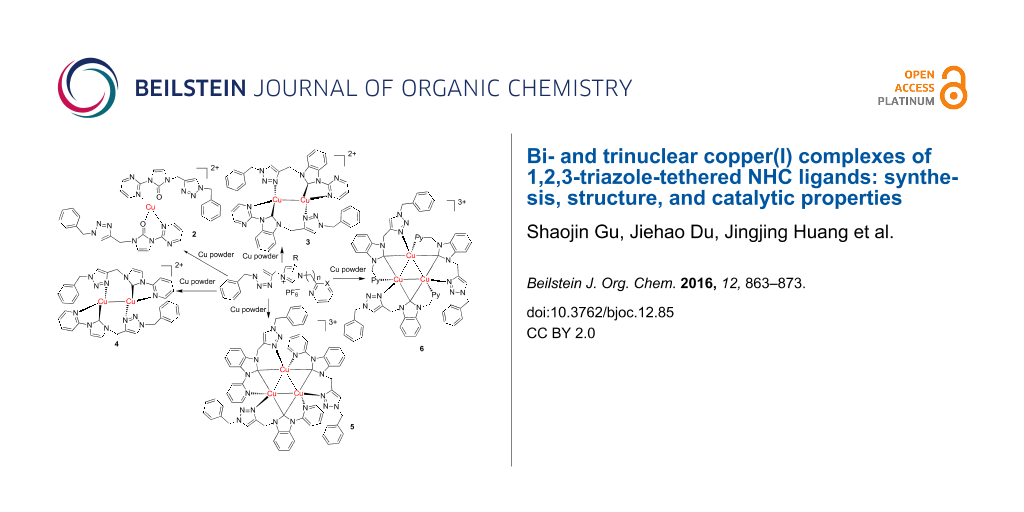
Abstract
A series of copper complexes (3–6) stabilized by 1,2,3-triazole-tethered N-heterocyclic carbene ligands have been prepared via simple reaction of imidazolium salts with copper powder in good yields. The structures of bi- and trinuclear copper complexes were fully characterized by NMR, elemental analysis (EA), and X-ray crystallography. In particular, [Cu2(L2)2](PF6)2 (3) and [Cu2(L3)2](PF6)2 (4) were dinuclear copper complexes. Complexes [Cu3(L4)2](PF6)3 (5) and [Cu3(L5)2](PF6)3 (6) consist of a triangular Cu3 core. These structures vary depending on the imidazolium backbone and N substituents. The copper–NHC complexes tested are highly active for the Cu-catalyzed azide–alkyne cycloaddition (CuAAC) reaction in an air atmosphere at room temperature in a CH3CN solution. Complex 4 is the most efficient catalyst among these polynuclear complexes in an air atmosphere at room temperature.

Graphical Abstract
Introduction
N-Heterocyclic carbene (NHC) have interesting electronic and structural properties. This resulted in their use as versatile ligands in organometallic chemistry and homogeneous catalysis [1-12]. A number of transition metal complexes of NHCs containing pyridine [13], pyrimidine [14], pyrazole [15,16], naphthyridine [17], pyridazine [18], and phenanthroline [19,20] donating groups have been studied in metal-catalyzed organic transformations. Recently, the easy synthesis and versatile coordination ability of 1,2,3-triazoles have led to an explosion of interest in coordination chemistry [21] and homogeneous catalysis [22-26]. Although a number of metal complexes containing 1,4-disubstituted-1,2,3-triazole ligands were well studied, reports concerning their preparation and use of 1,4-disubstituted-1,2,3-triazoles bearing NHC ligands are rare [22,23]. Elsevier et al. [23] reported several of palladium(II) complexes containing a heterobidentate N-heterocyclic carbene-triazolyl ligand. These palladium(II) complexes are active precatalysts in the transfer semihydrogenation of alkynes to Z-alkenes. Messerle et al. [26] synthesized a series of new cationic Rh(I), Rh(III) and Ir(III) complexes containing hybrid bidentate N-heterocyclic carbene-1,2,3-triazolyl donors. We [27] have synthesized a series of nonsymmetrical pincer palladium and platinum complexes containing 1,2,3-triazole-tethered NHC ligands. The obtained palladium complexes displayed high activity in aqueous Suzuki–Miyaura cross-coupling reactions.
We are interested in the synthesis and use of functionalized NHC ligands [20,28-31]. Herein, the synthesis, structural characterization, and catalytic properties of a few copper-1,2,3-triazole-tethered NHC complexes is reported.
Results and Discussion
Synthesis and spectroscopic characterization
The imidazolium salts (1a–e) were prepared according to the reported procedure in 61–90% yields [27]. These imidazolium salts have been characterized by NMR spectroscopy. The 1H NMR spectra of these imidazolium salts show singlet peaks between 10.04 and 10.89 ppm in DMSO-d6. As seen in Scheme 1, copper–NHC complexes 3–6 can be obtained in 52–90% yields via directly reacting the corresponding imidazolium salts with an excess of copper powder in CH3CN at 50 °C for 5 h.
As shown in Scheme 1, reactions of the pyrimidine imidazolium salt 1a with copper powder in acetonitrile afforded a light yellow Cu(II) complex. In complex 2, the carbenic carbon atom was oxidized into carbonyl, which is similar with the reported pyrimidyl-imidazole complex [32]. However, a red binuclear Cu(I) complex 3 was obtained in 57% yield when we reacted pyrimidyl benzimidazolium salt 1b with copper powder. Furtherly, we got a yellow Cu(I)–NHC complex 4 in about 70% yield from pyridine imidazolium salt 1c and copper powder (Scheme 1). In addition, a triangular Cu(I) complex 6 can be obtained when a flexible ligand was used. Complex 6 consists of a triangular Cu3 core bridged by three NHCs, which is similar with the published Cu3 complexes containing flexible ligands [33]. Interestingly, we can also obtain a similar triangular Cu3 complex 5 rather than a binuclear copper complex using a rigid pyridine benzimidazolium salt 1d. These results demonstrated that the structures vary depending on the N substituents and on the imidazolium backbone. Fine adjustment of the structure of the ligand can lead to different structures.
![[1860-5397-12-85-i1]](/bjoc/content/inline/1860-5397-12-85-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of copper complexes 2–6.
Scheme 1: Synthesis of copper complexes 2–6.
All of the prepared copper–NHC complexes are stable in air. They were fully characterized by NMR, elemental analysis (EA), and X-ray crystallography. The generation of these copper–NHC complexes were confirmed by the absence of the 1H NMR resonance signal of the acidic imidazolium protons between 10.04 and 10.89 ppm. The 1H NMR spectra of all the complexes display only one set of resonance signals assignable to the corresponding ligands, indicating two or three magnetically equivalent ligands. 13C NMR spectra of the copper(I) complexes showed their carbenic carbon resonances at 177.6–191.2 ppm, which are in the normal range of 157.6–216 ppm [34,35].
Single crystal X-ray diffraction studies
To obtain additional insight into the coordination and supramolecular properties, suitable single crystals of all the copper complexes were obtained for single-crystal X-ray diffraction analysis. Crystals were grown by slow diffusion of diethyl ether into an acetonitrile solution of the copper complex at room temperature.
Green-yellow single crystals of complex 2 suitable for an X-ray diffraction study were grown from acetonitrile solution and diethyl ether. The molecular structure of complex 2 in the solid state is depicted in Figure 1 along with the principal bond lengths and angles. Complex 2 crystallizes in the orthorhombic space group Pnna. The remaining atoms of the cation are related by a crystallographic 2-fold symmetry. In complex 2, the copper ion is four-coordinate in a distorted square planar ligand environment of two nitrogen atoms and two oxyen atoms. The Cu–O bonds are in trans configuration and Cu–O distances are shorter than Cu–N distances. The two ligands are arranged in head-to-tail manner. And the Ntriazole did not participate in the corrdination.
![[1860-5397-12-85-1]](/bjoc/content/figures/1860-5397-12-85-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray diffraction structure of copper(II) complex 2 with thermal ellipsoids drawn at 30% probability. The anion and hydrogen atoms are omitted for clarity. Selected bond distances (Å) and angles (°): Cu1-O1 1.931(4), Cu1-N6 2.042(5); O1-Cu1-O1A 180.0(3), O1-Cu1-N6A 90.5(2), O1-Cu1-N6 89.5(2), N6-Cu1-N6A 180.00(8). Symmetry transformations used to generate equivalent atoms: −X, Y, 0.5−Z.
Figure 1: X-ray diffraction structure of copper(II) complex 2 with thermal ellipsoids drawn at 30% probabilit...
Single crystals of complex 3 suitable for an X-ray diffraction study were grown from acetonitrile solution and diethyl ether. The molecular structure of complex 3 is depicted in Figure 2. Complex 3 crystallizes in the monoclinic space group C2/c. The Cu(I) complex contains two crystallographically equivalent Cu centers, which are doubly bridged by two L2 ligands. The two ligands are arranged in head-to-tail manner. The copper ions are each tri-coordinated by one carbene carbon atom, one nitrogen from pyrimidine, and one nitrogen atom of the triazole rings from two different L2 ligands. The Cu–carbene bond distances are 1.896(6) and 1.899(5) Å, which are comparable to the known Cu(I)–NHC complexes [36-39]. The Cu1–Cu2 separation is 2.7867(7) Å, showing a weak metal−metal interaction.
![[1860-5397-12-85-2]](/bjoc/content/figures/1860-5397-12-85-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ORTEP the cationic section of [Cu2(L2)2](PF6)2 (3). Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms and anions have been removed for clarity. Selected bond distances (Å) and angles (°): Cu2-C5 1.896(3), Cu2-N14 1.911(3), Cu2-N1 2.362(3), Cu2-Cu1 2.7867(7), Cu1-C26 1.898(3), Cu1-N7 1.915(3), Cu1-N8 2.340(3); C5-Cu2-N14 173.37(13), C5-Cu2-N1 77.64(13), N14-Cu2-N1 108.15(12), C5-Cu2-Cu1 69.80(9), N14-Cu2-Cu1 111.70(8), N1-Cu2-Cu1 98.90(7), C26-Cu1-N7 167.08(15), C26-Cu1-N8 78.22(13), N7-Cu1-N8 111.77(13), C26-Cu1-Cu2 73.57(9).
Figure 2: ORTEP the cationic section of [Cu2(L2)2](PF6)2 (3). Thermal ellipsoids are drawn at the 30% probabi...
The molecular structure of complex 4 is depicted in Figure 3. Complex 4 consists of the cation unit [Cu2(L3)2]2+ and two hexafluorophosphate anions. Complex 4 crystallizes in the triclinic space group P-1. The two ligands are also arranged in head-to-tail manner. Each copper ion is three-coordinate in a trigonal planar ligand environment of two nitrogen atoms and one NHC carbon center. The Cu–carbene bond distances are 1.888(6) and 1.899(5) Å which are similar with reported copper-carbene complexes (1.85–2.18 Å) [40]. The Cu1–Cu2 separation is 2.6413(12) Å is shorter than in complex 3, and slightly higher than reported Cu–Cu separations (2.4907 to 2.5150 Å) of the triangular Cu(I)–NHC clusters [33], showing a weak metal–metal interaction.
![[1860-5397-12-85-3]](/bjoc/content/figures/1860-5397-12-85-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: ORTEP drawing of [Cu2(L3)2](PF6)2 (4). Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms and anions have been removed for clarity. Selected bond distances (Å) and angles (°): Cu1-C26 1.888(6), Cu1-N5 1.912(5), Cu1-N7 2.289(5), Cu1-Cu2 2.6413(12), Cu2-C8 1.899(5), Cu2-N11 1.922(4), Cu2-N1 2.311(5); C26-Cu1-N5 159.2(2), C26-Cu1-N7 79.0(2), N5-Cu1-N7 116.65(19), C26-Cu1-Cu2 72.49(17), N5-Cu1-Cu2 113.20(15), N7-Cu1-Cu2 105.13(12), C8-Cu2-N11 166.1(2), C8-Cu2-N1 78.6(2), N11-Cu2-N1 110.5(2), C8-Cu2-Cu1 70.45(16), N11-Cu2-Cu1-116.14(15), N1- Cu2-Cu1 102.03(13).
Figure 3: ORTEP drawing of [Cu2(L3)2](PF6)2 (4). Thermal ellipsoids are drawn at the 30% probability level. H...
Complex 5 was also characterized via X-ray diffraction. It's structure is shown in Figure 4. Complex 5 consists of two independent molecules in the unit cell. Here, only one molecule was given in Figure 4. The molecule structure consists of a triangular Cu3 core bridged by three NHCs ligands. Each NHC forms the 3c-2e bond with two Cu(I) ions with almost equal bond distances (average 2.085 Å), longer than normal Cu–NHC bonds and reported triangular Cu3 complexes [33,41]. The Cu3 cores of complex 5 possess nearly equilateral angles close to 60°, whereas in complex 6, the core is crystallographically restrained to an equilateral triangle. The Cu–Cu distances are around 2.4887 Å and are shorten than that of complexe 6, which may be attributed to more rigid ligand.
![[1860-5397-12-85-4]](/bjoc/content/figures/1860-5397-12-85-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: ORTEP drawing of [Cu3(L4)3](PF6)3 (5). Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms and anions have been removed for clarity. Selected bond distances (Å) and angles (°): Cu2-N17 2.015(5), Cu2-C50 2.074(6), Cu2-N19 2.107(6), Cu2-C72 2.142(6), Cu2-Cu4 2.4899(11), Cu2-Cu3 2.4928(11), Cu3-N11 2.038(5), Cu3-C27 2.069(6), Cu3-C50 2.113(6), Cu3-N13 2.133(5), Cu3-Cu4 2.4833(10), Cu4-N22 2.043(5), Cu4-C72 2.044(7), Cu4-C27 2.071(6), Cu4-N7 2.086(6); N17-Cu2-C50 94.8(2), N17-Cu2-N19 112.1(2), C50-Cu2-N19 106.8(2), N17-Cu2-C72 93.5(2), C50- Cu2-C72 165.4(2), N19-Cu2-C72 81.0(2), N17-Cu2-Cu4 125.46(18), C50-Cu2-Cu4 113.86(16), N19-Cu2-Cu4 102.90(19), C72-Cu2-Cu4 51.72(17), N17-Cu2-Cu3 129.13(16), C50-Cu2-Cu3 54.18(16), N19-Cu2-Cu3 115.04(17), C72-Cu2-Cu3 111.50(17), Cu4-Cu2-Cu3 59.79(3), N11-Cu3-C27 94.1(2), N11-Cu3-C50 90.9(2), C27-Cu3-C50 163.7(2), N11-Cu3-N13 112.2(2), C27-Cu3-N13 112.1(2), C50-Cu3-N13 80.1(2), N11-Cu3-Cu4 131.09(16), C27-Cu3-Cu4 53.18(17), C50-Cu3-Cu4 112.70(16), N13-Cu3-Cu4 113.70(15), N11-Cu3-Cu2 128.00(15), C27-Cu3-Cu2 112.99(17), C50-Cu3-Cu2 52.76(16), N13-Cu3-Cu2 97.90(16), Cu4-Cu3-Cu2 60.05(3), N22-Cu4-C72 94.4(2), N22-Cu4-C27 92.5(2), C72-Cu4-C27 167.8(2), N22 -Cu4-N7 99.9(2), C72-Cu4-N7 107.5(2), C27-Cu4-N7 81.1(2), N22-Cu4-Cu3 134.20(17), C72-Cu4-Cu3 115.49(17), C27-Cu4-Cu3 53.10(17), N7-Cu4-Cu3 102.67(18), N22-Cu4-Cu2 134.48(17), C72-Cu4-Cu2 55.33(17), C27-Cu4-Cu2 113.02(17), N7-Cu4 -Cu2 120.11(16), Cu3- Cu4- Cu2 60.16(3).
Figure 4: ORTEP drawing of [Cu3(L4)3](PF6)3 (5). Thermal ellipsoids are drawn at the 30% probability level. H...
Complex 6 has also been characterized by single crystal X-ray diffraction (Figure 5). Complex 6 crystallizes in the hexagonal space group R3c, which is different to the reported trinuclear copper(I) complex containing the symmetric 1,3-bis(2-pyridinylmethyl)benzimidazolylidene ligand (monoclinic, P21/c) [33] and to the trinuclear copper(I) complex containing a symmetric 1,3-bis(triazole)benzimidazolylidene ligand (monoclinic, C2/c) [38]. Three copper atoms are bridged by three NpyridineCNtriazole NHC ligands forming a Cu3 ring with three Cu–Cu–Cu angles of 60.0. The geometry of the copper center can be described as distorted trigonal planar. Each copper ion is coordinated by one pyridine, one triazole, and two benzimidazolylidene ligands displaying a distorted tetrahedral geometry. The Cu–Cu distance is around 2.5145(12) Å showing a weak metal–metal interaction, which is similar with the reported triangle Cu(I) complexes and is shorter than in complexes 3 and 4. The Cu–N and Cu–C bond distances fall in the range of 2.092(5)–2.152(5) Å and 2.024(6)–2.092(6) Å, respectively, which are slightly longer than in dinuclear complexes 3 and 4. Benzimidazolylidene acts as a bridging ligand in a u2 mode and bonded equally to two Cu(I) ions, which is only observed in a few silver(I) and copper(I) complexes.
![[1860-5397-12-85-5]](/bjoc/content/figures/1860-5397-12-85-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: ORTEP drawing of [Cu3(L5)3](PF6)3 (6). Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms and anions have been removed for clarity. Selected bond distances (Å) and angles (°): Cu1-C26 2.092(6), Cu1-N5 2.092(5) , Cu1-C26A 2.024(6), Cu1- N9A 2. 152(5), Cu1-Cu1A 2.5141(11), Cu1-Cu1B 2.5141(11); C26-Cu1-N5 101.8(2), C26-Cu1-C26A 163.7(2), N5-Cu1-C26 92.5(2), C26A-Cu1-N9 92.0(2), Cu1-Cu1A-Cu1B 60.0. Symmetry transformations used to generate equivalent atoms: 1−x, 1−y, −z.
Figure 5: ORTEP drawing of [Cu3(L5)3](PF6)3 (6). Thermal ellipsoids are drawn at the 30% probability level. H...
Catalytic application in CuAAC reactions
Inspired by the catalytic activity of Cu(I) species supported by NHC ligand in Cu-catalyzed azide–alkyne cycloaddition (CuAAC) reaction under mild conditions, copper complexes 2–6 were investigated in the CuAAC reaction of azide and phenylacetylene. Firstly, we compared the catalytic activity of different complexes with a complex loading of 0.5 mol %. The reactions were monitored by 1H NMR analysis at different time points within 4 h (Figure 6). As seen in Figure 6, the yield increased with the extension of reaction time. The results showed that complex 4 displays the best activities for the CuAAC reaction of benzyl azide and phenylacetylene giving a conversion of 95%. To further examine the catalytic efficiency of complex 4, a variation of the catalyst loading from 0.1 to 0.25 to 0.5 mol % within 5 h was performed to give the expected product in yields of 17%, 48%, and 100%. As expected, the coupling reaction with low catalyst loading results in incomplete conversion.
![[1860-5397-12-85-6]](/bjoc/content/figures/1860-5397-12-85-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Yield vs reaction time of different copper complex. The reaction was carried out in acetonitrile-d3 at 25 °C using 0.5 mol % copper complex, yields were determined by 1H NMR spectra, hexamethylbenzene was used as internal standard.
Figure 6: Yield vs reaction time of different copper complex. The reaction was carried out in acetonitrile-d3...
Subsequently the catalytic activity of different solvents was tested at a Cu loading of 0.5 mol % (Table 1). Moderate catalytic activities were obtained for DMSO or without solvent. When CH3CN was used, the reaction gave an excellent yield (Table 1, entry 4). However, only a moderate yield was obtained when a CH3CN/H2O solvent mixture was used (Table 1, entry 6). Thus, CH3CN was selected as the optimal solvent.
Table 1: CuAAC reaction with different solventsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-85-i2.svg?max-width=637&scale=1.0)
|
|||
| entry | solvent | cat. | yield %b |
|---|---|---|---|
| 1 | neat | 4 | 50 |
| 2 | H2O | 4 | 22 |
| 3 | DMSO | 4 | 53 |
| 4 | CH3CN | 4 | 95 |
| 5 | t-BuOH/H2O (1:1) | 4 | trace |
| 6 | CH3CN/H2O (1:1) | 4 | 59 |
aReaction carried out using 0.5 mol % of complex 4 with different solvents. bYields were determined by 1H NMR spectra and are reported after 4 h, hexamethylbenzene was used as internal standard.
Having optimized the reaction conditions, we extended the CuAAC reaction to other azides and alkynes at room temperature in CH3CN. As shown in Table 2 (entries 1–5), (azidomethyl)benzene, azidobenzene, (2-azidoethyl)benzene, and 2-(azidomethyl)pyridine could react with phenylacetylene in more than 83% yield (Table 2, entries 1–4). What is more, methyl 1-benzyl-1H-1,2,3-triazole-4-carboxylate could be afforded in 85% yield via reacting methyl propiolate with (azidomethyl)benzene. This promising catalytic behavior of complex 4 prompted us to extend our studies toward a one-pot synthesis of 1,2,3-triazoles from alkyl halides, sodium azide, and alkynes. The three-component version has already been successfully performed and described in previous work [20]. As displayed in Table 2, the reactions proceeded smoothly to completion, and the products were isolated in good to excellent yields (83–95%).
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-85-i3.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-85-i4.svg?max-width=637&scale=1.0)
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-85-i5.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-12-85-i6.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-12-85-i7.svg?max-width=637&scale=1.0)
![[Graphic 7]](/bjoc/content/inline/1860-5397-12-85-i8.svg?max-width=637&scale=1.0)
![[Graphic 8]](/bjoc/content/inline/1860-5397-12-85-i9.svg?max-width=637&scale=1.0)
![[Graphic 9]](/bjoc/content/inline/1860-5397-12-85-i10.svg?max-width=637&scale=1.0)
![[Graphic 10]](/bjoc/content/inline/1860-5397-12-85-i11.svg?max-width=637&scale=1.0)
![[Graphic 11]](/bjoc/content/inline/1860-5397-12-85-i12.svg?max-width=637&scale=1.0)
![[Graphic 12]](/bjoc/content/inline/1860-5397-12-85-i13.svg?max-width=637&scale=1.0)
![[Graphic 13]](/bjoc/content/inline/1860-5397-12-85-i14.svg?max-width=637&scale=1.0)
![[Graphic 14]](/bjoc/content/inline/1860-5397-12-85-i15.svg?max-width=637&scale=1.0)
![[Graphic 15]](/bjoc/content/inline/1860-5397-12-85-i16.svg?max-width=637&scale=1.0)
![[Graphic 16]](/bjoc/content/inline/1860-5397-12-85-i17.svg?max-width=637&scale=1.0)
![[Graphic 17]](/bjoc/content/inline/1860-5397-12-85-i18.svg?max-width=637&scale=1.0)
![[Graphic 18]](/bjoc/content/inline/1860-5397-12-85-i19.svg?max-width=637&scale=1.0)
![[Graphic 19]](/bjoc/content/inline/1860-5397-12-85-i20.svg?max-width=637&scale=1.0)
![[Graphic 20]](/bjoc/content/inline/1860-5397-12-85-i21.svg?max-width=637&scale=1.0)
![[Graphic 21]](/bjoc/content/inline/1860-5397-12-85-i22.svg?max-width=637&scale=1.0)
![[Graphic 22]](/bjoc/content/inline/1860-5397-12-85-i23.svg?max-width=637&scale=1.0)
![[Graphic 23]](/bjoc/content/inline/1860-5397-12-85-i24.svg?max-width=637&scale=1.0)
![[Graphic 24]](/bjoc/content/inline/1860-5397-12-85-i25.svg?max-width=637&scale=1.0)
![[Graphic 25]](/bjoc/content/inline/1860-5397-12-85-i26.svg?max-width=637&scale=1.0)
Conclusion
In summary, a series of di-, and trinuclear copper(I) complexes (3–6) stablized by 1,2,3-triazole-tethered N-heterocyclic carbene ligands have been prepared via simple reactions of imidazolium salts with copper powder in good yields. These complexes have been fully characterized by NMR, elemental analysis (EA) and X-ray crystallography. Fine adjustment of the structure of the ligand can lead to different structures. All the Cu–NHC complexes showed high catalyst activity in CuAAC reactions at room temperature. Among these complexes, complex 4 is the most efficient catalyst in an air atmosphere at room temperature.
Experimental
All the chemicals were obtained from commercial suppliers and were used without further purification. Elemental analyses were performed on a Flash EA1112 instrument. 1H and 13C NMR spectra were recorded on a Bruker Avance-400 (400 MHz) spectrometer or a Varian 600 MHz NMR spectrometer. Chemical shifts (δ) are expressed in ppm downfield to TMS at δ = 0 ppm and coupling constants (J) are expressed in Hz.
Synthesis of 3-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)-1-(pyrimidin-2-yl)-1H-benzo[d]imidazol-3-ium hexafluorophosphate [(HL2)PF6] (1b): Analogously as described in a published work [27], (azidomethyl)benzene (160 mg, 1.2 mmol), copper sulfate pentahydrate (12.5 mg, 0.05 mmol), sodium ascorbate (20 mg, 0.1 mmol), and 3-(prop-2-yn-1-yl)-1-(pyrimidin-2-yl)-1H-benzo[d]imidazol-3-ium bromide (314 mg, 1 mmol) were added to a Schlenk tube containing 2 mL of water and tert-butyl alcohol (1:1). After the heterogeneous mixture was stirred vigorously for 24 h at 50 °C, the reaction mixture was diluted with water (20 mL). The obtained yellow solution was dropwise added to the aqueous solution of NH4PF6. A white precipitate was collected by filtration and dried. Yield: 315 mg, 61%. 1H NMR (400 MHz, DMSO-d6) δ 10.89 (s, 1H), 9.14 (d, J = 4.9 Hz, 2H), 8.83 (d, J = 8.1 Hz, 1H), 8.39 (s, 1H), 8.17 (d, J = 8.0 Hz, 1H), 7.79 (br, 3H), 7.34–7.30 (m, 5H), 6.06 (s, 2H), 5.63 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 160.34, 159.78, 143.41, 140.45, 136.12, 132.14, 129.62, 129.26, 128.69, 128.49, 127.79, 125.52, 53.47, 42.95.
Synthesis of 3-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)-1-(pyridin-2-yl)-1H-imidazol-3-ium hexafluorophosphate [(HL3)PF6] (1c): Similarly as described in a previous procedure [27], a mixture of (azidomethyl)benzene (160 mg, 1.2 mmol), copper sulfate pentahydrate (12.5 mg, 0.05 mmol) and sodium ascorbate (20 mg, 0.1 mmol), 3-(prop-2-ynyl)-1-(pyridin-2-yl)-1H-imidazol-3-ium bromide (265 mg, 1 mmol) was added to 2 mL of water and tert-butyl alcohol (1:1). The heterogeneous mixture was stirred vigorously for 24 h at 50 °C. The reaction mixture was diluted with water (20 mL), and the yellow solution was dropwise added to the aqueous solution of NH4PF6. A white precipitate was collected by filtration and dried. Yield: 415 mg, 90%. 1H NMR (400 MHz, DMSO-d6) δ 10.19 (s, 1H), 8.69–8.63 (m, 1H), 8.54 (t, J = 1.9 Hz, 1H), 8.35 (d, J = 2.4 Hz, 1H), 8.31–8.17 (m, 1H), 8.03 (q, J = 4.3, 3.5 Hz, 2H), 7.70–7.61 (m, 1H), 7.36 (qd, J = 7.0, 6.5, 2.5 Hz, 5H), 5.65 (dd, J = 5.1, 2.4 Hz, 4H), 3.39 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 149.69, 146.78, 141.06, 141.02, 135.68, 129.29, 128.76, 128.55, 125.77, 125.23, 124.14, 120.07, 114.75, 53.49, 44.85.
Synthesis of 3-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)-1-(pyridin-2-yl)-1H-benzo[d]imidazol-3-ium hexafluorophosphate [(HL4)PF6] (1d): Similarly as described in a previous procedure [27], the imidazolium salt was prepared similarly as for [(HL3)PF6] from (azidomethyl)benzene (160 mg, 1.2 mmol), copper sulfate pentahydrate (12.5 mg, 0.05 mmol), sodium ascorbate (20 mg, 0.1 mmol), and 3-(prop-2-yn-1-yl)-1-(pyridin-2-yl)-1H-benzo[d]imidazol-3-ium bromide (314 mg, 1 mmol). Yield: 317 mg, 62%. 1H NMR (400 MHz, DMSO-d6) δ 10.60 (s, 1H), 8.75 (d, J = 4.7 Hz, 1H), 8.46–8.31 (m, 2H), 8.25 (t, J = 8.0 Hz, 1H), 8.19–8.10 (m, 1H), 8.02 (d, J = 8.2 Hz, 1H), 7.79–7.65 (m, 3H), 7.31 (dt, J = 21.5, 7.4 Hz, 7H), 5.95 (s, 2H), 5.59 (d, J = 5.1 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ 149.97, 147.61, 143.07, 141.07, 140.46, 136.13, 131.72, 130.09, 129.27, 128.74, 128.50, 128.30, 127.73, 125.73, 125.50, 117.68, 116.38, 114.80, 53.42, 42.83.
Synthesis of 3-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)-1-(pyridin-2-ylmethyl)-1H-benzo[d]imidazol-3-ium hexafluorophosphate [(HL5)PF6] (1e): Similarly as described in previous procedure [27], the imidazolium salt was prepared similarly as for [(HL3)PF6] from (azidomethyl)benzene (160 mg, 1.2 mmol), and 3-(prop-2-yn-1-yl)-1-(pyridin-2-ylmethyl)-1H-benzo[d]imidazol-3-ium bromide (328 g, 1 mmol). Yield: 390 mg, 74%.1H NMR (400 MHz, DMSO-d6) δ 10.04 (s, 1H, NCHN), 8.46 (d, J = 4.8 Hz, 1H, 2-Py), 8.39 (s, 1H, triazole), 8.10 (d, J = 8.0 Hz, 1H, 4-Py), 7.94–7.90 (m, 3H), 7.73–7.59 (m, 3H), 7.46–7.27 (m, 6H, phenyl+benzene), 5.95 (s, 4H, CH2), 5.63 (s, 2H, CH2);13C NMR (101 MHz, DMSO-d6) δ 153.34, 150.04, 143.67, 140.07, 138.02, 136.14, 131.80, 131.29, 129.27, 128.74, 128.48, 127.33, 127.16, 125.33, 124.19, 123.20, 114.48, 114.41, 53.5, 51.4, 42.3.
General procedure for the preparation of Cu(I)–NHC complexes and Cu(II) complex: Analogously as described in [39], all the copper complexes were prepared by the following route: imidazolium salt (0.2 mmol) and an excess of copper powder (64 mg, 1.0 mmol) were placed in 3 mL of MeCN to form a heterogeneous mixture solution. After the mixture was stirred at 50 °C for 10 h under air, the solution was filtered through Celite. Single crystals suitable for X-ray diffraction analysis were grown from acetonitrile solution and diethyl ether.
Synthesis of [Cu-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)-3-(pyrimidin-2-yl)-1,3-dihydro-2H-imidazol-2-one)2](PF6)2 (2): This complex was synthesized by the reaction of [H(L1)](PF6) (1a; 93 mg, 0.2 mmol) with copper powder (64 mg, 1.0 mmol) at 50 °C for 10 h. Yield: 79 mg (75%), light green crystals. Anal. calcd for C34H30CuF12N14O2P2. 0.5 CH3CN: C, 40.39; H, 3.05; N, 19.52; found: C, 40.73; H, 2.95; N, 19.15.
Synthesis of [Cu2(L2)2](PF6)2 (3): This complex was synthesized by the reaction of [HL2](PF6) (1b; 102 mg, 0.2 mmol) with copper powder (64 mg, 1.0 mmol) at 50 oC for 10 h. Yield: 66 mg (57%), red crystals. 1H NMR (600 MHz, acetonitrile-d3) δ 8.75 (d, J = 8.1 Hz, 2H, benzimidazole), 8.70 (d, J = 4.9 Hz, 4H, pyrimidine), 7.88 (s, 2H, triazole), 7.79 (d, J = 7.8 Hz, 2H, benzimidazole), 7.59–7.54 (m, 2H, benzimidazole), 7.52 (t, J = 7.2 Hz, 2H, benzimidazole), 7.36 (t, J = 4.9 Hz, 2H, pyrimidine), 7.31–7.29 (m, 6H, phenyl), 7.19–7.18 (m, 4H, phenyl), 5.61 (s, 4H, -CH2-), 5.38 (s, 4H, -CH2-); 13C NMR (151 MHz, acetonitrile-d3) δ 191.23 (Cu-C), 158.73, 157.33 142.39, 136.17 135.75, 132.61, 129.94, 129.67, 129.17, 126.34, 126.19, 125.58, 120.66, 116.95, 112.75, 54.12, 43.41; Anal. calcd for C42H34Cu2F12N14P2: C, 43.80; H, 2.98; N, 17.02; found: C, 43.51; H, 2.85; N, 16.95.
Synthesis of [Cu2(L3)2](PF6)2 (4): The compound was prepared similarly as for complex 3 from [HL3](PF6) (90 mg, 0.20 mmol) with copper powder (64 mg, 1.0 mmol) at 50 °C for 10 h, orange yellow solid. Yield: 71 mg, 68%. 1H NMR (600 MHz, acetonitrile-d3) δ 7.96 (s, 1H, triazole), 7.90 (br, 1H, 2-py), 7.83 (br, 1H, imidazole), 7.76 (s, 1H, 4-py), 7.57 (br, 1H, imidazole), 7.41 (s, 1H, 5-py), 7.37 (d, J = 7.8 Hz, 3H, phenyl), 7.30–7.22 (m, 3H, phenyl + 3-py), 5.47 (s, 2H), 5.38 (s, 2H); 13C NMR (151 MHz, acetonitrile-d3) δ 181.20 (Cu-C), 149.68, 147.26, 140.72, 138.41, 134.71, 129.02, 128.89, 128.80, 128.28, 128.04, 123.96, 123.48, 112.13, 54.36, 45.69; Anal. calcd for C36H32Cu2F12N12P2: C, 41.19; H, 3.07; N, 16.01; found: C, 41.25; H, 3.31; N, 15.46.
Synthesis of [Cu3(L4)3](PF6)3 (5): The compound was prepared similarly as for complex 3 from [HL4](PF6) (106 mg, 0.20 mmol) with copper powder (64 mg, 1.0 mmol) at 50 °C for 10 h, light yellow solid. Yield: 93 mg, 52%. 1H NMR (600 MHz, acetone-d6) δ 8.77 (d, J = 8.1 Hz, 1H, 2-py), 8.46 (td, J = 8.0, 1.8 Hz, 1H, 4-py), 8.33 (s, 1H, triazole), 8.23–8.20 (m, 1H), 7.90 (dd, J = 5.1, 1.5 Hz, 1H, benzimidazole), 7.84–7.81 (m, 1H, benzimidazole), 7.68–7.60 (m, 2H, benzimidazole), 7.44 (dd, J = 7.6, 5.0 Hz, 1H, 5-py), 7.33–7.31 (m, 3H, phenyl), 6.94–6.92 (m, 2H, phenyl), 5.84 (d, J = 15.8 Hz, 1H), 5.37 (d, J = 15.8 Hz, 1H), 5.18 (s, 2H); 13C NMR (151 MHz, DMSO-d6) δ 178.99 (Cu-C), 149.61, 148.44, 142.25, 140.55, 136.19, 134.91, 133.34, 129.31, 128.93, 128.09, 126.05, 125.82, 124.48, 124.27, 119.09, 118.51 (CH3CN), 112.74, 111.59, 54.13, 41.17, 1.56(CH3CN); Anal. calcd for C132H108Cu6F36N36P6. 3CH3CN: C, 46.39; H, 3.30; N, 15.29; found: C, 45.87; H, 3.40; N, 15.30.
Synthesis of [Cu3(L5)3](PF6)3 (6): The compound was prepared similarly as for complex 3 from [HL5](PF6) (106 mg, 0.20 mmol) with copper powder (64 mg, 1.0 mmol) at 50 °C for 10 h, light yellow solid. Yield: 106 mg, 90%. 1H NMR (600 MHz, nitromethane-d3) δ 8.13 (s, 1H, triazole), 7.92 (td, J = 7.8, 1.8 Hz, 1H, pyridine), 7.77 (d, J = 7.8 Hz, 1H, pyridine), 7.59 (d, J = 7.8, 1H, pyridine), 7.61–7.36 (m, 6H, phenyl + benzimidazole), 7.14–7.08 (m, 2H, benzimidazole), 6.95 (ddd, J = 7.5, 5.2, 1.1 Hz, 1H, pyridine), 6.49–6.45 (d, J = 4.8 Hz,1H, benzimidazole), 5.40 (d, J = 15.0 Hz, 1H, -CH2-), 5.30 (d, J = 15.0 Hz, 1H, -CH2-), 5.27 (d, J = 15.0 Hz, 1H, -CH2-), 5.26 (d, J = 15.6 Hz, 1H, -CH2-), 5.21 (d, J = 15.0 Hz, 1H, -CH2-), 4.98 (d, J = 15.6 Hz, 1H, -CH2-); 13C NMR (150 MHz, nitromethane-d3) 177.57 (Cu-C), 151.99, 148.85, 141.78, 139.93, 135.15, 134.68, 133.85, 129.16, 128.98, 128.81, 128.12, 125.04, 124.62, 124.35, 123.79, 110.58, 110.26, 54.49, 51.29, 40.72; Anal. calcd for C69H60Cu3F18N18P3: C, 46.90; H, 3.42; Cu, 10.79; N, 14.27; found: C, 46.35; H, 3.31; N, 13.95.
General procedure for the copper-catalyzed CuAAC reaction: Analogously as described in [31], in a 10 mL Schlenk tube, azide (0.5 mmol), alkyne (0.6 mmol), and 0.5 mol % copper complex were dissolved in 3.0 mL of CH3CN. After the mixture was stirred at rt under air for a desired time, the reaction was stopped by the addition of H2O (2 mL) to the resultant mixture. Then the mixture was extracted with CH2Cl2. The organic layer was separated from the aqueous phase. After the organic phase was dried over MgSO4, the solution was filtered and concentrated under vacuum. The residue was purified by flash chromatography (silica gel, petroleum ether/ethyl acetate, 3:1) to give the desired product
X-ray diffraction analysis
Analogously as described in [27], single-crystal X-ray diffraction data were collected at 298(2) K on a Siemens Smart/CCD area-detector or Oxford Diffraction Gemini A Ultra diffractometer with a Mo Kα radiation (λ = 0.71073 Å) by using an ω-2θ scan mode. Unit-cell dimensions were obtained with least-squares refinement. Data collection and reduction were performed using the SMART and SAINT software [46]. The structures were solved by direct methods, and the non-hydrogen atoms were subjected to anisotropic refinement by full-matrix least squares on F2 using the SHELXTXL package [47]. Hydrogen atom positions for all of the structures were calculated and allowed to ride on their respective C atoms with the C–H distances of 0.93–0.97 Å and Uiso(H) = 1.2 − 1.5Ueq(C). Disordered solvent molecules that could not be modeled successfully were removed with SQUEEZE [48]. Further details of the structural analysis are summarized in Table 3.
Table 3: Crystallographic data for complexes 2–6.
| 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|
| CCDC number | 1424013 | 1424014 | 1424015 | 1424016 | 1424017 |
| formula | C70H63Cu2F24N29O4P4 | C42H34Cu2F12N14P2 | C36H32Cu2F12N12P2 | C278H237Cu12F72N79P12 | C69H60Cu3F18N18P3 |
| Fw. | 2081.47 | 1161.93 | 1049.76 | 7186.59 | 1766.88 |
| crystal system | orthorhombic | monoclinic | triclinic | hexagonal | hexagonal |
| space group | Pnna | C2/c | P-1 | R3c | R3c |
| a/Å | 14.0847(15) | 34.298(3) | 12.9626(10) | 28.178(3) | 21.4692(13) |
| b/Å | 14.5426(16) | 13.2602(12) | 13.0382(9) | 28.178(3) | 21.4692(13) |
| c/Å | 22.599(2) | 26.605(4) | 13.1663(10) | 16.3997(19) | 71.858(9) |
| β/deg | 90.00 | 129.8310(10) | 80.661(6) | 90.00 | 90.00 |
| V/Å3 | 4629.0(9) | 9291.9(19) | 2111.7(3) | 45655(10) | 28684(4) |
| Z | 2 | 8 | 2 | 6 | 12 |
| D/g cm−3 | 1.493 | 1.661 | 1.651 | 1.568 | 1.228 |
| Reflns collected | 18121 | 10632 | 7571 | 21009 | 5624 |
| ind reflns, Rint | 12510, 0.0545 | 7828, 0.0250 | 4679, 0.0467 | 15722, 0.0869 | 3716, 0.0650 |
| goodness-of-fit on F2 | 1.063 | 1.018 | 1.023 | 1.031 | 1.157 |
| R1, wR2 [I > 2σ(I)] | 0.0869, 0.2496 | 0.0562, 0.1652 | 0.0696, 0.1774 | 0.0622, 0.1353 | 0.0688, 0.2044 |
| R1, wR2 (all data) | 0.1338, 0.3001 | 0.0779, 0.1858 | 0.1122, 0.2172 | 0.0917, 0.1543 | 0.1200, 0.2623 |
Supporting Information
| Supporting Information File 1: X-ray crystallographic data in cif format CCDC 1424013–1424017. | ||
| Format: CIF | Size: 146.7 KB | Download |
References
-
Díez-González, S.; Marion, N.; Nolan, S. P. Chem. Rev. 2009, 109, 3612–3676. doi:10.1021/Cr900074m
Return to citation in text: [1] -
Poyatos, M.; Mata, J. A.; Peris, E. Chem. Rev. 2009, 109, 3677–3707. doi:10.1021/Cr800501s
Return to citation in text: [1] -
Bernhammer, J. C.; Han Vinh, H. Organometallics 2014, 33, 1266–1275. doi:10.1021/om500083r
Return to citation in text: [1] -
Farrell, K.; Albrecht, M. Late Transition Metal Complexes with Pincer Ligands that Comprise N-Heterocyclic Carbene Donor Sites. In The Privileged Pincer-Metal Platform: Coordination Chemistry & Applications; van Koten, G.; Gossage, R. A., Eds.; Springer: Berlin, Germany, 2016; pp 45–91.
Return to citation in text: [1] -
Velazquez, H. D.; Verpoort, F. Chem. Soc. Rev. 2012, 41, 7032–7060. doi:10.1039/c2cs35102a
Return to citation in text: [1] -
Gaillard, S.; Cazin, C. S. J.; Nolan, S. P. Acc. Chem. Res. 2012, 45, 778–787. doi:10.1021/ar200188f
Return to citation in text: [1] -
Lazreg, F.; Nahra, F.; Cazin, C. S. J. Coord. Chem. Rev. 2015, 293, 48–79. doi:10.1016/j.ccr.2014.12.019
Return to citation in text: [1] -
Prakasham, A. P.; Ghosh, P. Inorg. Chim. Acta 2015, 431, 61–100. doi:10.1016/j.ica.2014.11.005
Return to citation in text: [1] -
Mata, J. A.; Hahn, F. E.; Peris, E. Chem. Sci. 2014, 5, 1723–1732. doi:10.1039/c3sc53126k
Return to citation in text: [1] -
Fortman, G. C.; Nolan, S. P. Chem. Soc. Rev. 2011, 40, 5151–5169. doi:10.1039/c1cs15088j
Return to citation in text: [1] -
Mejuto, C.; Royo, B.; Guisado-Barrios, G.; Peris, E. Beilstein J. Org. Chem. 2015, 11, 2584–2590. doi:10.3762/bjoc.11.278
Return to citation in text: [1] -
Schulte to Brinke, C.; Hahn, F. E. Dalton Trans. 2015, 44, 14315–14322. doi:10.1039/c5dt02115d
Return to citation in text: [1] -
Ibrahim, H.; Bala, M. D. J. Organomet. Chem. 2015, 794, 301–310. doi:10.1016/j.jorganchem.2015.07.015
Return to citation in text: [1] -
Chen, C.; Lu, C.; Zheng, Q.; Ni, S.; Zhang, M.; Chen, W. Beilstein J. Org. Chem. 2015, 11, 1786–1795. doi:10.3762/bjoc.11.194
Return to citation in text: [1] -
Liu, B.; Liu, B.; Zhou, Y.; Chen, W. Organometallics 2010, 29, 1457–1464. doi:10.1021/om100009u
Return to citation in text: [1] -
Guo, T.; Dechert, S.; Meyer, F. Organometallics 2014, 33, 5145–5155. doi:10.1021/om500351j
Return to citation in text: [1] -
Zhang, X.; Xi, Z.; Liu, A.; Chen, W. Organometallics 2008, 27, 4401–4406. doi:10.1021/om8003674
Return to citation in text: [1] -
Liu, X.; Chen, W. Organometallics 2012, 31, 6614–6622. doi:10.1021/om300644h
Return to citation in text: [1] -
Gu, S.; Liu, B.; Chen, J.; Wu, H.; Chen, W. Dalton Trans. 2012, 41, 962–970. doi:10.1039/c1dt11269d
Return to citation in text: [1] -
Gu, S.; Xu, D.; Chen, W. Dalton Trans. 2011, 40, 1576–1583. doi:10.1039/c0dt01211d
Return to citation in text: [1] [2] [3] -
Crowley, J.; McMorran, D. “Click-Triazole” Coordination Chemistry: Exploiting 1,4-Disubstituted-1,2,3-Triazoles as Ligands. In Click Triazoles; Košmrlj, J., Ed.; Springer: Berlin, Germany, 2012; pp 31–83.
Return to citation in text: [1] -
Sluijter, S. N.; Elsevier, C. J. Organometallics 2014, 33, 6389–6397. doi:10.1021/om5007038
Return to citation in text: [1] [2] -
Warsink, S.; Drost, R. M.; Lutz, M.; Spek, A. L.; Elsevier, C. J. Organometallics 2010, 29, 3109–3116. doi:10.1021/om100435x
Return to citation in text: [1] [2] [3] -
Huang, D.; Zhao, P.; Astruc, D. Coord. Chem. Rev. 2014, 272, 145–165. doi:10.1016/j.ccr.2014.04.006
Return to citation in text: [1] -
Zamora, M. T.; Ferguson, M. J.; McDonald, R.; Cowie, M. Organometallics 2012, 31, 5463–5477.
Return to citation in text: [1] -
Vuong, K. Q.; Timerbulatova, M. G.; Peterson, M. B.; Bhadbhade, M.; Messerle, B. A. Dalton Trans. 2013, 42, 14298–14308. doi:10.1039/C3DT51440D
Return to citation in text: [1] [2] -
Gu, S.; Xu, H.; Zhang, N.; Chen, W. Chem. – Asian J. 2010, 5, 1677–1686. doi:10.1002/asia.201000071
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Lu, C.; Gu, S.; Liu, X. Inorg. Chem. Commun. 2014, 47, 45–47. doi:10.1016/j.inoche.2014.07.004
Return to citation in text: [1] -
Gu, S.; Xu, W.; Huang, J. Prog. Chem. 2013, 25, 330–339. doi:10.7536/PC120746
Return to citation in text: [1] -
Gu, S.; Huang, J.; Chen, W. Chin. J. Org. Chem. 2013, 33, 715–737. doi:10.6023/cjoc201301075
Return to citation in text: [1] -
Gu, S.; Huang, J.; Liu, X.; Liu, H.; Zhou, Y.; Xu, W. Inorg. Chem. Commun. 2012, 21, 168–172. doi:10.1016/j.inoche.2012.05.007
Return to citation in text: [1] [2] -
Liu, B.; Zhang, Y.; Xu, D.; Chen, W. Chem. Commun. 2011, 47, 2883–2885. doi:10.1039/c0cc05260d
Return to citation in text: [1] -
Chen, C.; Qiu, H.; Chen, W. J. Organomet. Chem. 2012, 696, 4166–4172. doi:10.1016/j.jorganchem.2011.09.008
Return to citation in text: [1] [2] [3] [4] -
Lin, J. C. Y.; Huang, R. T. W.; Lee, C. S.; Bhattacharyya, A.; Hwang, W. S.; Lin, I. J. B. Chem. Rev. 2009, 109, 3561–3598. doi:10.1021/cr8005153
Return to citation in text: [1] -
Charra, V.; de Frémont, P.; Breuil, P.-A. R.; Olivier-Bourbigou, H.; Braunstein, P. J. Organomet. Chem. 2015, 795, 25–33. doi:10.1016/j.jorganchem.2015.01.025
Return to citation in text: [1] -
Liu, B.; Pan, S.; Liu, B.; Chen, W. Inorg. Chem. 2014, 53, 10485–10497. doi:10.1021/ic501544d
Return to citation in text: [1] -
Collins, L. R.; Lowe, J. P.; Mahon, M. F.; Poulten, R. C.; Whittlesey, M. K. Inorg. Chem. 2014, 53, 2699–2707. doi:10.1021/ic4031014
Return to citation in text: [1] -
Liu, B.; Ma, X.; Wu, F.; Chen, W. Dalton Trans. 2015, 44, 1836–1844. doi:10.1039/C4DT02986K
Return to citation in text: [1] [2] -
Liu, B.; Chen, C.; Zhang, Y.; Liu, X.; Chen, W. Organometallics 2013, 32, 5451–5460. doi:10.1021/om400738c
Return to citation in text: [1] [2] -
Pouy, M. J.; Delp, S. A.; Uddin, J.; Ramdeen, V. M.; Cochrane, N. A.; Fortman, G. C.; Gunnoe, T. B.; Cundari, T. R.; Sabat, M.; Myers, W. H. ACS Catal. 2012, 2, 2182–2193. doi:10.1021/cs300544w
Return to citation in text: [1] -
Catalano, V. J.; Munro, L. B.; Strasser, C. E.; Samin, A. F. Inorg. Chem. 2011, 50, 8465–8476. doi:10.1021/ic201053t
Return to citation in text: [1] -
Appukkuttan, P.; Dehaen, W.; Fokin, V. V.; Van der Eycken, E. Org. Lett. 2004, 6, 4223–4225. doi:10.1021/ol048341v
Return to citation in text: [1] [2] [3] -
Urankar, D.; Pevec, A.; Turel, I.; Košmrlj, J. Cryst. Growth Des. 2010, 10, 4920–4927. doi:10.1021/cg100993k
Return to citation in text: [1] -
Campbell-Verduyn, L. S.; Mirfeizi, L.; Dierckx, R. A.; Elsinga, P. H.; Feringa, B. L. Chem. Commun. 2009, 2139–2141. doi:10.1039/B822994E
Return to citation in text: [1] [2] [3] -
Chassaing, S.; Sani Souna Sido, A.; Alix, A.; Kumarraja, M.; Pale, P.; Sommer, J. Chem. – Eur. J. 2008, 14, 6713–6721. doi:10.1002/chem.200800479
Return to citation in text: [1] -
SMART-CCD Software, Version 4.05; Siemens Analytical X-ray Instruments: Madison, WI, U.S.A., 1996.
Return to citation in text: [1] -
SHELXS-97 and SHELXL-97, Program for X-ray Crystal Structure Refinement; G. M. Sheldrick: University of Göttingen, Germany, 1997.
Return to citation in text: [1] -
Spek, A. L. PLATON, A Multipurpose Crystallographic Tool; University of Utrecht: Netherlands, 1998.
Return to citation in text: [1]
| 44. | Campbell-Verduyn, L. S.; Mirfeizi, L.; Dierckx, R. A.; Elsinga, P. H.; Feringa, B. L. Chem. Commun. 2009, 2139–2141. doi:10.1039/B822994E |
| 42. | Appukkuttan, P.; Dehaen, W.; Fokin, V. V.; Van der Eycken, E. Org. Lett. 2004, 6, 4223–4225. doi:10.1021/ol048341v |
| 44. | Campbell-Verduyn, L. S.; Mirfeizi, L.; Dierckx, R. A.; Elsinga, P. H.; Feringa, B. L. Chem. Commun. 2009, 2139–2141. doi:10.1039/B822994E |
| 1. | Díez-González, S.; Marion, N.; Nolan, S. P. Chem. Rev. 2009, 109, 3612–3676. doi:10.1021/Cr900074m |
| 2. | Poyatos, M.; Mata, J. A.; Peris, E. Chem. Rev. 2009, 109, 3677–3707. doi:10.1021/Cr800501s |
| 3. | Bernhammer, J. C.; Han Vinh, H. Organometallics 2014, 33, 1266–1275. doi:10.1021/om500083r |
| 4. | Farrell, K.; Albrecht, M. Late Transition Metal Complexes with Pincer Ligands that Comprise N-Heterocyclic Carbene Donor Sites. In The Privileged Pincer-Metal Platform: Coordination Chemistry & Applications; van Koten, G.; Gossage, R. A., Eds.; Springer: Berlin, Germany, 2016; pp 45–91. |
| 5. | Velazquez, H. D.; Verpoort, F. Chem. Soc. Rev. 2012, 41, 7032–7060. doi:10.1039/c2cs35102a |
| 6. | Gaillard, S.; Cazin, C. S. J.; Nolan, S. P. Acc. Chem. Res. 2012, 45, 778–787. doi:10.1021/ar200188f |
| 7. | Lazreg, F.; Nahra, F.; Cazin, C. S. J. Coord. Chem. Rev. 2015, 293, 48–79. doi:10.1016/j.ccr.2014.12.019 |
| 8. | Prakasham, A. P.; Ghosh, P. Inorg. Chim. Acta 2015, 431, 61–100. doi:10.1016/j.ica.2014.11.005 |
| 9. | Mata, J. A.; Hahn, F. E.; Peris, E. Chem. Sci. 2014, 5, 1723–1732. doi:10.1039/c3sc53126k |
| 10. | Fortman, G. C.; Nolan, S. P. Chem. Soc. Rev. 2011, 40, 5151–5169. doi:10.1039/c1cs15088j |
| 11. | Mejuto, C.; Royo, B.; Guisado-Barrios, G.; Peris, E. Beilstein J. Org. Chem. 2015, 11, 2584–2590. doi:10.3762/bjoc.11.278 |
| 12. | Schulte to Brinke, C.; Hahn, F. E. Dalton Trans. 2015, 44, 14315–14322. doi:10.1039/c5dt02115d |
| 17. | Zhang, X.; Xi, Z.; Liu, A.; Chen, W. Organometallics 2008, 27, 4401–4406. doi:10.1021/om8003674 |
| 27. | Gu, S.; Xu, H.; Zhang, N.; Chen, W. Chem. – Asian J. 2010, 5, 1677–1686. doi:10.1002/asia.201000071 |
| 27. | Gu, S.; Xu, H.; Zhang, N.; Chen, W. Chem. – Asian J. 2010, 5, 1677–1686. doi:10.1002/asia.201000071 |
| 15. | Liu, B.; Liu, B.; Zhou, Y.; Chen, W. Organometallics 2010, 29, 1457–1464. doi:10.1021/om100009u |
| 16. | Guo, T.; Dechert, S.; Meyer, F. Organometallics 2014, 33, 5145–5155. doi:10.1021/om500351j |
| 32. | Liu, B.; Zhang, Y.; Xu, D.; Chen, W. Chem. Commun. 2011, 47, 2883–2885. doi:10.1039/c0cc05260d |
| 39. | Liu, B.; Chen, C.; Zhang, Y.; Liu, X.; Chen, W. Organometallics 2013, 32, 5451–5460. doi:10.1021/om400738c |
| 14. | Chen, C.; Lu, C.; Zheng, Q.; Ni, S.; Zhang, M.; Chen, W. Beilstein J. Org. Chem. 2015, 11, 1786–1795. doi:10.3762/bjoc.11.194 |
| 27. | Gu, S.; Xu, H.; Zhang, N.; Chen, W. Chem. – Asian J. 2010, 5, 1677–1686. doi:10.1002/asia.201000071 |
| 27. | Gu, S.; Xu, H.; Zhang, N.; Chen, W. Chem. – Asian J. 2010, 5, 1677–1686. doi:10.1002/asia.201000071 |
| 13. | Ibrahim, H.; Bala, M. D. J. Organomet. Chem. 2015, 794, 301–310. doi:10.1016/j.jorganchem.2015.07.015 |
| 20. | Gu, S.; Xu, D.; Chen, W. Dalton Trans. 2011, 40, 1576–1583. doi:10.1039/c0dt01211d |
| 28. | Lu, C.; Gu, S.; Liu, X. Inorg. Chem. Commun. 2014, 47, 45–47. doi:10.1016/j.inoche.2014.07.004 |
| 29. | Gu, S.; Xu, W.; Huang, J. Prog. Chem. 2013, 25, 330–339. doi:10.7536/PC120746 |
| 30. | Gu, S.; Huang, J.; Chen, W. Chin. J. Org. Chem. 2013, 33, 715–737. doi:10.6023/cjoc201301075 |
| 31. | Gu, S.; Huang, J.; Liu, X.; Liu, H.; Zhou, Y.; Xu, W. Inorg. Chem. Commun. 2012, 21, 168–172. doi:10.1016/j.inoche.2012.05.007 |
| 27. | Gu, S.; Xu, H.; Zhang, N.; Chen, W. Chem. – Asian J. 2010, 5, 1677–1686. doi:10.1002/asia.201000071 |
| 22. | Sluijter, S. N.; Elsevier, C. J. Organometallics 2014, 33, 6389–6397. doi:10.1021/om5007038 |
| 23. | Warsink, S.; Drost, R. M.; Lutz, M.; Spek, A. L.; Elsevier, C. J. Organometallics 2010, 29, 3109–3116. doi:10.1021/om100435x |
| 24. | Huang, D.; Zhao, P.; Astruc, D. Coord. Chem. Rev. 2014, 272, 145–165. doi:10.1016/j.ccr.2014.04.006 |
| 25. | Zamora, M. T.; Ferguson, M. J.; McDonald, R.; Cowie, M. Organometallics 2012, 31, 5463–5477. |
| 26. | Vuong, K. Q.; Timerbulatova, M. G.; Peterson, M. B.; Bhadbhade, M.; Messerle, B. A. Dalton Trans. 2013, 42, 14298–14308. doi:10.1039/C3DT51440D |
| 23. | Warsink, S.; Drost, R. M.; Lutz, M.; Spek, A. L.; Elsevier, C. J. Organometallics 2010, 29, 3109–3116. doi:10.1021/om100435x |
| 44. | Campbell-Verduyn, L. S.; Mirfeizi, L.; Dierckx, R. A.; Elsinga, P. H.; Feringa, B. L. Chem. Commun. 2009, 2139–2141. doi:10.1039/B822994E |
| 21. | Crowley, J.; McMorran, D. “Click-Triazole” Coordination Chemistry: Exploiting 1,4-Disubstituted-1,2,3-Triazoles as Ligands. In Click Triazoles; Košmrlj, J., Ed.; Springer: Berlin, Germany, 2012; pp 31–83. |
| 26. | Vuong, K. Q.; Timerbulatova, M. G.; Peterson, M. B.; Bhadbhade, M.; Messerle, B. A. Dalton Trans. 2013, 42, 14298–14308. doi:10.1039/C3DT51440D |
| 27. | Gu, S.; Xu, H.; Zhang, N.; Chen, W. Chem. – Asian J. 2010, 5, 1677–1686. doi:10.1002/asia.201000071 |
| 19. | Gu, S.; Liu, B.; Chen, J.; Wu, H.; Chen, W. Dalton Trans. 2012, 41, 962–970. doi:10.1039/c1dt11269d |
| 20. | Gu, S.; Xu, D.; Chen, W. Dalton Trans. 2011, 40, 1576–1583. doi:10.1039/c0dt01211d |
| 42. | Appukkuttan, P.; Dehaen, W.; Fokin, V. V.; Van der Eycken, E. Org. Lett. 2004, 6, 4223–4225. doi:10.1021/ol048341v |
| 22. | Sluijter, S. N.; Elsevier, C. J. Organometallics 2014, 33, 6389–6397. doi:10.1021/om5007038 |
| 23. | Warsink, S.; Drost, R. M.; Lutz, M.; Spek, A. L.; Elsevier, C. J. Organometallics 2010, 29, 3109–3116. doi:10.1021/om100435x |
| 45. | Chassaing, S.; Sani Souna Sido, A.; Alix, A.; Kumarraja, M.; Pale, P.; Sommer, J. Chem. – Eur. J. 2008, 14, 6713–6721. doi:10.1002/chem.200800479 |
| 36. | Liu, B.; Pan, S.; Liu, B.; Chen, W. Inorg. Chem. 2014, 53, 10485–10497. doi:10.1021/ic501544d |
| 37. | Collins, L. R.; Lowe, J. P.; Mahon, M. F.; Poulten, R. C.; Whittlesey, M. K. Inorg. Chem. 2014, 53, 2699–2707. doi:10.1021/ic4031014 |
| 38. | Liu, B.; Ma, X.; Wu, F.; Chen, W. Dalton Trans. 2015, 44, 1836–1844. doi:10.1039/C4DT02986K |
| 39. | Liu, B.; Chen, C.; Zhang, Y.; Liu, X.; Chen, W. Organometallics 2013, 32, 5451–5460. doi:10.1021/om400738c |
| 33. | Chen, C.; Qiu, H.; Chen, W. J. Organomet. Chem. 2012, 696, 4166–4172. doi:10.1016/j.jorganchem.2011.09.008 |
| 31. | Gu, S.; Huang, J.; Liu, X.; Liu, H.; Zhou, Y.; Xu, W. Inorg. Chem. Commun. 2012, 21, 168–172. doi:10.1016/j.inoche.2012.05.007 |
| 34. | Lin, J. C. Y.; Huang, R. T. W.; Lee, C. S.; Bhattacharyya, A.; Hwang, W. S.; Lin, I. J. B. Chem. Rev. 2009, 109, 3561–3598. doi:10.1021/cr8005153 |
| 35. | Charra, V.; de Frémont, P.; Breuil, P.-A. R.; Olivier-Bourbigou, H.; Braunstein, P. J. Organomet. Chem. 2015, 795, 25–33. doi:10.1016/j.jorganchem.2015.01.025 |
| 27. | Gu, S.; Xu, H.; Zhang, N.; Chen, W. Chem. – Asian J. 2010, 5, 1677–1686. doi:10.1002/asia.201000071 |
| 46. | SMART-CCD Software, Version 4.05; Siemens Analytical X-ray Instruments: Madison, WI, U.S.A., 1996. |
| 42. | Appukkuttan, P.; Dehaen, W.; Fokin, V. V.; Van der Eycken, E. Org. Lett. 2004, 6, 4223–4225. doi:10.1021/ol048341v |
| 43. | Urankar, D.; Pevec, A.; Turel, I.; Košmrlj, J. Cryst. Growth Des. 2010, 10, 4920–4927. doi:10.1021/cg100993k |
| 38. | Liu, B.; Ma, X.; Wu, F.; Chen, W. Dalton Trans. 2015, 44, 1836–1844. doi:10.1039/C4DT02986K |
| 20. | Gu, S.; Xu, D.; Chen, W. Dalton Trans. 2011, 40, 1576–1583. doi:10.1039/c0dt01211d |
| 33. | Chen, C.; Qiu, H.; Chen, W. J. Organomet. Chem. 2012, 696, 4166–4172. doi:10.1016/j.jorganchem.2011.09.008 |
| 41. | Catalano, V. J.; Munro, L. B.; Strasser, C. E.; Samin, A. F. Inorg. Chem. 2011, 50, 8465–8476. doi:10.1021/ic201053t |
| 33. | Chen, C.; Qiu, H.; Chen, W. J. Organomet. Chem. 2012, 696, 4166–4172. doi:10.1016/j.jorganchem.2011.09.008 |
| 40. | Pouy, M. J.; Delp, S. A.; Uddin, J.; Ramdeen, V. M.; Cochrane, N. A.; Fortman, G. C.; Gunnoe, T. B.; Cundari, T. R.; Sabat, M.; Myers, W. H. ACS Catal. 2012, 2, 2182–2193. doi:10.1021/cs300544w |
| 47. | SHELXS-97 and SHELXL-97, Program for X-ray Crystal Structure Refinement; G. M. Sheldrick: University of Göttingen, Germany, 1997. |
| 33. | Chen, C.; Qiu, H.; Chen, W. J. Organomet. Chem. 2012, 696, 4166–4172. doi:10.1016/j.jorganchem.2011.09.008 |
| 48. | Spek, A. L. PLATON, A Multipurpose Crystallographic Tool; University of Utrecht: Netherlands, 1998. |
© 2016 Gu et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)