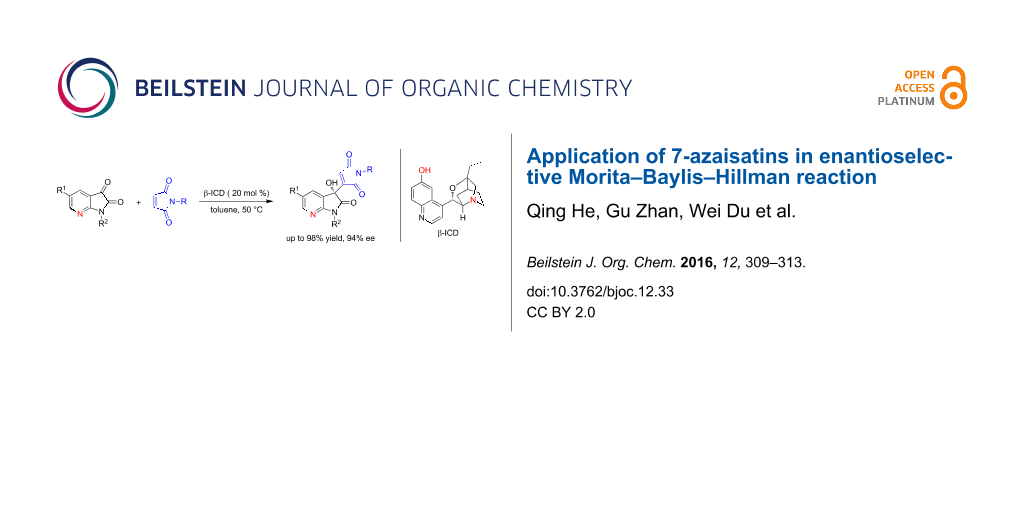
Abstract
7-Azaisatin and 7-azaoxindole skeletons are valuable building blocks in diverse biologically active substances. Here 7-azaisatins turned out to be more efficient electrophiles than the analogous isatins in the enantioselective Morita–Baylis–Hillman (MBH) reactions with maleimides using a bifunctional tertiary amine, β-isocupreidine (β-ICD), as the catalyst. This route allows a convenient approach to access multifunctional 3-hydroxy-7-aza-2-oxindoles with high enantiopurity (up to 94% ee). Other types of activated alkenes, such as acrylates and acrolein, could also be efficiently utilized.

Graphical Abstract
Introduction
The asymmetric Morita–Baylis–Hillman (MBH) reaction is one of the most powerful synthetic methods in organic chemistry, as it directly constructs carbon–carbon bonds in an atom-economical manner and provides densely functionalized molecules [1-4]. In particular, the direct formation of stereogenic quaternary carbon centers by enantioselective MBH reactions has been a fascinating and challenging area, because the compatible electrophiles are always limited to aldehydes or derivatives thereof. Since the first elegant work on the enantioselective MBH reaction between isatins and acrolein catalyzed by β-isocupreidine (β-ICD) was reported by the Zhou group [5], isatin derivatives, as highly activated electrophiles, have been utilized by other groups for similar transformations with acrylates or acrylamides, affording the 3-hydroxyoxindole derivatives with moderate to excellent stereocontrol [6-12]. Maleimides are also good nucleophilic precursors in the MBH reactions and in 2013, Chimni developed an asymmetric MBH reaction of isatins and maleimides with excellent enantioselectivity [13]. Later, the same group expanded this strategy to isatin-derived ketimines under the identical catalytic conditions [14]. Remarkably, these reactions were usually promoted by bifunctional catalysts, such as β-ICD, whose C6’-OH group served as a H-bond donor to facilitate the proton-transfer step and to stabilize the transition state in MBH reactions [11-14]. Nevertheless, all these cases suffered from low reactivity and long reaction times were always required (usually > 48 h) for better conversions.
7-Azaisatins bearing an additional nitrogen atom at the 7-position of the 2-oxindole scaffold might be better electrophiles than isatins owing to the electron-withdrawing effect of the pyridine motif. More importantly, a number of 7-azaisatins and 7-azaoxindoles were shown to exhibit significant anticancer or TrkA kinase inhibitory activities (Figure 1) [15-17]. This would render their derivatives as pharmaceutically interesting compounds with high potential. In addition, 7-azaisatins were also applied in asymmetric synthesis [18,19]. Therefore, with our continuing interest in the catalytic application of bifunctional β-ICD [20-23], herein we report the enantioselective MBH reaction of 7-azaisatins with maleimides. A series of 3-hydroxy-7-aza-2-oxindoles have been synthesized in good to excellent yields and with moderate to high enantioselectivity in a shorter time (for most cases, the reaction time is 24 h).
![[1860-5397-12-33-1]](/bjoc/content/figures/1860-5397-12-33-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Bioactive 7-azaisatins and their derivatives.
Figure 1: Bioactive 7-azaisatins and their derivatives.
Results and Discussion
The enantioselective MBH reaction was first investigated with N-methyl-7-azaisatin (1a) and N-phenylmaleimide (2a) in toluene catalyzed by β-ICD. To our delight, the desired product 3a was obtained in 45% yield and with excellent enantioselectivity at 50 °C after 72 h (Table 1, entry 1). The reaction could be accelerated significantly by increasing the amounts of 2a (Table 1, entries 2–5), and almost a quantitative yield could be obtained after 24 h by employing 6 equiv of 2a (Table 1, entry 5). Other solvents, such as MeCN, THF and CHCl3, were also tested but provided inferior results (Table 1, entries 6–8). The attempt to improve the enantioselectivity by lowering the temperature failed (Table 1, entry 9), and the enantiocontrol was diminished at 70 °C (Table 1, entry 10). On the other hand, when α-isocupreine (α-IC) [24,25] was employed as the catalyst instead of β-ICD, unfortunately no desired product was obtained even after 120 h (Table 1, entry 11). Finally, we compared the reaction of 7-azaisatin 1a and maleimide 2a under the catalytic conditions developed by Chimni [13]. The reaction proceeded slowly, but a high yield with an outstanding ee value could be obtained after 72 h (Table 1, entry 12).
Table 1: Screening conditions of the enantioselective MBH reaction of 1a and 2a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-33-i2.svg?max-width=637&scale=1.0)
|
||||||
| Entrya | 2a (equiv) | Solvent | Temperature (°C) | Time (h) | Yield (%)b | ee (%)c |
|---|---|---|---|---|---|---|
| 1 | 1.5 | toluene | 50 | 72 | 45 | 91 |
| 2 | 3.0 | toluene | 50 | 72 | 66 | 92 |
| 3 | 4.0 | toluene | 50 | 48 | 83 | 92 |
| 4 | 5.0 | toluene | 50 | 48 | 89 | 91 |
| 5 | 6.0 | toluene | 50 | 24 | 98 | 94 |
| 6 | 6.0 | MeCN | 50 | 24 | 98 | 86 |
| 7 | 6.0 | THF | 50 | 24 | 98 | 88 |
| 8 | 6.0 | CHCl3 | 50 | 96 | 90 | 91 |
| 9 | 6.0 | toluene | rt | 48 | 91 | 91 |
| 10 | 6.0 | toluene | 70 | 24 | 85 | 88 |
| 11d | 6.0 | toluene | 50 | >120 | – | – |
| 12 | 2.0 | CHCl3 | rt | 72 | 90 | 95 |
aUnless noted otherwise, reactions were performed with 1a (0.05 mmol), 2a and catalyst (0.01 mmol) in solvent (0.5 mL). bIsolated yield. cDetermined by HPLC analysis on a chiral stationary phase. dα-IC was used as the catalyst.
With the optimal conditions in hand, next, the substrate scope of the MBH reaction was studied under the catalysis of β-ICD (Table 2). At first, a variety of N-substituted maleimides 2 were explored in the reaction with 7-azaisatin 1a in toluene. Maleimides bearing an electron-rich N-aryl ring generally afforded the corresponding 3-hydroxy-7-aza-2-oxindoles in high yields and with excellent enantioselectivity (Table 2, entries 2–4) while good results were obtained in a mixture of THF and DCM for a maleimide with an electron-deficient N-aryl group because of better solubility (Table 2, entry 5). In addition, N-alkylated maleimides provided the desired products in good yields (Table 2, entries 6–9), while only moderate enantioselectivity was observed for products 3h and 3i (Table 2, entries 8 and 9). 7-Azaisatins with different N-protecting groups were also applied to the MBH reaction with N-phenylmaleimide (2a), including methoxymethyl (MOM), benzyl (Bn) and 4-chlorophenyl substituents. All of them showed a lower reactivity and enantioselectivity than that of the methyl-substituted one, and better results were generally obtained in a mixture of THF and DCM (Table 2, entries 10–12). The C5-phenyl substituted or halogenated 7-azaisatins could be smoothly applied, although a longer reaction time was required for the halogenated substrates (Table 2, entries 13–15).
Table 2: Substrate scope of the enantioselective MBH reaction.
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-33-i3.svg?max-width=637&scale=1.0)
|
||||||
| Entrya | R1 | R2 | R | Time (h) | Yield (%)b | ee (%)c |
|---|---|---|---|---|---|---|
| 1 | H | 1a, Me | 2a, Ph | 24 | 3a, 98 | 94 |
| 2 | H | 1a,Me | 2b, 4-MePh | 24 | 3b, 87 | 90 |
| 3 | H | 1a, Me | 2c, 4-MeOPh | 24 | 3c, 88 | 92 |
| 4 | H | 1a, Me | 2e, 2,4,6-MePh | 24 | 3e, 90 | 79 |
| 5d | H | 1a, Me | 2d, 4-ClPh | 24 | 3d, 87 | 92 |
| 6 | H | 1a, Me | 2f, Me | 24 | 3f, 84 | 89 |
| 7 | H | 1a, Me | 2g, Bn | 24 | 3g, 86 | 89 |
| 8 | H | 1a, Me | 2h, n-Bu | 24 | 3h, 86 | 66 |
| 9 | H | 1a, Me | 2i, cyclohexyl | 24 | 3i, 84 | 61 |
| 10d | H | 1b, MOM | 2a, Ph | 24 | 3j, 92 | 91 |
| 11d | H | 1c, Bn | 2a, Ph | 48 | 3k, 93 | 87 |
| 12d | H | 1d, 4-ClPh | 2a, Ph | 96 | 3l, 37 | 71 |
| 13d | Ph | 1e, Me | 2a, Ph | 24 | 3m, 88 | 92 |
| 14 | Cl | 1f, Me | 2a, Ph | 48 | 3n, 81 | 85 |
| 15 | Br | 1g, Me | 2a, Ph | 48 | 3o, 82 | 88 |
aUnless otherwise noted, all reactions were performed with 1 (0.1 mmol), 2 (0.6 mmol) and β-ICD (0.02 mmol) in toluene (1.0 mL) at 50 °C. bIsolated yield. cDetermined by HPLC analysis on a chiral stationary phase. The absolute configuration of the chiral products was assigned by analogy to Chimni’s work [13]. dIn a mixture of THF and DCM (0.5/0.5 mL).
To further assess the high electrophilicity of 7-azaisatins in MBH reactions, more activated alkenes were explored with 1a. Methyl and ethyl acrylates afforded products 3p and 3q, respectively, with comparable results to Wu’s work [7], albeit in a shorter time (24 h vs 2–3 d). Notably, the MBH reaction of acrolein and 7-azaisatin 1a was carried out under the same conditions as described in Zhou’s work [5], providing the highly enantio-enriched product 3r in an excellent yield after 1.5 h. The reaction with 1a was much more efficient, as it took 4 d to afford the product 5 in a high yield by using N-methylisatin (4) as the substrate (Scheme 1).
![[1860-5397-12-33-i1]](/bjoc/content/inline/1860-5397-12-33-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Further exploration with 7-azaisatin 1a and comparison with the previous work by Zhou [5].
Scheme 1: Further exploration with 7-azaisatin 1a and comparison with the previous work by Zhou [5].
Conclusion
In summary, we have developed an efficient and enantioselective Morita–Baylis–Hillman reaction between 7-azaisatins and maleimides and other activated alkenes in the presence of the bifunctional catalyst β-ICD. 7-Azaisatins were proven to be better electrophiles than the analogous isatins, and furnished a convenient protocol to enantio-enriched multifunctional 3-hydroxy-7-aza-2-oxindoles. Such substances might be further applied in organic synthesis for potential biological and pharmaceutical studies in the future.
Experimental
General procedure for the synthesis of 3-hydroxy-7-aza-2-oxindoles: A solution of N-protected 7-azaisatin 1 (0.1 mmol), N-substituted maleimide 2 (0.6 mmol) and β-ICD (20 mol %) was stirred in dry toluene (1.0 mL) at 50 °C. The progress of the reaction was monitored by TLC. After completion, the MBH reaction product 3 was purified by flash chromatography on silica gel using petroleum ether/EtOAc 6:1–3:1 as the eluent.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures and analytical data for the compounds. | ||
| Format: PDF | Size: 3.0 MB | Download |
References
-
Basavaiah, D.; Rao, K. V.; Reddy, R. J. Chem. Soc. Rev. 2007, 36, 1581–1588. doi:10.1039/B613741P
Return to citation in text: [1] -
Basavaiah, D.; Reddy, B. S.; Badsara, S. S. Chem. Rev. 2010, 110, 5447–5674. doi:10.1021/cr900291g
Return to citation in text: [1] -
Cran, J. W.; Krafft, M. E.; Seibert, K. A.; Haxell, T. F. N.; Wright, J. A.; Hirosawa, C.; Abboud, K. A. Tetrahedron 2011, 67, 9922–9943. doi:10.1016/j.tet.2011.09.061
Return to citation in text: [1] -
Wei, Y.; Shi, M. Chem. Rev. 2013, 113, 6659–6690. doi:10.1021/cr300192h
Return to citation in text: [1] -
Liu, Y.-L.; Wang, B.-L.; Cao, J.-J.; Chen, L.; Zhang, Y.-X.; Wang, C.; Zhou, J. J. Am. Chem. Soc. 2010, 132, 15176–15178. doi:10.1021/ja107858z
Return to citation in text: [1] [2] [3] -
Guan, X.-Y.; Wei, Y.; Shi, M. Chem. – Eur. J. 2010, 16, 13617–13621. doi:10.1002/chem.201002240
Return to citation in text: [1] -
Qian, J.-Y.; Wang, C.-C.; Sha, F.; Wu, X.-Y. RSC Adv. 2012, 2, 6042–6048. doi:10.1039/C2RA20521A
Return to citation in text: [1] [2] -
Duan, Z.; Zhang, Z.; Qian, P.; Han, J.; Pan, Y. RSC Adv. 2013, 3, 10127–10130. doi:10.1039/C3RA41115J
Return to citation in text: [1] -
Dong, Z.; Yan, C.; Gao, Y.; Dong, C.; Qiu, G.; Zhou, H.-B. Adv. Synth. Catal. 2015, 357, 2132–2142. doi:10.1002/adsc.201500110
Return to citation in text: [1] -
Zhao, X.; Li, T.-Z.; Qian, J.-Y.; Sha, F.; Wu, X.-Y. Org. Biomol. Chem. 2014, 12, 8072–8078. doi:10.1039/C4OB01358A
Return to citation in text: [1] -
Zhong, F.; Chen, G.-Y.; Lu, Y. Org. Lett. 2011, 13, 82–85. doi:10.1021/ol102597s
Return to citation in text: [1] [2] -
Yoshida, Y.; Sako, M.; Kishi, K.; Sasai, H.; Hatakeyama, S.; Takizawa, S. Org. Biomol. Chem. 2015, 13, 9022–9028. doi:10.1039/C5OB00874C
Return to citation in text: [1] [2] -
Chauhan, P.; Chimni, S. S. Asian J. Org. Chem. 2013, 2, 586–592. doi:10.1002/ajoc.201300093
Return to citation in text: [1] [2] [3] [4] -
Kumar, A.; Sharma, V.; Kaur, J.; Kumar, N.; Chimni, S. S. Org. Biomol. Chem. 2015, 13, 5629–5635. doi:10.1039/C5OB00182J
Return to citation in text: [1] [2] -
Wood, E. R.; Kuyper, L.; Petrov, K. G.; Hunter, R. N., III; Harris, P. A.; Lackey, K. Bioorg. Med. Chem. Lett. 2004, 14, 953–957. doi:10.1016/j.bmcl.2003.12.002
Return to citation in text: [1] -
Kritsanida, M.; Magiatis, P.; Skaltsounis, A.-L.; Peng, Y.; Li, P.; Wennogle, L. P. J. Nat. Prod. 2009, 72, 2199–2202. doi:10.1021/np9003905
Return to citation in text: [1] -
Rekulapally, S.; Jarapula, R.; Gangarapu, K.; Manda, S.; Vaidya, J. R. Med. Chem. Res. 2015, 24, 3412–3422. doi:10.1007/s00044-015-1390-0
Return to citation in text: [1] -
Liu, Y.-L.; Wang, X.; Zhao, Y.-L.; Zhu, F.; Zeng, X.-P.; Chen, L.; Wang, C.-H.; Zhao, X.-L.; Zhou, J. Angew. Chem. 2013, 125, 13980–13984. doi:10.1002/ange.201307250
Return to citation in text: [1] -
Yin, X.-P.; Zeng, X.-P.; Liu, Y.-L.; Liao, F.-M.; Yu, J.-S.; Zhou, F.; Zhou, J. Angew. Chem., Int. Ed. 2014, 53, 13740–13745. doi:10.1002/anie.201407677
Return to citation in text: [1] -
Peng, J.; Huang, X.; Cui, H.-L.; Chen, Y.-C. Org. Lett. 2010, 12, 4260–4263. doi:10.1021/ol101668z
Return to citation in text: [1] -
Peng, J.; Huang, X.; Jiang, L.; Cui, H.-L.; Chen, Y.-C. Org. Lett. 2011, 13, 4584–4587. doi:10.1021/ol201776h
Return to citation in text: [1] -
Peng, J.; Huang, X.; Zheng, P.-F.; Chen, Y.-C. Org. Lett. 2013, 15, 5534–5537. doi:10.1021/ol402694b
Return to citation in text: [1] -
Peng, J.; Ran, G.-Y.; Du, W.; Chen, Y.-C. Org. Lett. 2015, 17, 4490–4493. doi:10.1021/acs.orglett.5b02157
Return to citation in text: [1] -
Nakamoto, Y.; Urabe, F.; Takahashi, K.; Ishihara, J.; Hatakeyama, S. Chem. – Eur. J. 2013, 19, 12653–12656. doi:10.1002/chem.201302665
Return to citation in text: [1] -
Peng, J.; Ran, G.-Y.; Du, W.; Chen, Y.-C. Synthesis 2015, 47, 2538–2544. doi:10.1055/s-0034-1378716
Return to citation in text: [1]
| 1. | Basavaiah, D.; Rao, K. V.; Reddy, R. J. Chem. Soc. Rev. 2007, 36, 1581–1588. doi:10.1039/B613741P |
| 2. | Basavaiah, D.; Reddy, B. S.; Badsara, S. S. Chem. Rev. 2010, 110, 5447–5674. doi:10.1021/cr900291g |
| 3. | Cran, J. W.; Krafft, M. E.; Seibert, K. A.; Haxell, T. F. N.; Wright, J. A.; Hirosawa, C.; Abboud, K. A. Tetrahedron 2011, 67, 9922–9943. doi:10.1016/j.tet.2011.09.061 |
| 4. | Wei, Y.; Shi, M. Chem. Rev. 2013, 113, 6659–6690. doi:10.1021/cr300192h |
| 14. | Kumar, A.; Sharma, V.; Kaur, J.; Kumar, N.; Chimni, S. S. Org. Biomol. Chem. 2015, 13, 5629–5635. doi:10.1039/C5OB00182J |
| 5. | Liu, Y.-L.; Wang, B.-L.; Cao, J.-J.; Chen, L.; Zhang, Y.-X.; Wang, C.; Zhou, J. J. Am. Chem. Soc. 2010, 132, 15176–15178. doi:10.1021/ja107858z |
| 13. | Chauhan, P.; Chimni, S. S. Asian J. Org. Chem. 2013, 2, 586–592. doi:10.1002/ajoc.201300093 |
| 6. | Guan, X.-Y.; Wei, Y.; Shi, M. Chem. – Eur. J. 2010, 16, 13617–13621. doi:10.1002/chem.201002240 |
| 7. | Qian, J.-Y.; Wang, C.-C.; Sha, F.; Wu, X.-Y. RSC Adv. 2012, 2, 6042–6048. doi:10.1039/C2RA20521A |
| 8. | Duan, Z.; Zhang, Z.; Qian, P.; Han, J.; Pan, Y. RSC Adv. 2013, 3, 10127–10130. doi:10.1039/C3RA41115J |
| 9. | Dong, Z.; Yan, C.; Gao, Y.; Dong, C.; Qiu, G.; Zhou, H.-B. Adv. Synth. Catal. 2015, 357, 2132–2142. doi:10.1002/adsc.201500110 |
| 10. | Zhao, X.; Li, T.-Z.; Qian, J.-Y.; Sha, F.; Wu, X.-Y. Org. Biomol. Chem. 2014, 12, 8072–8078. doi:10.1039/C4OB01358A |
| 11. | Zhong, F.; Chen, G.-Y.; Lu, Y. Org. Lett. 2011, 13, 82–85. doi:10.1021/ol102597s |
| 12. | Yoshida, Y.; Sako, M.; Kishi, K.; Sasai, H.; Hatakeyama, S.; Takizawa, S. Org. Biomol. Chem. 2015, 13, 9022–9028. doi:10.1039/C5OB00874C |
| 7. | Qian, J.-Y.; Wang, C.-C.; Sha, F.; Wu, X.-Y. RSC Adv. 2012, 2, 6042–6048. doi:10.1039/C2RA20521A |
| 5. | Liu, Y.-L.; Wang, B.-L.; Cao, J.-J.; Chen, L.; Zhang, Y.-X.; Wang, C.; Zhou, J. J. Am. Chem. Soc. 2010, 132, 15176–15178. doi:10.1021/ja107858z |
| 5. | Liu, Y.-L.; Wang, B.-L.; Cao, J.-J.; Chen, L.; Zhang, Y.-X.; Wang, C.; Zhou, J. J. Am. Chem. Soc. 2010, 132, 15176–15178. doi:10.1021/ja107858z |
| 20. | Peng, J.; Huang, X.; Cui, H.-L.; Chen, Y.-C. Org. Lett. 2010, 12, 4260–4263. doi:10.1021/ol101668z |
| 21. | Peng, J.; Huang, X.; Jiang, L.; Cui, H.-L.; Chen, Y.-C. Org. Lett. 2011, 13, 4584–4587. doi:10.1021/ol201776h |
| 22. | Peng, J.; Huang, X.; Zheng, P.-F.; Chen, Y.-C. Org. Lett. 2013, 15, 5534–5537. doi:10.1021/ol402694b |
| 23. | Peng, J.; Ran, G.-Y.; Du, W.; Chen, Y.-C. Org. Lett. 2015, 17, 4490–4493. doi:10.1021/acs.orglett.5b02157 |
| 13. | Chauhan, P.; Chimni, S. S. Asian J. Org. Chem. 2013, 2, 586–592. doi:10.1002/ajoc.201300093 |
| 18. | Liu, Y.-L.; Wang, X.; Zhao, Y.-L.; Zhu, F.; Zeng, X.-P.; Chen, L.; Wang, C.-H.; Zhao, X.-L.; Zhou, J. Angew. Chem. 2013, 125, 13980–13984. doi:10.1002/ange.201307250 |
| 19. | Yin, X.-P.; Zeng, X.-P.; Liu, Y.-L.; Liao, F.-M.; Yu, J.-S.; Zhou, F.; Zhou, J. Angew. Chem., Int. Ed. 2014, 53, 13740–13745. doi:10.1002/anie.201407677 |
| 13. | Chauhan, P.; Chimni, S. S. Asian J. Org. Chem. 2013, 2, 586–592. doi:10.1002/ajoc.201300093 |
| 15. | Wood, E. R.; Kuyper, L.; Petrov, K. G.; Hunter, R. N., III; Harris, P. A.; Lackey, K. Bioorg. Med. Chem. Lett. 2004, 14, 953–957. doi:10.1016/j.bmcl.2003.12.002 |
| 16. | Kritsanida, M.; Magiatis, P.; Skaltsounis, A.-L.; Peng, Y.; Li, P.; Wennogle, L. P. J. Nat. Prod. 2009, 72, 2199–2202. doi:10.1021/np9003905 |
| 17. | Rekulapally, S.; Jarapula, R.; Gangarapu, K.; Manda, S.; Vaidya, J. R. Med. Chem. Res. 2015, 24, 3412–3422. doi:10.1007/s00044-015-1390-0 |
| 11. | Zhong, F.; Chen, G.-Y.; Lu, Y. Org. Lett. 2011, 13, 82–85. doi:10.1021/ol102597s |
| 12. | Yoshida, Y.; Sako, M.; Kishi, K.; Sasai, H.; Hatakeyama, S.; Takizawa, S. Org. Biomol. Chem. 2015, 13, 9022–9028. doi:10.1039/C5OB00874C |
| 13. | Chauhan, P.; Chimni, S. S. Asian J. Org. Chem. 2013, 2, 586–592. doi:10.1002/ajoc.201300093 |
| 14. | Kumar, A.; Sharma, V.; Kaur, J.; Kumar, N.; Chimni, S. S. Org. Biomol. Chem. 2015, 13, 5629–5635. doi:10.1039/C5OB00182J |
| 24. | Nakamoto, Y.; Urabe, F.; Takahashi, K.; Ishihara, J.; Hatakeyama, S. Chem. – Eur. J. 2013, 19, 12653–12656. doi:10.1002/chem.201302665 |
| 25. | Peng, J.; Ran, G.-Y.; Du, W.; Chen, Y.-C. Synthesis 2015, 47, 2538–2544. doi:10.1055/s-0034-1378716 |
© 2016 He et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)