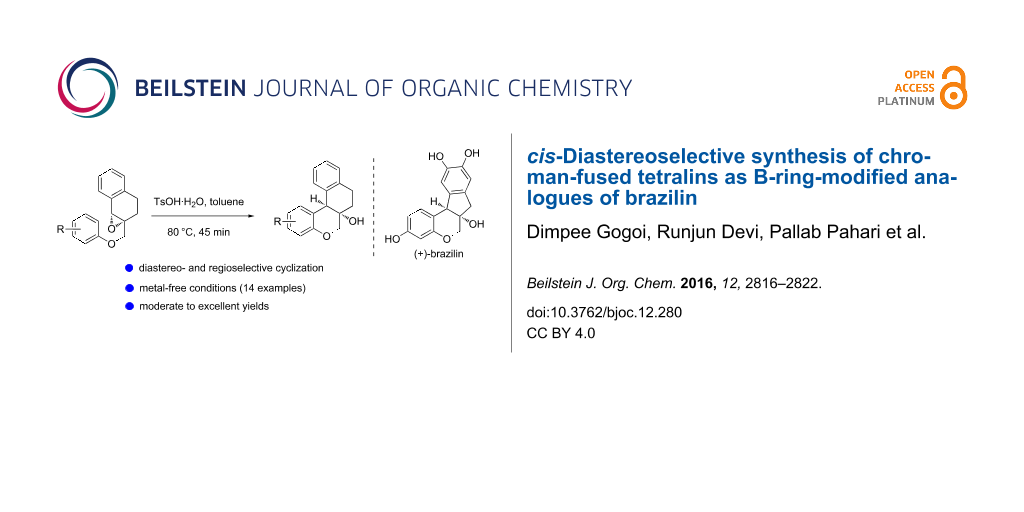
Abstract
We have synthesized a series of cis-6a,7,8,12b-tetrahydro-6H-naphtho[2,1-c]chromen-6a-ols as B-ring-modified analogues of (±)-brazilin. A completely regio- and cis-diastereoselective intramolecular Friedel–Crafts epoxy–arene cyclization of 1-tetralone-derived glycidyl ethers catalyzed by Brønsted acids was used as the key step. Our worries concerning the formation of cis–trans product mixtures and their probable conversion to naphthopyran derivatives via dehydration of the tertiary hydroxy group were laid to rest. Additionally, the angular hydroxy group of one of the synthesized products has been reductively removed by a diastereoselective method which should be useful in future for preparing libraries of chroman-fused tetralins with trans-stereochemistry at the ring junction.

Graphical Abstract
Findings
The chroman unit occurs widely as a privileged framework in a large number of natural products (NPs), natural product-like molecules (NPLMs) and pharmaceuticals, possessing diverse biological activities [1]. (+)-Brazilin (1) and (−)-haematoxylin (2) are two structurally-related tetracyclic homoisoflavonoid (containing a chroman unit) natural products (Figure 1) [2-4]. Their synthetic des-(angular)hydroxy derivatives (+)-brazilane (3) and haematoxylane (4) have also been mentioned in the literature [2-4]. Among them, brazilin (1), the most-studied member of this group, has been proved to have a number of potentially important biological activities including the abilities to act as telomerase inhibitor and to produce DNA nicks [5-7]. Also, haematoxylin has recently been demonstrated to be a potent inhibitor of protein tyrosine kinase [8]. Given the broad and interesting biological activities of these two natural products, significant efforts have been devoted for their stereoselective syntheses [9-16]. Nevertheless, many possibilities remain unexplored especially for executing new design and synthetic strategies to generate their analogues [17,18].
![[1860-5397-12-280-1]](/bjoc/content/figures/1860-5397-12-280-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chroman-based tetracyclic natural products 1–4 of the brazilin family and our designed, B-ring-modified analogues of brazilin 5.
Figure 1: Chroman-based tetracyclic natural products 1–4 of the brazilin family and our designed, B-ring-modi...
On the other hand, the fusion of two or more privileged scaffolds leads to geometrically well-defined rigid polycyclic structures with enhanced receptor-binding selectivity. Thus, the design and synthesis of structurally diverse, privileged structure-based polycyclic molecules with multiple chiral centers has been intensely studied during the past 15 years [19]. Moreover, on many occasions natural product-like molecules exhibit more potent biological activities than the parent natural products [20].
Meanwhile, the intramolecular Friedel–Crafts epoxy–arene (IFCEA) cyclization has been well-established as a powerful tool for the synthesis of carbo- and heterocyclic compounds [21-24]. The easy accessibility of racemic and enantiomerically pure epoxy substrates through a number of well-established epoxidation methods coupled with procedural simplicity, high atom economy, regio- and stereoselectivity of IFCEA cyclization make this methodology highly practical for the use in organic synthesis. Recently, we have reported the synthesis of diverse trans-4-arylchroman-3-ols via Brønsted acid catalysed regio- and stereoselective IFCEA cyclization of 2-(aryloxymethyl)-3-aryloxiranes [22,23]. The use of IFCEA cyclization as the key step is part of our long term objective of synthesizing chroman-based NPs and NPLMs and their subsequent application in medicinal chemistry.
Like chroman, the tetralin unit has been recognized as privileged structure. We were interested to synthesize cis-6a,7,8,12b-tetrahydro-6H-naphtho[2,1-c]chromen-6a-ols 5 (Figure 1) as B-ring-modified analogues of brazilin through the fusion of chroman and tetralin motifs to generate new bioactive molecules. To the best of our knowledge, the stereoselective synthesis of such chroman-fused tetralins has never been reported. Based on the continuous literature reports on bioactivities of brazilin, haematoxylin and their analogues and our fruitful experience [22,23,25,26] in intramolecular epoxide ring-opening chemistry including IFCEA cyclization, we became interested in the possibility of extending the IFCEA cyclization protocol to the diastereoselective synthesis of 5. In this communication, we describe our synthetic study along this line.
We envisioned that compounds 5 could be achieved from tetralin-based epoxy ethers 6 via the IFCEA cyclization strategy (Scheme 1). Compounds 6 could be synthesized from tetralin-based epoxy alcohols 7 and different phenols. Commercially available 1-tetralone (8) could be the starting materials to obtain compound 7.
![[1860-5397-12-280-i1]](/bjoc/content/inline/1860-5397-12-280-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis of the designed B-ring-modified analogues of brazilin.
Scheme 1: Retrosynthetic analysis of the designed B-ring-modified analogues of brazilin.
One major synthetic challenge associated with 5 is the preferential formation of the cis-fused [6,6] ring system over the corresponding trans-isomer. While the stereochemical obligation makes sure that the [6,5]-ring system in brazilin and related natural products 1–4 remains cis-fused, its corresponding [6,6]-ring system in 5 can have cis or trans stereochemistry at the ring junction. In our previous work, we observed a cis relationship (both in concerted or/and stepwise-epoxide ring opening) between the 4-aryl group and H atom at the C-3 position of 4-arylchroman-3-ols (thus giving rise to trans-4-arylchroman-3-ols) [22,23]. But it was not obvious how the planned IFCEA cyclization onto the pre-existing 6-membered ring, in case of stepwise-epoxide ring opening, would influence the product diastereoselectivity (Scheme 2, upper panel). Thus, we were concerned about the possibility of getting a mixture of 5 and 9 under the planned IFCEA cyclization of 6. However, a literature survey indicated that cis-diastereoselective synthesis of related tetracyclic molecules via intramolecular Friedel–Crafts cyclization of tetralins (Scheme 2, lower panel) has been described by Lautens and co-workers [27,28].
![[1860-5397-12-280-i2]](/bjoc/content/inline/1860-5397-12-280-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The synthetic challenge associated with the synthesis of 5 by IFCEA of 6 (above) and recent literature reports of cis-diastereoselective synthesis of related tetracyclic molecules via intramolecular Friedel–Crafts cyclization.
Scheme 2: The synthetic challenge associated with the synthesis of 5 by IFCEA of 6 (above) and recent literat...
Nonetheless, a second major issue came to our mind before the synthesis of 5 could be started. The Lewis/Brønsted acidic conditions employed during the IFCEA cyclization might activate the tertiary –OH group of 5 and/or 9, leading to the formation of a stable 3°carbocation that can undergo further dehydration reactions until full aromatization to the naphthalene ring is achieved. This study would serve to help us to find the real scenario. Finally, in the presence of Lewis/Brønsted acids, substrates 6 might undergo a competitive semipinacol rearrangement to give ring contracted product 11.
With all these concerns in our mind, we started this work first with the synthesis of compound (±)-5a from glycidyl ether (±)-6a as a model reaction for validation and optimization studies. (±)-6a was synthesized from commercially available 1-tetralone (8) in six steps (Scheme S1, Supporting Information File 1). Substrate (±)-6a bearing a pendant 4-methoxyphenoxy group with a moderately electron-rich reacting site was expected to undergo IFCEA cyclization in high regio- and diastereoselectivity to give the tetracyclic product (±)-5a (see the reaction scheme in Table 1). A series of reactions was carried out with varying acid catalysts, solvents, reaction temperature, reaction time as well as the amount of catalysts. The results are summarized in Table 1. We first investigated the IFCEA cyclization of (±)-6a using 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) as reaction medium as well as the reaction promoter. The first choice of HFIP was based on its recently found ability to promote high-yielding IFCEA cyclization of benzylic epoxides [29]. Unfortunately, however, refluxing a solution of (±)-6a in HFIP for 4 h (entry 1, Table 1) led to the formation of a mixture of two major isolable compounds, out of which one was the expected product (±)-5 (50%) and the remaining one was hexafluoroisopropyl ether (generated from the nucleophilic addition of HFIP on the benzylic position). Meanwhile, we found that triggering the reaction with other well-known Brønsted and Lewis acids possessing different activating abilities also led to the formation of the desired product (±)-5a with varying product yields (entries 1–15, Table 1).
Table 1: Screening of reaction conditions on the IFCEA cyclization of (±)-6a leading to (±)-5aa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-280-i4.svg?max-width=637&scale=1.0)
|
|||||
| entry |
catalyst
(mol %) |
solvent |
temp
(°C) |
time |
yieldb
(%) |
|---|---|---|---|---|---|
| 1 | – | HFIP | reflux | 4 h | 50 |
| 2 | TsOH·H2O (20) | toluene | 80 | 45 min | 81 |
| 3 | TsOH·H2O (20) | toluene | 115 | 45 min | 75 |
| 4 | TsOH·H2O (20) | toluene | 50 | 2 h | 65 |
| 5 | TsOH·H2O (20) | MeCN | 80 | 45 min | 79 |
| 6 | TsOH·H2O (20) | DCE | 80 | 45 min | 70 |
| 7 | TsOH·H2O (20) | MeNO2 | 80 | 45 min | 75 |
| 8 | TFA (20) | toluene | rt | 45 min | 74 |
| 9 | TfOH (20) | toluene | rt | 45 min | 70 |
| 10 | H2SO4 (20) | toluene | rt | 45 min | 75 |
| 11 | Sc(OTf)2 (20) | DCM | rt | 60 min | 78 |
| 12 | BF3·Et2O (100) | DCM | 0 | 30 min | 81 |
| 13 | FeBr3 (20) | DCM | rt | 60 min | 78 |
| 14 | AgSbF6 (20) | DCM | reflux | 60 min | 79 |
| 15 | TiCl4 (20) | DCM | reflux | 60 min | 78 |
aReaction conditions: (±)-6a (0.4 mmol), acid catalyst, solvent (8 mL). bIsolated yields after silica gel column chromatography.
For example, when we conducted the reaction in toluene in the presence of 20 mol % of TsOH·H2O in AR-grade toluene at 80 °C (the reaction conditions that have been previously found by us [23] to be optimal for IFCEA cyclization), (±)-5 was isolated in much better yield of 81% (Table 1, entry 2). With 20 mol % of TsOH·H2O in toluene, a significant increase or decrease in temperature caused noticeable changes in the product yields (Table 1, entries 3 and 4). We observed that the reaction efficiency was also dependent on the reaction medium (Table 1, entries 5–7) and toluene appeared to be the best one. Lower yields of (±)-5 were obtained when stronger Brønsted acids like TFA, H2SO4 and TfOH were used as catalysts (Table 1, entries 8–10) in toluene as reaction medium. Among the five different Lewis acids we screened (Table 1, entries 11–15), the best result was achieved by using BF3·OEt2 (Table 1, entry 12).
Undoubtedly, the IFCEA cyclization proceeded almost equally well with different Lewis acids (Table 1, entries 11–15). However, all of these protocols required strict anhydrous conditions and some special attention to handle small amounts of the catalyst. On the other hand, TsOH·H2O is easily accessible, cheap, air stable, and even a minute amount can be weighed comfortably in an open atmosphere. It is important to mention that, among various acid catalyzed/promoted reactions described in the literature, on many occasions Brønsted acids have appeared as catalysts of choice under metal-free reaction conditions. On the basis of this fact and above investigations, we selected TsOH·H2O (20 mol %) as catalyst in toluene at 80 °C as the optimized reaction conditions.
With the optimized reaction conditions in hand, we next synthesized additional substrates (±)-6b–n with varying aryloxy groups (Scheme S1, Supporting Information File 1), and subsequently subjected them to the optimized reaction conditions with no precautions to exclude air or moisture. The results are summarized in Figure 2.
![[1860-5397-12-280-2]](/bjoc/content/figures/1860-5397-12-280-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Assessment of the IFCEA cyclization on additional substrates (±)-6b–n leading to (±)-5b–n. Reaction conditions: (±)-6b–h (0.4 mmol), TsOH·H2O (20 mol %), toluene (8 mL), 80 °C, 45 min. The numbers in parentheses represent isolated yields after silica gel column chromatography. aYield of isolated mixture of the inseparable regioisomers.
Figure 2: Assessment of the IFCEA cyclization on additional substrates (±)-6b–n leading to (±)-5b–n. Reaction...
All of the synthesized substrates (±)-6b–n could be effectively cyclized to give the corresponding chroman-fused tetralins (±)-5b–n, although the product yields were varied. Among the synthesized products, high yields were observed for (±)-5b (96%) and (±)-5k (95%), attributable to the high reactivity of symmetrical 3,5-dimethoxyphenoxy and 3,5-dimethylphenoxy groups in substrates (±)-6b and (±)-6k, respectively. The synthesis of (±)-5j was much significant as it has an allyl group which could be functionalized by diverse alkene chemistry. We believe that a large number of molecules similar to (±)-5j can also be synthesized using diverse 2-allylphenols (which are easily accessible via the well-known Claisen rearrangement). Also, compound (±)-5l seems to have important synthetic potential for further diversification due to the presence of a bromo substituent on the chroman-arene ring. Meanwhile, we witnessed somewhat lower (75–79%), but still synthetically useful, yields of products (±)-5g, (±)-5h, (±)-5i, (±)-5j and (±)-5l from the corresponding substrates, bearing moderately reactive arenoxy groups. But naphthochroman-fused tetralins (±)-5m and (±)-5n were obtained again in higher yields.
It is to be mentioned that, in each of the above-mentioned IFCEA cyclization reactions, the product was entirely the cis-isomer and no traces of the corresponding trans-isomers were detected in the 1H NMR of the crude reaction mixtures. Stereochemical assignment at the ring junction of the products posed some initial challenge as it was not easy to confirm the cis relationship between the angular hydrogen and hydroxy substituents with simple 1D NMR spectroscopy. Alternative concrete evidences for the structural assignment were sought from X-ray crystallography. Fortunately, we got the single crystals of (±)-5k by slow evaporation of a solution of 5k in hexane/EtOAc, and its molecular structure was confirmed by X-ray diffraction analysis (Figure 3). The 1H and 13C NMR spectra of all the products were clean (Supporting Information File 1) and the cis-stereochemistry at the ring junction of the remaining products were tentatively confirmed by comparing their 1H and 13C NMR spectra with that of (±)-5k.
![[1860-5397-12-280-3]](/bjoc/content/figures/1860-5397-12-280-3.png?scale=2.0&max-width=1024&background=FFFFFF)
The obsereved cis-diastereoselectivity in this work is consistent with the reported synthesis of related-tetracyclic molecules via Friedel–Crafts cyclization (Scheme 2, lower panel) promoted by metal-based Lewis acids [27,28]. Noteably, none of the synthesized molecules described in these two reports contains an angular –OH group. We feel that our synthesis involves more challenging Friedel–Crafts cyclization (Scheme 2, upper panel) and the products are relevant in the field of natural product-like molecules, because the synthesized molecules are close analogs of a natural product (brazilin). Moreover, the use of TsOH·H2O as an easily-accesible Brønsted acid catalyst with low loading under metal-free conditions and the operational simplicity render this transformation an attractive approach.
Nevertheless, in an additional work, the angular –OH group of (±)-5k was reductively removed on treatment with Et3SiH and boron trifluoride etherate to get chroman-fused tetralin (±)-14 with the two angular H atoms being positioned in trans fashion (Scheme 3); no cis-isomer was formed. The stereochemical assignment was confirmed on the vicinal coupling constant between the two angular H atoms (the benzylic methine on the chroman ring in 14 appeared as a doublet with J = 10.5 Hz at δ 3.97) and by comparison with reported data of related tetracyclic molecules [30-32]. Such a stereoselective reductive removal of an –OH group should be useful in preparing a library of chroman-fused tetralins with trans-stereochemistry at the ring junction.
![[1860-5397-12-280-i3]](/bjoc/content/inline/1860-5397-12-280-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Stereoselective conversion of (±)-5k into (±)-14.
Scheme 3: Stereoselective conversion of (±)-5k into (±)-14.
Conclusion
In conclusion, we herein report a convenient Brønsted acid-catalyzed, metal-free, stereoselective synthesis of 6a,7,8,12b-tetrahydro-6H-naphtho[2,1-c]chromen-6a-ols as B-ring-modified analogues of brazilin using starting materials derived from inexpensive 1-tetralone and phenol derivatives. Our worries concerning the formation cis–trans mixture of 6a,7,8,12b-tetrahydro-6H-naphtho[2,1-c]chromen-6a-ols and their probable conversion to of naphthopyran derivatives via dehydration of tertiary –OH group were laid to rest. To the best of our knowledge, this is the first example of the generation of such type of chroman-fused tetralins. The easy accessibility of the starting materials, the mild reaction conditions, and the importance of products as B-ring-modified analogues of brazilin should make this synthetic work a useful addition in the diversity-oriented synthesis of natural-product like molecules. Moreover, since enantiomerically pure/enriched epoxides are compatible under IFCEA cyclization (as demonstarted previously by us [22,23] and others [29,33]), this methodology should also be applicable with the enantioenriched substrates. Further work is in progress aimed at synthesizing such type of fused hybrid molecules with different ring sizes and heteroatoms and performing mechanistic studies which will be reported in due course as a full paper.
References
-
Ellis, G. P. Chromenes, Chromanones, and Chromones—Introduction. In Chemistry of Heterocyclic Compounds: Chromenes, Chromanones, and Chromones; Ellis, G. P., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; Vol. 31, pp 1–1196. doi:10.1002/9780470187012.ch1
Return to citation in text: [1] -
Robinson, R. Bull. Soc. Chim. Fr. 1958, 125–134.
Return to citation in text: [1] [2] -
Xu, J.; Yadan, J. C. Tetrahedron Lett. 1996, 37, 2421–2424. doi:10.1016/0040-4039(96)00288-2
Return to citation in text: [1] [2] -
Lin, C.-C.; Teng, T.-M.; Tsai, C.-C.; Liao, H.-Y.; Liu, R.-S. J. Am. Chem. Soc. 2008, 130, 16417–16423. doi:10.1021/ja806415t
Return to citation in text: [1] [2] -
Chin, R. L.; Tolman, A. C. Telomerase inhibitors and methods of their use. WO. Patent 0,193,864, Dec 13, 2001.
Return to citation in text: [1] -
Mar, W.; Lee, H.-T.; Je, K.-H.; Choi, H.-Y.; Seo, E.-K. Arch. Pharmacal Res. 2003, 26, 147–150. doi:10.1007/BF02976661
Return to citation in text: [1] -
Yen, C.-T.; Nakagawa-Goto, K.; Hwang, T.-L.; Wu, P.-C.; Morris-Natschke, S.-L.; Lai, W.-C.; Bastow, K. F.; Chang, F.-R.; Wu, Y.-C.; Lee, K.-H. Bioorg. Med. Chem. Lett. 2010, 20, 1037–1039. doi:10.1016/j.bmcl.2009.12.041
Return to citation in text: [1] -
Lin, L.-G.; Xie, H.; Li, H.-L.; Tong, L.-J.; Tang, C.-P.; Ke, C.-Q.; Liu, Q.-F.; Lin, L.-P.; Geng, M.-Y.; Jiang, H.; Zhao, W.-M.; Ding, J.; Ye, Y. J. Med. Chem. 2008, 51, 4419–4429. doi:10.1021/jm701501x
Return to citation in text: [1] -
Jung, Y.; Kim, I. J. Org. Chem. 2015, 80, 2001–2005. doi:10.1021/jo502745j
Return to citation in text: [1] -
Jung, Y.; Kim, I. Org. Biomol. Chem. 2015, 13, 4331–4335. doi:10.1039/C5OB00216H
Return to citation in text: [1] -
Yadav, J. S.; Mishra, A. K.; Das, S. Tetrahedron 2014, 70, 7560–7566. doi:10.1016/j.tet.2014.08.001
Return to citation in text: [1] -
Javed, U.; Karim, M.; Jahng, K. C.; Park, J.-G.; Jahng, Y. Tetrahedron: Asymmetry 2014, 25, 1270–1274. doi:10.1016/j.tetasy.2014.08.009
Return to citation in text: [1] -
Wang, X.; Zhang, H.; Yang, X.; Zhao, J.; Pan, C. Chem. Commun. 2013, 5405–5407. doi:10.1039/c3cc42385a
Return to citation in text: [1] -
Li, L.-Q.; Li, M.-M.; Wang, K.; Qin, H.-B. Tetrahedron Lett. 2013, 54, 6029–6031. doi:10.1016/j.tetlet.2013.08.081
Return to citation in text: [1] -
Huang, Y.; Zhang, J.; Pettus, T. R. R. Org. Lett. 2005, 7, 5841–5844. doi:10.1021/ol0523749
Return to citation in text: [1] -
Davis, F. A.; Chen, B.-C. J. Org. Chem. 1993, 58, 1751–1753. doi:10.1021/jo00059a026
Return to citation in text: [1] -
Pan, C.; Zeng, X.; Guan, Y.; Jiang, X.; Li, L.; Zhang, H. Synlett 2011, 425–429. doi:10.1055/s-0030-1259511
Return to citation in text: [1] -
Fies, M.; Friedrich, K. J. Prakt. Chem./Chem.-Ztg. 1995, 337, 50–54. doi:10.1002/prac.19953370111
Return to citation in text: [1] -
Kim, J.; Kim, H.; Park, S. B. J. Am. Chem. Soc. 2014, 136, 14629–14638. doi:10.1021/ja508343a
Return to citation in text: [1] -
Crane, E. A.; Gademann, K. Angew. Chem., Int. Ed. 2016, 55, 3882–3902. doi:10.1002/anie.201505863
Return to citation in text: [1] -
Ling, J.; Lam, S. K.; Lo, B.; Lam, S.; Wong, W.-T.; Sun, J.; Chen, G.; Chiu, P. Org. Chem. Front. 2016, 3, 457–461. doi:10.1039/C5QO00333D
And related references cited therein.
Return to citation in text: [1] -
Devi, R.; Kalita, T.; Das, S. K. RSC Adv. 2015, 5, 39692–39696. doi:10.1039/C5RA02193F
Return to citation in text: [1] [2] [3] [4] [5] -
Devi, R.; Borah, P.; Das, S. K. Tetrahedron 2016, 72, 4878–4888. doi:10.1016/j.tet.2016.06.059
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Chen, D.; Liu, H. M.; Li, M. M.; Yan, Y. M.; Xu, W. D.; Li, X. N.; Cheng, Y. X.; Qin, H. B. Chem. Commun. 2015, 14594–14596.
Return to citation in text: [1] -
Dinda, S. K.; Das, S. K.; Panda, G. Synthesis 2009, 1886–1896. doi:10.1055/s-0028-1088077
Return to citation in text: [1] -
Das, S. K.; Panda, G. Tetrahedron 2008, 64, 4162–4173. doi:10.1016/j.tet.2008.03.001
Return to citation in text: [1] -
Tsoung, J.; Kramer, K.; Zajdlik, A.; Liebert, C.; Lautens, M. J. Org. Chem. 2011, 76, 9031–9045. doi:10.1021/jo201781x
Return to citation in text: [1] [2] -
Liebert, C.; Brinks, M. K.; Capacci, A. G.; Fleming, M. J.; Lautens, M. Org. Lett. 2011, 13, 3000–3003. doi:10.1021/ol2008236
Return to citation in text: [1] [2] -
Li, G.-X.; Qu, J. Chem. Commun. 2010, 46, 2653–2655. doi:10.1039/b926684d
Return to citation in text: [1] [2] -
In the related cis-fused tetracycles, benzylic methine appeared as a doublet with coupling constant J = 4–6 Hz, whereas that of trans-fused ones appeared as a doublet with coupling constant J = 9–11 Hz.
Return to citation in text: [1] -
Lee, Y. R.; Kim, Y. M.; Kim, S. H. Tetrahedron 2009, 65, 101–108. doi:10.1016/j.tet.2008.10.101
Return to citation in text: [1] -
LaLonde, R. T.; Ramdayal, F.; Sarko, A.; Yanai, K.; Zhang, M. J. Med. Chem. 2003, 46, 1180–1190. doi:10.1021/jm020158p
Return to citation in text: [1] -
Marcos, R.; Rodrıguez-Escrich, C.; Herrerıas, C.; Pericas, M. A. J. Am. Chem. Soc. 2008, 130, 16838–16839. doi:10.1021/ja8062887
Return to citation in text: [1]
| 22. | Devi, R.; Kalita, T.; Das, S. K. RSC Adv. 2015, 5, 39692–39696. doi:10.1039/C5RA02193F |
| 23. | Devi, R.; Borah, P.; Das, S. K. Tetrahedron 2016, 72, 4878–4888. doi:10.1016/j.tet.2016.06.059 |
| 27. | Tsoung, J.; Kramer, K.; Zajdlik, A.; Liebert, C.; Lautens, M. J. Org. Chem. 2011, 76, 9031–9045. doi:10.1021/jo201781x |
| 28. | Liebert, C.; Brinks, M. K.; Capacci, A. G.; Fleming, M. J.; Lautens, M. Org. Lett. 2011, 13, 3000–3003. doi:10.1021/ol2008236 |
| 30. | In the related cis-fused tetracycles, benzylic methine appeared as a doublet with coupling constant J = 4–6 Hz, whereas that of trans-fused ones appeared as a doublet with coupling constant J = 9–11 Hz. |
| 31. | Lee, Y. R.; Kim, Y. M.; Kim, S. H. Tetrahedron 2009, 65, 101–108. doi:10.1016/j.tet.2008.10.101 |
| 32. | LaLonde, R. T.; Ramdayal, F.; Sarko, A.; Yanai, K.; Zhang, M. J. Med. Chem. 2003, 46, 1180–1190. doi:10.1021/jm020158p |
| 1. | Ellis, G. P. Chromenes, Chromanones, and Chromones—Introduction. In Chemistry of Heterocyclic Compounds: Chromenes, Chromanones, and Chromones; Ellis, G. P., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; Vol. 31, pp 1–1196. doi:10.1002/9780470187012.ch1 |
| 8. | Lin, L.-G.; Xie, H.; Li, H.-L.; Tong, L.-J.; Tang, C.-P.; Ke, C.-Q.; Liu, Q.-F.; Lin, L.-P.; Geng, M.-Y.; Jiang, H.; Zhao, W.-M.; Ding, J.; Ye, Y. J. Med. Chem. 2008, 51, 4419–4429. doi:10.1021/jm701501x |
| 5. | Chin, R. L.; Tolman, A. C. Telomerase inhibitors and methods of their use. WO. Patent 0,193,864, Dec 13, 2001. |
| 6. | Mar, W.; Lee, H.-T.; Je, K.-H.; Choi, H.-Y.; Seo, E.-K. Arch. Pharmacal Res. 2003, 26, 147–150. doi:10.1007/BF02976661 |
| 7. | Yen, C.-T.; Nakagawa-Goto, K.; Hwang, T.-L.; Wu, P.-C.; Morris-Natschke, S.-L.; Lai, W.-C.; Bastow, K. F.; Chang, F.-R.; Wu, Y.-C.; Lee, K.-H. Bioorg. Med. Chem. Lett. 2010, 20, 1037–1039. doi:10.1016/j.bmcl.2009.12.041 |
| 23. | Devi, R.; Borah, P.; Das, S. K. Tetrahedron 2016, 72, 4878–4888. doi:10.1016/j.tet.2016.06.059 |
| 2. | Robinson, R. Bull. Soc. Chim. Fr. 1958, 125–134. |
| 3. | Xu, J.; Yadan, J. C. Tetrahedron Lett. 1996, 37, 2421–2424. doi:10.1016/0040-4039(96)00288-2 |
| 4. | Lin, C.-C.; Teng, T.-M.; Tsai, C.-C.; Liao, H.-Y.; Liu, R.-S. J. Am. Chem. Soc. 2008, 130, 16417–16423. doi:10.1021/ja806415t |
| 22. | Devi, R.; Kalita, T.; Das, S. K. RSC Adv. 2015, 5, 39692–39696. doi:10.1039/C5RA02193F |
| 23. | Devi, R.; Borah, P.; Das, S. K. Tetrahedron 2016, 72, 4878–4888. doi:10.1016/j.tet.2016.06.059 |
| 2. | Robinson, R. Bull. Soc. Chim. Fr. 1958, 125–134. |
| 3. | Xu, J.; Yadan, J. C. Tetrahedron Lett. 1996, 37, 2421–2424. doi:10.1016/0040-4039(96)00288-2 |
| 4. | Lin, C.-C.; Teng, T.-M.; Tsai, C.-C.; Liao, H.-Y.; Liu, R.-S. J. Am. Chem. Soc. 2008, 130, 16417–16423. doi:10.1021/ja806415t |
| 27. | Tsoung, J.; Kramer, K.; Zajdlik, A.; Liebert, C.; Lautens, M. J. Org. Chem. 2011, 76, 9031–9045. doi:10.1021/jo201781x |
| 28. | Liebert, C.; Brinks, M. K.; Capacci, A. G.; Fleming, M. J.; Lautens, M. Org. Lett. 2011, 13, 3000–3003. doi:10.1021/ol2008236 |
| 20. | Crane, E. A.; Gademann, K. Angew. Chem., Int. Ed. 2016, 55, 3882–3902. doi:10.1002/anie.201505863 |
| 22. | Devi, R.; Kalita, T.; Das, S. K. RSC Adv. 2015, 5, 39692–39696. doi:10.1039/C5RA02193F |
| 23. | Devi, R.; Borah, P.; Das, S. K. Tetrahedron 2016, 72, 4878–4888. doi:10.1016/j.tet.2016.06.059 |
| 19. | Kim, J.; Kim, H.; Park, S. B. J. Am. Chem. Soc. 2014, 136, 14629–14638. doi:10.1021/ja508343a |
| 22. | Devi, R.; Kalita, T.; Das, S. K. RSC Adv. 2015, 5, 39692–39696. doi:10.1039/C5RA02193F |
| 23. | Devi, R.; Borah, P.; Das, S. K. Tetrahedron 2016, 72, 4878–4888. doi:10.1016/j.tet.2016.06.059 |
| 25. | Dinda, S. K.; Das, S. K.; Panda, G. Synthesis 2009, 1886–1896. doi:10.1055/s-0028-1088077 |
| 26. | Das, S. K.; Panda, G. Tetrahedron 2008, 64, 4162–4173. doi:10.1016/j.tet.2008.03.001 |
| 17. | Pan, C.; Zeng, X.; Guan, Y.; Jiang, X.; Li, L.; Zhang, H. Synlett 2011, 425–429. doi:10.1055/s-0030-1259511 |
| 18. | Fies, M.; Friedrich, K. J. Prakt. Chem./Chem.-Ztg. 1995, 337, 50–54. doi:10.1002/prac.19953370111 |
| 29. | Li, G.-X.; Qu, J. Chem. Commun. 2010, 46, 2653–2655. doi:10.1039/b926684d |
| 33. | Marcos, R.; Rodrıguez-Escrich, C.; Herrerıas, C.; Pericas, M. A. J. Am. Chem. Soc. 2008, 130, 16838–16839. doi:10.1021/ja8062887 |
| 9. | Jung, Y.; Kim, I. J. Org. Chem. 2015, 80, 2001–2005. doi:10.1021/jo502745j |
| 10. | Jung, Y.; Kim, I. Org. Biomol. Chem. 2015, 13, 4331–4335. doi:10.1039/C5OB00216H |
| 11. | Yadav, J. S.; Mishra, A. K.; Das, S. Tetrahedron 2014, 70, 7560–7566. doi:10.1016/j.tet.2014.08.001 |
| 12. | Javed, U.; Karim, M.; Jahng, K. C.; Park, J.-G.; Jahng, Y. Tetrahedron: Asymmetry 2014, 25, 1270–1274. doi:10.1016/j.tetasy.2014.08.009 |
| 13. | Wang, X.; Zhang, H.; Yang, X.; Zhao, J.; Pan, C. Chem. Commun. 2013, 5405–5407. doi:10.1039/c3cc42385a |
| 14. | Li, L.-Q.; Li, M.-M.; Wang, K.; Qin, H.-B. Tetrahedron Lett. 2013, 54, 6029–6031. doi:10.1016/j.tetlet.2013.08.081 |
| 15. | Huang, Y.; Zhang, J.; Pettus, T. R. R. Org. Lett. 2005, 7, 5841–5844. doi:10.1021/ol0523749 |
| 16. | Davis, F. A.; Chen, B.-C. J. Org. Chem. 1993, 58, 1751–1753. doi:10.1021/jo00059a026 |
| 21. |
Ling, J.; Lam, S. K.; Lo, B.; Lam, S.; Wong, W.-T.; Sun, J.; Chen, G.; Chiu, P. Org. Chem. Front. 2016, 3, 457–461. doi:10.1039/C5QO00333D
And related references cited therein. |
| 22. | Devi, R.; Kalita, T.; Das, S. K. RSC Adv. 2015, 5, 39692–39696. doi:10.1039/C5RA02193F |
| 23. | Devi, R.; Borah, P.; Das, S. K. Tetrahedron 2016, 72, 4878–4888. doi:10.1016/j.tet.2016.06.059 |
| 24. | Chen, D.; Liu, H. M.; Li, M. M.; Yan, Y. M.; Xu, W. D.; Li, X. N.; Cheng, Y. X.; Qin, H. B. Chem. Commun. 2015, 14594–14596. |
© 2016 Gogoi et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)