Abstract
Mycothiol is found in Gram-positive bacteria, where it helps in maintaining a reducing intracellular environment and it plays an important role in protecting the cell from toxic chemicals. The inhibition of the mycothiol biosynthesis is considered as a treatment for tuberculosis. Mycothiol contains an α-aminoglycoside, which is difficult to prepare stereoselectively by a conventional glycosylation reaction. In this study, mycothiol was synthesized by an anomerization reaction from an easily prepared β-aminoglycoside through endocyclic cleavage.

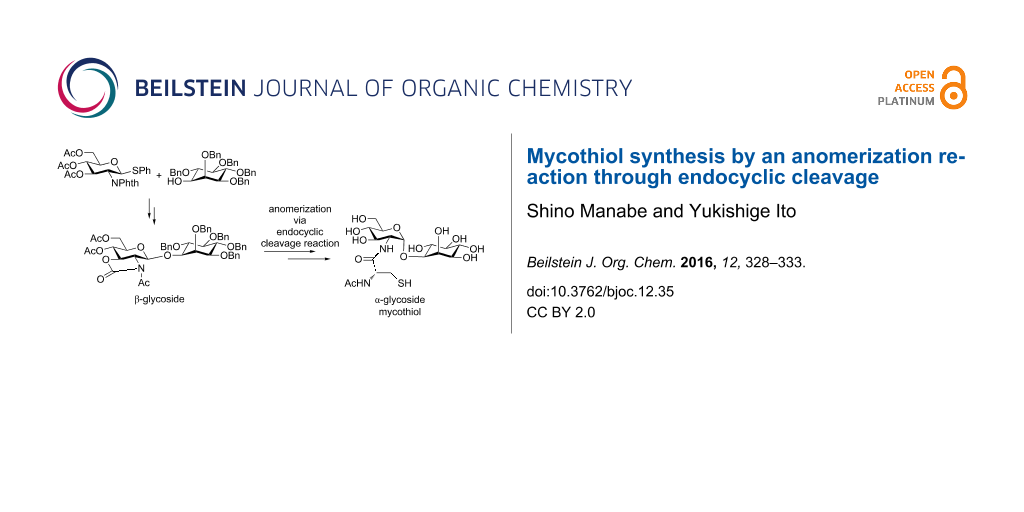
Graphical Abstract
Introduction
Tuberculosis is an infectious disease and has had a high death rate over the past few decades [1-4]. The occurrence of multiple-drug-resistant (MDR), extensive-drug-resistant (EDR), and totally drug-resistant (TDR) pathogens has increased the need for new drug candidates for treating tuberculosis.
Mycothiol (MSH) 1 is the main low-molecular-weight thiol found in most actinomycetes, including Mycobacteria and Streptomycetes [5-10]. It consists of an N-acetylcysteine, a D-glucosamine, and a D-myo-inositol moiety (Figure 1). D-Glucosamine is α-linked to D-myo-inositol at the 1-position, and N-acetylcysteine is linked to the amino group of D-glucosamine. The conformation of MSH has been investigated by NMR analyses and computational calculations [11,12]. Recently, N-acyl variants of MSH homologs, such as formyl, propanoyl, and succinoyl, have been reported [13-15].
![[1860-5397-12-35-1]](/bjoc/content/figures/1860-5397-12-35-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Gram-negative bacteria and most Eukaryotes utilize glutathione as a low-molecular-weight thiol for maintaining a reducing environment in the cytosol. Gram-positive bacteria including actinomycetes lack glutathione, instead, MSH is found as the major low-molecular-weight thiol. It is considered that MSH is required for maintaining a reducing intracellular environment in Gram-positive bacteria, similar to glutathione in eukaryotes and Gram-negative bacteria. MSH undergoes metal-catalyzed autoxidation more rapidly than glutathione [16]. The biosynthetic pathway of MSH has been well investigated; MSH is synthesized from 1-inositol phosphate and uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) in five steps [15]. It is used by mycobacteria for protection against foreign electrophilic agents such as oxidants, radicals, and drugs. In the detoxification pathway, MSH reacts with alkylating reagents and the resulting S-conjugates are subsequently cleaved at the amide bond by MSH S-conjugate amidase (Scheme 1) [4-10]. After cleavage, the N-acetylcysteine S-conjugate is transported out of the cell, while the inositol–glucosamine conjugate is recycled to afford MSH. MSH also plays an important role in the growth and survival of Mycobacterium tuberculosis. Because MSH-dependent pathways are not found in eukaryotes, the enzymes involved may be considered as novel antimicrobacterial targets, especially for tuberculosis, and several compounds with inhibitory activity have been synthesized [17,18]. In addition, another function of MSH was recently reported; it is involved in the biosynthesis of lincomycin A, a sulfur-containing lincosamide antibiotic [19].
![[1860-5397-12-35-i1]](/bjoc/content/inline/1860-5397-12-35-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Detoxification pathway mediated by MSH.
Scheme 1: Detoxification pathway mediated by MSH.
Due to the limited availability of MSH from M. smegmatis cell culture (<1.5 mg of MSH from 1 L culture) [20], the chemical synthesis of MSH is highly desired. Bewley et al. and Lee and Rosazza independently reported the synthesis of MSH and determined the absolute stereochemistry of the glucosamine and cysteine moieties [21,22]. Hung also reported the utility of the 2-azido-2-deoxyglycosyl donor and resolved inositol isomers in a recent MSH synthesis [23]. Knapp et al. reported an intramolecular aglycone delivery method in order to achieve complete α-stereoselectivity [24]. The Ni(4-F-PhCN)4(OTf)2-catalyzed synthesis of N-substituted benzylideneaminoglycosides has also been achieved to fabricate MSH [25]. The α-stereoselective formation of aminoglycoside is the crucial step in MSH synthesis. However, except for the intramolecular aglycone delivery method developed by Knapp et al., complete α-stereoselective glycosylation reactions were difficult in mycothiol synthesis. The complete α-stereoselective glycosylation reaction of aminoglycosides is still generally difficult at this moment [26-28].
Oscarson and our group recently demonstrated that reactions of pyranosides with N-acetyl 2,3-trans-carbamate groups exhibited complete anomerization from β-glycoside to α-glycoside in the presence of a weak Lewis acid through an endocyclic cleavage reaction [29-33]. We showed evidence of the endocyclic cleavage reaction by trapping linear cations through reduction, and intramolecular Friedel–Crafts reaction [29,30] (Scheme 2). In particular, the reaction of pyranosides bearing acetyl substituents on the carbamate groups showed complete anomerization [32]. We expected that anomerization via endocyclic cleavage would be useful for mycothiol synthesis.
![[1860-5397-12-35-i2]](/bjoc/content/inline/1860-5397-12-35-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Anomerization via endocyclic cleavage.
Scheme 2: Anomerization via endocyclic cleavage.
Results and Discussion
Based on the results of our previous study, we expected that an anomerization would be useful for the stereoselective synthesis of α-aminoglycosides, which is normally difficult by conventional glycosylation reactions. β-Glycoside 2, which is synthesized by assistance from the phthalimide group in the 2-position, was converted to α-glycoside 4, by introducing an N-acetyl 2,3-trans-carbamate group (Scheme 3) and by conducting an anomerization reaction.
![[1860-5397-12-35-i3]](/bjoc/content/inline/1860-5397-12-35-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Outline of mycothiol synthesis by anomerization.
Scheme 3: Outline of mycothiol synthesis by anomerization.
The glycosylation reaction of phthalimido-protected glucosamine thioglycoside 5 with inositol 6 [24] afforded β-linked pseudo-disaccharide 2 in 90% yield (Scheme 4). After removing the phthaloyl and acetyl groups by using ethylenediamine in dimethylformamide (DMF), a carbamate group was introduced using triphosgene in the presence of NaHCO3. Acetylation of both the hydroxy and carbamate groups was carried out using acetic anhydride in pyridine in the presence of 4-dimethylaminopyridine (DMAP). Finally, β-glycoside 3 was completely anomerized to the corresponding α-anomer 4 in quantitative yield in the presence of two equivalents of BF3·OEt2 in CH3CN [33] within 30 min at −30 °C.
![[1860-5397-12-35-i4]](/bjoc/content/inline/1860-5397-12-35-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of a pseudodisaccharide by an anomerization reaction.
Scheme 4: Synthesis of a pseudodisaccharide by an anomerization reaction.
With the α-linked inositol 4 in hand, the synthesis of MSH was completed as follows (Scheme 5): the carbamate and acetyl groups were removed by alkaline hydrolysis to give known compound 8 [24]; then, benzyl groups were removed by H2/Pd(OH)2/C in AcOH/dioxane/H2O. Although it was reported that the cysteine moiety was introduced by 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) [23,24], we found that purification of the product was rather difficult, especially for >50 mg-scale reactions. The reason for the low yield is probably the low solubility of the product in the reported solvent system during column chromatography (CHCl3/MeOH/AcOH), and azabenzotriazole from HATU was difficult to remove. Instead, ethyl (hydroxyimino)cyanoacetate (COMU) [34] coupling gave product 9 in 80% yield after reversed-phase column chromatography purification (H2O/MeOH). Finally, Boc removal and subsequent acetyl migration, as reported previously, gave mycothiol 1 [23,24].
![[1860-5397-12-35-i5]](/bjoc/content/inline/1860-5397-12-35-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Mycothiol synthesis from pseudo-disaccharide 4.
Scheme 5: Mycothiol synthesis from pseudo-disaccharide 4.
Conclusion
We demonstrated a MSH synthesis using an anomerization reaction through endocyclic cleavage in the presence of a weak Lewis acid. Murphy also reported the utility of the anomerization of glucuronic acid for preparing 1,2-cis-linked glycolipids [35-37]. Sulfated sugars are isomerized from pyranosides to furanosides [38]. The anomerization reaction would be a useful methodology to prepare 1,2-cis-glycosides such as heparin and glycosylphosphatidylinositol (GPI) anchors.
Supporting Information
| Supporting Information File 1: Experimental procedures, spectral data of new compounds, including 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 3.4 MB | Download |
Acknowledgements
S. M. received support through a Grant-in-Aid for Scientific Research (C) (Grant No. 24590041) from the Japan Society for Promotion of Science, Yamada Science Foundation, Takeda Science Foundation, and A-STEP from JST. S. M. thanks Dr. Kaori Otsuki, Dr. Masaya Usui, and Dr. Aya Abe of the Research Resource Center at the Brain Science Center, RIKEN for help with HRMS measurements, and Ms. Akemi Takahashi for technical assistance.
References
-
Zumla, A.; Nahid, P.; Cole, S. T. Nat. Rev. Drug Discovery 2013, 12, 388–404. doi:10.1038/nrd4001
Return to citation in text: [1] -
Almeida Da Silva, P. E.; Palomino, J. C. J. Antimicrob. Chemother. 2011, 66, 1417–1430. doi:10.1093/jac/dkr173
Return to citation in text: [1] -
Velayati, A. A.; Farnia, P.; Masjedi, M. R. Int. J. Clin. Exp. Med. 2013, 6, 307–309.
Return to citation in text: [1] -
Nilewar, S. S.; Kathiravan, M. K. Bioorg. Chem. 2014, 52, 62–68. doi:10.1016/j.bioorg.2013.11.004
Return to citation in text: [1] [2] -
Newton, G. L.; Fahey, R. C.; Cohen, G.; Aharonowitz, Y. J. Bacteriol. 1993, 175, 2734–2742.
Return to citation in text: [1] [2] -
Sakuda, S.; Zhou, Z.-Y.; Yamada, Y. Biosci., Biotechnol., Biochem. 1994, 58, 1347–1348. doi:10.1271/bbb.58.1347
Return to citation in text: [1] [2] -
Spies, H. S. C.; Steenkamp, D. J. Eur. J. Biochem. 1994, 224, 203–213. doi:10.1111/j.1432-1033.1994.tb20013.x
Return to citation in text: [1] [2] -
Newton, G. L.; Bewley, C. A.; Dwyer, T. J.; Horn, R.; Aharonowitz, Y.; Cohen, G.; Davies, J.; Faulkner, D. J.; Fahey, R. C. Eur. J. Biochem. 1995, 230, 821–825. doi:10.1111/j.1432-1033.1995.0821h.x
Return to citation in text: [1] [2] -
Newton, G. L.; Av-Gay, Y.; Fahey, R. C. Biochemistry 2000, 39, 10739–10746. doi:10.1021/bi000356n
Return to citation in text: [1] [2] -
Rawat, M.; Newton, G. L.; Ko, M.; Martinez, G. J.; Fahey, R. C.; Av-Gay, Y. Antimicrob. Agents Chemother. 2002, 46, 3348–3355. doi:10.1128/AAC.46.11.3348-3355.2002
Return to citation in text: [1] [2] -
Mahadevan, J.; Nicholas, G. M.; Bewley, C. A. J. Org. Chem. 2003, 68, 3380–3386. doi:10.1021/jo026872w
Return to citation in text: [1] -
Hand, C. E.; Auzanneau, F.-I.; Honek, J. F. Carbohydr. Res. 2006, 341, 1164–1173. doi:10.1016/j.carres.2006.03.020
Return to citation in text: [1] -
Newton, G. L.; Ta, P.; Fahey, R. C. J. Bacteriol. 2005, 187, 7309–7316. doi:10.1128/JB.187.21.7309-7316.2005
Return to citation in text: [1] -
Newton, G. L.; Jensen, P. R.; MacMillan, J. B.; Fenical, W.; Fahey, R. C. Arch. Microbiol. 2008, 190, 547–557. doi:10.1007/s00203-008-0405-3
Return to citation in text: [1] -
Newton, G. L.; Buchmeier, N.; Fahey, R. C. Microbiol. Mol. Biol. Rev. 2008, 72, 471–494. doi:10.1128/MMBR.00008-08
Return to citation in text: [1] [2] -
Held, K. D.; Biaglow, J. E. Radiat. Res. 1994, 139, 15–23. doi:10.2307/3578727
Return to citation in text: [1] -
Gammon, D. W.; Hunter, R.; Steenkamp, D. J.; Mudzunga, T. T. Bioorg. Med. Chem. Lett. 2003, 13, 2045–2049. doi:10.1016/S0960-894X(03)00157-4
Return to citation in text: [1] -
Knapp, S.; Amorelli, B.; Darout, E.; Ventocilla, C. C.; Goldman, L. M.; Huhn, R. A.; Minnihan, E. C. J. Carbohydr. Chem. 2005, 24, 103–130. doi:10.1081/CAR-200059965
Return to citation in text: [1] -
Zhao, Q.; Wang, M.; Xu, D.; Zhang, Q.; Liu, W. Nature 2015, 518, 115–119. doi:10.1038/nature14137
Return to citation in text: [1] -
Newton, G. L.; Arnold, K.; Price, M. S.; Sherrill, C.; Delcardayre, S. B.; Aharonowitz, Y.; Cohen, G.; Davis, J.; Fahey, R. C.; Davis, C. J. Bacteriol. 1996, 178, 1990–1995.
Return to citation in text: [1] -
Nicholas, G. M.; Kováč, P.; Bewley, C. A. J. Am. Chem. Soc. 2002, 124, 3492–3493. doi:10.1021/ja017891a
Return to citation in text: [1] -
Lee, S.; Rosazza, J. P. N. Org. Lett. 2004, 6, 365–368. doi:10.1021/ol0362008
Return to citation in text: [1] -
Chung, C.-C.; Zulueta, M. M. L.; Padiyar, L. T.; Hung, S.-C. Org. Lett. 2011, 13, 5496–5499. doi:10.1021/ol202218n
Return to citation in text: [1] [2] [3] -
Ajayi, K.; Thakur, V. V.; Lapo, R. C.; Knapp, S. Org. Lett. 2010, 12, 2630–2633. doi:10.1021/ol1008334
Return to citation in text: [1] [2] [3] [4] [5] -
McConnell, M. S.; Yu, F.; Nguyen, H. M. Chem. Commun. 2013, 49, 4313–4315. doi:10.1039/C2CC35823A
Return to citation in text: [1] -
Paulsen, H.; Kolář, Č.; Stenzel, W. Chem. Ber. 1978, 111, 2358–2369. doi:10.1002/cber.19781110630
Return to citation in text: [1] -
Lemieux, R. U.; Ratcliffe, R. M. Can. J. Chem. 1979, 57, 1244–1251. doi:10.1139/v79-203
Return to citation in text: [1] -
Ngoje, G.; Li, Z. Org. Biomol. Chem. 2013, 11, 1879–1886. doi:10.1039/c3ob26994a
And references cited therein.
Return to citation in text: [1] -
Manabe, S.; Ito, Y. Chem. Rec. 2014, 14, 502–515. doi:10.1002/tcr.201402004
Return to citation in text: [1] [2] -
Manabe, S.; Ishii, K.; Hashizume, D.; Koshino, H.; Ito, Y. Chem. – Eur. J. 2009, 15, 6894–6901. doi:10.1002/chem.200900064
Return to citation in text: [1] [2] -
Olsson, J. D. M.; Eriksson, L.; Lahmann, M.; Oscarson, S. J. Org. Chem. 2008, 73, 7181–7188. doi:10.1021/jo800971s
Return to citation in text: [1] -
Manabe, S.; Satoh, H.; Hutter, J.; Lüthi, H. P.; Laino, T.; Ito, Y. Chem. – Eur. J. 2014, 20, 124–132. doi:10.1002/chem.201303474
Return to citation in text: [1] [2] -
Manabe, S.; Ito, Y. Tetrahedron Lett. 2009, 50, 4827–4829. doi:10.1016/j.tetlet.2009.05.119
Return to citation in text: [1] [2] -
El-Faham, A.; Funosas, R. S.; Prohens, R.; Albericio, F. Chem. – Eur. J. 2009, 15, 9404–9416. doi:10.1002/chem.200900615
Return to citation in text: [1] -
Pilgrim, W.; Murphy, P. V. Org. Lett. 2009, 11, 939–942. doi:10.1021/ol802915h
Return to citation in text: [1] -
O’Reilly, C.; Murphy, P. V. Org. Lett. 2011, 13, 5168–5171. doi:10.1021/ol202042h
Return to citation in text: [1] -
Pilgrim, W.; O’Reilly, C.; Murphy, P. V. Molecules 2013, 18, 11198–11218. doi:10.3390/molecules180911198
Return to citation in text: [1] -
Krylov, V. B.; Argunov, D. A.; Vinnitskiy, D. Z.; Verkhnyatskaya, S. A.; Gerbst, A. G.; Ustyuzhanina, N. E.; Dmitrenok, A. S.; Huebner, J.; Holst, O.; Siebert, H.-C.; Nifantiev, N. E. Chem. – Eur. J. 2014, 20, 16516–16522. doi:10.1002/chem.201405083
Return to citation in text: [1]
| 1. | Zumla, A.; Nahid, P.; Cole, S. T. Nat. Rev. Drug Discovery 2013, 12, 388–404. doi:10.1038/nrd4001 |
| 2. | Almeida Da Silva, P. E.; Palomino, J. C. J. Antimicrob. Chemother. 2011, 66, 1417–1430. doi:10.1093/jac/dkr173 |
| 3. | Velayati, A. A.; Farnia, P.; Masjedi, M. R. Int. J. Clin. Exp. Med. 2013, 6, 307–309. |
| 4. | Nilewar, S. S.; Kathiravan, M. K. Bioorg. Chem. 2014, 52, 62–68. doi:10.1016/j.bioorg.2013.11.004 |
| 16. | Held, K. D.; Biaglow, J. E. Radiat. Res. 1994, 139, 15–23. doi:10.2307/3578727 |
| 26. | Paulsen, H.; Kolář, Č.; Stenzel, W. Chem. Ber. 1978, 111, 2358–2369. doi:10.1002/cber.19781110630 |
| 27. | Lemieux, R. U.; Ratcliffe, R. M. Can. J. Chem. 1979, 57, 1244–1251. doi:10.1139/v79-203 |
| 28. |
Ngoje, G.; Li, Z. Org. Biomol. Chem. 2013, 11, 1879–1886. doi:10.1039/c3ob26994a
And references cited therein. |
| 13. | Newton, G. L.; Ta, P.; Fahey, R. C. J. Bacteriol. 2005, 187, 7309–7316. doi:10.1128/JB.187.21.7309-7316.2005 |
| 14. | Newton, G. L.; Jensen, P. R.; MacMillan, J. B.; Fenical, W.; Fahey, R. C. Arch. Microbiol. 2008, 190, 547–557. doi:10.1007/s00203-008-0405-3 |
| 15. | Newton, G. L.; Buchmeier, N.; Fahey, R. C. Microbiol. Mol. Biol. Rev. 2008, 72, 471–494. doi:10.1128/MMBR.00008-08 |
| 29. | Manabe, S.; Ito, Y. Chem. Rec. 2014, 14, 502–515. doi:10.1002/tcr.201402004 |
| 30. | Manabe, S.; Ishii, K.; Hashizume, D.; Koshino, H.; Ito, Y. Chem. – Eur. J. 2009, 15, 6894–6901. doi:10.1002/chem.200900064 |
| 31. | Olsson, J. D. M.; Eriksson, L.; Lahmann, M.; Oscarson, S. J. Org. Chem. 2008, 73, 7181–7188. doi:10.1021/jo800971s |
| 32. | Manabe, S.; Satoh, H.; Hutter, J.; Lüthi, H. P.; Laino, T.; Ito, Y. Chem. – Eur. J. 2014, 20, 124–132. doi:10.1002/chem.201303474 |
| 33. | Manabe, S.; Ito, Y. Tetrahedron Lett. 2009, 50, 4827–4829. doi:10.1016/j.tetlet.2009.05.119 |
| 11. | Mahadevan, J.; Nicholas, G. M.; Bewley, C. A. J. Org. Chem. 2003, 68, 3380–3386. doi:10.1021/jo026872w |
| 12. | Hand, C. E.; Auzanneau, F.-I.; Honek, J. F. Carbohydr. Res. 2006, 341, 1164–1173. doi:10.1016/j.carres.2006.03.020 |
| 24. | Ajayi, K.; Thakur, V. V.; Lapo, R. C.; Knapp, S. Org. Lett. 2010, 12, 2630–2633. doi:10.1021/ol1008334 |
| 5. | Newton, G. L.; Fahey, R. C.; Cohen, G.; Aharonowitz, Y. J. Bacteriol. 1993, 175, 2734–2742. |
| 6. | Sakuda, S.; Zhou, Z.-Y.; Yamada, Y. Biosci., Biotechnol., Biochem. 1994, 58, 1347–1348. doi:10.1271/bbb.58.1347 |
| 7. | Spies, H. S. C.; Steenkamp, D. J. Eur. J. Biochem. 1994, 224, 203–213. doi:10.1111/j.1432-1033.1994.tb20013.x |
| 8. | Newton, G. L.; Bewley, C. A.; Dwyer, T. J.; Horn, R.; Aharonowitz, Y.; Cohen, G.; Davies, J.; Faulkner, D. J.; Fahey, R. C. Eur. J. Biochem. 1995, 230, 821–825. doi:10.1111/j.1432-1033.1995.0821h.x |
| 9. | Newton, G. L.; Av-Gay, Y.; Fahey, R. C. Biochemistry 2000, 39, 10739–10746. doi:10.1021/bi000356n |
| 10. | Rawat, M.; Newton, G. L.; Ko, M.; Martinez, G. J.; Fahey, R. C.; Av-Gay, Y. Antimicrob. Agents Chemother. 2002, 46, 3348–3355. doi:10.1128/AAC.46.11.3348-3355.2002 |
| 25. | McConnell, M. S.; Yu, F.; Nguyen, H. M. Chem. Commun. 2013, 49, 4313–4315. doi:10.1039/C2CC35823A |
| 19. | Zhao, Q.; Wang, M.; Xu, D.; Zhang, Q.; Liu, W. Nature 2015, 518, 115–119. doi:10.1038/nature14137 |
| 21. | Nicholas, G. M.; Kováč, P.; Bewley, C. A. J. Am. Chem. Soc. 2002, 124, 3492–3493. doi:10.1021/ja017891a |
| 22. | Lee, S.; Rosazza, J. P. N. Org. Lett. 2004, 6, 365–368. doi:10.1021/ol0362008 |
| 17. | Gammon, D. W.; Hunter, R.; Steenkamp, D. J.; Mudzunga, T. T. Bioorg. Med. Chem. Lett. 2003, 13, 2045–2049. doi:10.1016/S0960-894X(03)00157-4 |
| 18. | Knapp, S.; Amorelli, B.; Darout, E.; Ventocilla, C. C.; Goldman, L. M.; Huhn, R. A.; Minnihan, E. C. J. Carbohydr. Chem. 2005, 24, 103–130. doi:10.1081/CAR-200059965 |
| 23. | Chung, C.-C.; Zulueta, M. M. L.; Padiyar, L. T.; Hung, S.-C. Org. Lett. 2011, 13, 5496–5499. doi:10.1021/ol202218n |
| 4. | Nilewar, S. S.; Kathiravan, M. K. Bioorg. Chem. 2014, 52, 62–68. doi:10.1016/j.bioorg.2013.11.004 |
| 5. | Newton, G. L.; Fahey, R. C.; Cohen, G.; Aharonowitz, Y. J. Bacteriol. 1993, 175, 2734–2742. |
| 6. | Sakuda, S.; Zhou, Z.-Y.; Yamada, Y. Biosci., Biotechnol., Biochem. 1994, 58, 1347–1348. doi:10.1271/bbb.58.1347 |
| 7. | Spies, H. S. C.; Steenkamp, D. J. Eur. J. Biochem. 1994, 224, 203–213. doi:10.1111/j.1432-1033.1994.tb20013.x |
| 8. | Newton, G. L.; Bewley, C. A.; Dwyer, T. J.; Horn, R.; Aharonowitz, Y.; Cohen, G.; Davies, J.; Faulkner, D. J.; Fahey, R. C. Eur. J. Biochem. 1995, 230, 821–825. doi:10.1111/j.1432-1033.1995.0821h.x |
| 9. | Newton, G. L.; Av-Gay, Y.; Fahey, R. C. Biochemistry 2000, 39, 10739–10746. doi:10.1021/bi000356n |
| 10. | Rawat, M.; Newton, G. L.; Ko, M.; Martinez, G. J.; Fahey, R. C.; Av-Gay, Y. Antimicrob. Agents Chemother. 2002, 46, 3348–3355. doi:10.1128/AAC.46.11.3348-3355.2002 |
| 15. | Newton, G. L.; Buchmeier, N.; Fahey, R. C. Microbiol. Mol. Biol. Rev. 2008, 72, 471–494. doi:10.1128/MMBR.00008-08 |
| 20. | Newton, G. L.; Arnold, K.; Price, M. S.; Sherrill, C.; Delcardayre, S. B.; Aharonowitz, Y.; Cohen, G.; Davis, J.; Fahey, R. C.; Davis, C. J. Bacteriol. 1996, 178, 1990–1995. |
| 24. | Ajayi, K.; Thakur, V. V.; Lapo, R. C.; Knapp, S. Org. Lett. 2010, 12, 2630–2633. doi:10.1021/ol1008334 |
| 29. | Manabe, S.; Ito, Y. Chem. Rec. 2014, 14, 502–515. doi:10.1002/tcr.201402004 |
| 30. | Manabe, S.; Ishii, K.; Hashizume, D.; Koshino, H.; Ito, Y. Chem. – Eur. J. 2009, 15, 6894–6901. doi:10.1002/chem.200900064 |
| 32. | Manabe, S.; Satoh, H.; Hutter, J.; Lüthi, H. P.; Laino, T.; Ito, Y. Chem. – Eur. J. 2014, 20, 124–132. doi:10.1002/chem.201303474 |
| 38. | Krylov, V. B.; Argunov, D. A.; Vinnitskiy, D. Z.; Verkhnyatskaya, S. A.; Gerbst, A. G.; Ustyuzhanina, N. E.; Dmitrenok, A. S.; Huebner, J.; Holst, O.; Siebert, H.-C.; Nifantiev, N. E. Chem. – Eur. J. 2014, 20, 16516–16522. doi:10.1002/chem.201405083 |
| 23. | Chung, C.-C.; Zulueta, M. M. L.; Padiyar, L. T.; Hung, S.-C. Org. Lett. 2011, 13, 5496–5499. doi:10.1021/ol202218n |
| 24. | Ajayi, K.; Thakur, V. V.; Lapo, R. C.; Knapp, S. Org. Lett. 2010, 12, 2630–2633. doi:10.1021/ol1008334 |
| 35. | Pilgrim, W.; Murphy, P. V. Org. Lett. 2009, 11, 939–942. doi:10.1021/ol802915h |
| 36. | O’Reilly, C.; Murphy, P. V. Org. Lett. 2011, 13, 5168–5171. doi:10.1021/ol202042h |
| 37. | Pilgrim, W.; O’Reilly, C.; Murphy, P. V. Molecules 2013, 18, 11198–11218. doi:10.3390/molecules180911198 |
| 23. | Chung, C.-C.; Zulueta, M. M. L.; Padiyar, L. T.; Hung, S.-C. Org. Lett. 2011, 13, 5496–5499. doi:10.1021/ol202218n |
| 24. | Ajayi, K.; Thakur, V. V.; Lapo, R. C.; Knapp, S. Org. Lett. 2010, 12, 2630–2633. doi:10.1021/ol1008334 |
| 34. | El-Faham, A.; Funosas, R. S.; Prohens, R.; Albericio, F. Chem. – Eur. J. 2009, 15, 9404–9416. doi:10.1002/chem.200900615 |
| 33. | Manabe, S.; Ito, Y. Tetrahedron Lett. 2009, 50, 4827–4829. doi:10.1016/j.tetlet.2009.05.119 |
| 24. | Ajayi, K.; Thakur, V. V.; Lapo, R. C.; Knapp, S. Org. Lett. 2010, 12, 2630–2633. doi:10.1021/ol1008334 |
© 2016 Manabe and Ito; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)