata.2014.10.031 |
| 43. | Kong, Y.; Cheng, M.; Ren, H.; Xu, S.; Song, H.; Yang, M.; Liu, B.; Wang, B. Organometallics 2011, 30, 1677–1681. doi:10.1021/om1011825 |
| 44. | Boydston, A. J.; Rice, J. D.; Sanderson, M. D.; Dykhno, O. L.; Bielawski, C. W. Organometallics 2006, 25, 6087–6098. doi:10.1021/om060494u |
| 45. |
Tao, W.-j.; Nakano, R.; Ito, S.; Nozaki, K. Angew. Chem. 2016, 128, 2885–2889. doi:10.1002/ange.201510077
Angew. Chem., Int. Ed. 2016, 55, 2835–2839. doi:10.1002/anie.201510077 |
| 40. | Burling, S.; Douglas, S.; Mahon, M. F.; Nama, D.; Pregosin, P. S.; Whittlesey, M. K. Organometallics 2006, 25, 2642–2648. doi:10.1021/om060202p |
| 29. | Benhamou, L.; Vujkovic, N.; César, V.; Gornitzka, H.; Lugan, N.; Lavigne, G. Organometallics 2010, 29, 2616–2630. doi:10.1021/om1003607 |
| 39. | Benhamou, L.; César, V.; Gornitzka, H.; Lugan, N.; Lavigne, G. Chem. Commun. 2009, 4720–4722. doi:10.1039/b907908d |
| 40. | Burling, S.; Douglas, S.; Mahon, M. F.; Nama, D.; Pregosin, P. S.; Whittlesey, M. K. Organometallics 2006, 25, 2642–2648. doi:10.1021/om060202p |
| 41. | Chaplin, A. B. Organometallics 2014, 33, 624–626. doi:10.1021/om401223w |
| 36. | Xu, X.; Kim, S. H.; Zhang, X.; Das, A. K.; Hirao, H.; Hong, S. H. Organometallics 2013, 32, 164–171. doi:10.1021/om3009603 |
| 37. | Jothibasu, R.; Huynh, H. V.; Koh, L. L. J. Organomet. Chem. 2008, 693, 374–380. doi:10.1016/j.jorganchem.2007.11.003 |
| 38. | Catalano, V. J.; Etogo, A. O. Inorg. Chem. 2007, 46, 5608–5615. doi:10.1021/ic070260i |
© 2016 Liu et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)