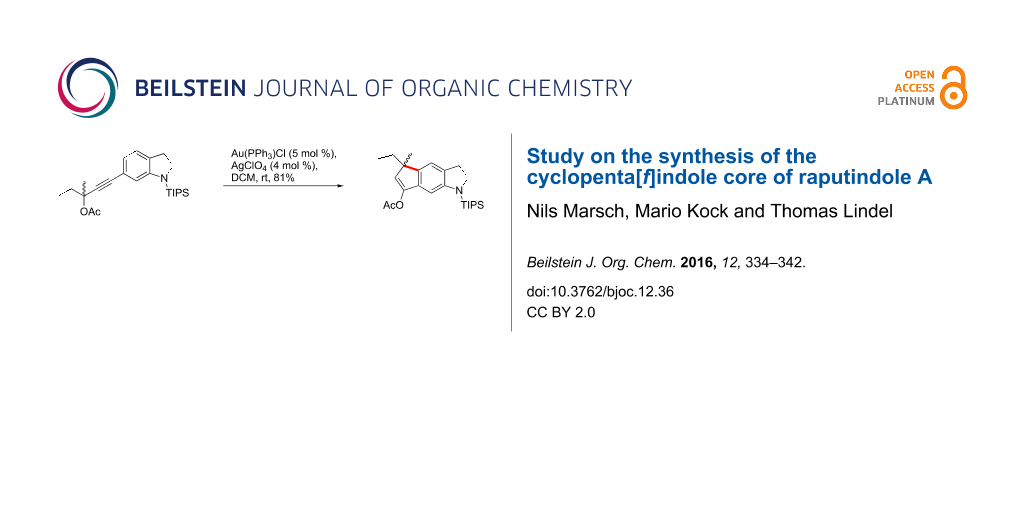
Abstract
The raputindoles from the rutaceous tree Raputia simulans share a cyclopenta[f]indole partial structure the synthesis of which is subject of this investigation. An efficient route to a series of 1,5-di(indol-6-yl)pentenones was developed via Mo/Au-catalyzed Meyer–Schuster rearrangement of tertiary propargylic alcohol precursors. However, none of the enones underwent the desired Nazarov cyclization to a cyclopenta[f]indole. More suitable were 6-hydroxyallylated indolines which gave good yields of cyclopenta[f]indolines after treatment with SnCl4, as soon as sterically demanding β-cyclocitral adducts were reacted. Most successful were Pt(II) and Au(I)-catalyzed cyclizations of N-TIPS-protected indolin-6-yl-substituted propargylacetates which provided the hydrogenated tricyclic cyclopenta[f]indole core system in high yield.

Graphical Abstract
Introduction
The raputindoles (1, raputindole A, Figure 1) from the rutaceous tree Raputia simulans Kallunki constitute a unique group of terpenoid bisindole natural products [1] sharing a linear cyclopenta[f]indole tricyclic partial structure. The cyclopenta[f]indole system also occurs as partial structure of the nodulisporic acids [2], the shearinines [3-6] and janthitrems [7-10]. While work has been done on the total syntheses of herbindoles [11] and trikentrines [12], which contain angular cyclopenta[g]indole partial structures [13-29], there is only little known on the assembly of linear cyclopenta[f]indole systems. Both existing approaches rely on the anellation of the pyrrole part to indene-based starting materials [30-33].
![[1860-5397-12-36-1]](/bjoc/content/figures/1860-5397-12-36-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Bisindole alkaloid raputindole A (1) from the Amazonian tree Raputia simulans.
Figure 1: Bisindole alkaloid raputindole A (1) from the Amazonian tree Raputia simulans.
In this paper we discuss our experiences with the assembly of cyclopenta[f]indole and -indoline systems (A, Scheme 1). A bond between the indole 5-position (4a in the resulting tricycle) and the quaternary center was to be formed. Ideally, anellation of a cyclopentane would be possible at an indole with an intact enamine partial structure (B, Scheme 1). Such an approach seemed possible, because we had already cyclized 6-prenoylindole to a mixture of cyclopenta[f]- and -[g]indoles in a Nazarov-type reaction [34]. By installation of a triflyloxy group in the indole 5-position, Pd-catalyzed cyclization also would become possible. As an alternative to the cyclization of enones, Lewis acid-induced cyclizations of allylic alcohols could afford the desired cyclopenta[f]indole system (C, D). Here, it was unclear whether the indole enamine section would be tolerated or have to be reduced prior to cyclization. Finally, propargyl alcohol derivatives (E) were to be investigated as substrates of Pt(II) and Au(I)-catalyzed reactions. All of the investigated building blocks were to be obtained from 6-iodoindole (2), which was synthesized via the Batcho–Leimgruber route and purified by sublimation [34].
![[1860-5397-12-36-i1]](/bjoc/content/inline/1860-5397-12-36-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Investigated synthetic precursors B–E of the cyclopenta[f]indole moiety (A) of raputindole A (1), all to be assembled from 6-iodoindole (2).
Scheme 1: Investigated synthetic precursors B–E of the cyclopenta[f]indole moiety (A) of raputindole A (1), a...
Results and Discussion
Di(indol-6-yl)pentenones. Regarding the investigation of 6-acryloylindoles, we aimed at the synthesis of methyl-branched di(indol-6-yl)pentenones from the beginning, which already included the second indole moiety of raputindole A (1). Boc protection of 6-iodoindole (2) [35], Sonogashira reaction of 3 with TMS-acetylene, and desilylation gave the N-protected alkynylindole 4 in excellent yield (Scheme 2). Boc-iodoindole 3 was also the precursor of the coupling partner, ketone 6, which was synthesized via Heck reaction with but-3-en-2-ol (5, 89%) in the presence of LiCl, inspired by a procedure by Camp and coworkers [36].
![[1860-5397-12-36-i2]](/bjoc/content/inline/1860-5397-12-36-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: 6-Iodoindole (2) serves twice as starting material towards indole-6-yl-substituted enone 8, obtained via Au-catalyzed Meyer–Schuster rearrangement.
Scheme 2: 6-Iodoindole (2) serves twice as starting material towards indole-6-yl-substituted enone 8, obtaine...
Propargyl bisindole 7 was obtained after conversion of 4 (2 equiv) to the magnesium acetylide (iPrMgCl in THF) and Grignard reaction with ketone 6. Due to the presence of Boc-protected indole nitrogens an acid-catalyzed Meyer–Schuster rearrangement was dismissed. Instead, a transition metal-catalyzed rearrangement employing 1 mol % of MoO2(acac)2/[Au(PPh3)Cl]/AgOTf [37] afforded the α,β-unsaturated ketone 8 as a 2:1 mixture of E/Z isomers (86%), which could not be separated by HPLC due to a reisomerization upon concentration of the fractions. When bisindole 8 was subjected to AlCl3, no cyclization product was observed and only deprotection occurred. This was somewhat surprising, because treatment of 6-prenoylindole with AlCl3 in 1,2-dichlorobenzene at 150 °C had induced Nazarov cyclization affording a mixture of regioisomeric cyclopenta[f]- and -[g]indolones [34]. Kern and coworkers had obtained an indanone under the same conditions [38]. Other attempts to cyclize 8 also failed.
We turned to Pd-catalyzed reductive cyclization. As precursor, a 5-triflyloxyindole was preferred over a 5-bromoindole, because 2-iodo-5-methyl-4-nitrophenol (11, Scheme 3), to be used in the Batcho–Leimgruber protocol, appeared to be more readily accessible than 1-bromo-2-iodo-5-methyl-4-nitrobenzene. Moreover, aryl triflates have been used in intramolecular cyclization reactions with α,β-unsaturated ketones to obtain indanones and dihydronaphthones [39,40]. Aminophenol 9 was converted to iodophenol 10 in good yield through a Sandmeyer reaction (Scheme 3) [41]. Various nitration conditions were tested, yet only the use of concentrated HNO3 in CH2Cl2, as reported by Chancellor and coworkers [42], gave nitrophenol 11 in a reasonable yield (45%). The Batcho–Leimgruber protocol failed for nitrophenol 11 [43]. However, after O-benzylation of 11 to 12, Boc-indole 15 was obtained in the very good yield of 91%. Coupling with TMS-acetylene was followed by desilylation to obtain the terminal alkyne 18 (97%). Magnesiation (iPrMgCl, THF) and reaction with ketone 6 afforded the benzyloxy-substituted bisindole 21. Transition metal-catalyzed Meyer–Schuster rearrangement afforded 24 (56%, E/Z mixture as above) albeit the catalyst loading had to be increased from 1 to 10 mol %, when compared to the synthesis of 8 (Scheme 2). We also observed elimination of water from 21. However, it proved to be impossible to debenzylate 24, which would have been necessary to access the corresponding triflate. For instance, we treated 24 with BCl3 in pentamethylbenzene/DCM which was used by Tokuyama and coworkers for benzyl ether cleavage in the presence of Boc-protected amines when studying the late stages of the total synthesis of the trisindole alkaloid yatakemycin [44].
![[1860-5397-12-36-i3]](/bjoc/content/inline/1860-5397-12-36-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Assembly of 5-oxygenated bisindolylpentenones. DMB: 3,4-dimethoxybenzyl, DMFDMA: N,N-dimethylformaldehyde dimethyl acetal.
Scheme 3: Assembly of 5-oxygenated bisindolylpentenones. DMB: 3,4-dimethoxybenzyl, DMFDMA: N,N-dimethylformal...
As an alternative, the 3,4-dimethoxybenzyl (DMB) protecting group was installed (13). We favored the DMB over the related PMB group, because the oxidation potential of DMB ethers is lower and the cleavage was expected to be more facile [45]. Indole synthesis from DMB-protected 13 and subsequent Boc protection afforded 16 (58%). Alkynylation and desilylation of 16 to 19 and coupling of 19 with ketone 6 proceeded smoothly providing the DMB-protected bisindole 22 in 18% overall yield over seven steps (Scheme 3). However, in the presence of the DMB group, the Meyer–Schuster rearrangement of alkyne 22 to the envisaged ketone 25 failed. Mixtures of elimination products dominated along with the cleavage of the DMB ether. Meyer–Schuster products could also be observed, but in small amounts and not as pure compounds.
It is worth mentioning that treatment of 22 with DDQ led to removal of the DMB group, affording the major product 27 (23%) exhibiting a keto group in the benzylic indole position (Scheme 4). This transformation might become useful in a future total synthesis of raputindole A (1) as a reduction–elimination sequence of the benzylic ketone could be used to introduce the olefinic double bond.
![[1860-5397-12-36-i4]](/bjoc/content/inline/1860-5397-12-36-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Benzylic oxidation as side reaction of DMB removal.
Scheme 4: Benzylic oxidation as side reaction of DMB removal.
Since neither from 22 nor from 24 the indole-5-OH group could be liberated, we turned back to the originally dismissed idea of installing the triflate group prior to indole assembly. There are no 5-triflyloxy-6-iodoindoles described in the literature. First, iodonitrophenol 11 was converted to triflate 14 in 96% yield. Subsequent Batcho–Leimgruber synthesis afforded the 6-iodo-5-triflyloxyindole, which was Boc-protected (17, 39%, three steps, Scheme 3). Sonogashira coupling occurred preferably at the iodinated 6-position affording 6-alkynylindole 20 after desilylation (56%). The reaction with ketone 6 after magnesiation led to propargylic alcohol 23 (50%). Interestingly, the Meyer–Schuster rearrangement of triflated alkyne 23 to ketone 26 proceeded in the highest yield of all our Meyer–Schuster rearrangements. With triflated α,β-unsaturated ketone 26 in hand, we attempted reductive Heck cyclizations to the raputindole core structure. For instance, we employed Pd(dba)2 (12 mol %), QPhos (24 mol %), and NEt3 (1.20 equiv) in DMF at 100 °C. Unfortunately, all reactions gave intractably complex mixtures. 1H and 19F NMR analysis suggested that cleavage of at least one Boc-protecting group had occurred. Additionally, the triflate was still visible in the 19F NMR spectrum, thus indicating that oxidative addition to the Pd catalyst had not taken place.
At this point, we abandoned our attempts of assembling the cyclopenta[f]indole unit of raputindole A (1) starting from Boc-protected indoles. None of the investigated bisindolylpentenones 8, 24, or 26 could be cyclized to a cyclopentaindole. At least, we learned how to synthesize the open-chain molecules and also their propargylic precursors.
Cyclization of allyl cations. Besides Nazarov and reductive Pd-catalyzed cyclizations there was the possibility of generating an allyl cation which would have to undergo cyclization to the cyclopenta[f]indole. There are not many examples of cyclopentene anellation by cyclization of aryl-substituted allyl cations. High yields were reported by Alvarez-Manzaneda et al. in the course of their total synthesis of taiwaniaquinone H. They induced the cyclization by treatment of arylvinylcarbinols with the mild Lewis acid SnCl4, which were synthesized by hydroxyalkylation with β-cyclocitral [46].
Hydroxyalkylation of Boc-protected 6-iodoindole (3) with 6-β-cyclocitral (30) was possible after iodine/magnesium exchange, affording adduct 31 (Scheme 5). However, treatment of 31 with SnCl4 in DCM did not afford any defined product. This changed after replacement of the Boc by a methyl group. The 6-hydroxyalkylated N-methylindole 32 (64%) was prepared from 6-iodo-N-methylindole (28) with β-cyclocitral (30). The subsequent treatment of 32 with SnCl4 in DCM afforded a 1:2 mixture of regioisomeric indeno[1,2-f]indole 34 (11%) and indeno[2,1-g]indole 35 (21%).
![[1860-5397-12-36-i5]](/bjoc/content/inline/1860-5397-12-36-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Hydroxyalkylation of N-protected indoles with β-cyclocitral and SnCl4-induced cyclization.
Scheme 5: Hydroxyalkylation of N-protected indoles with β-cyclocitral and SnCl4-induced cyclization.
For the corresponding N-TIPS-protected indole 33, obtained from 29, yields of tetracyclic products 36 (7%) and 37 (2%) were very low and we observed the loss of the TIPS protecting group. Noteworthy, we did not detect regioisomeric indeno[2,1-g]indole products when starting from 33, which points at a shielding effect of the TIPS group towards the indole 7-position.
Since the SnCl4-mediated cyclization yields with indoles were much lower than those obtained with benzene derivatives, we investigated the behavior of indolines which lack the reactive enamine moiety. We also abandoned the use of Boc-protecting groups. Reduction of 6-iodoindole with NaBH3CN in HOAc afforded 6-iodoindoline (38, 90%) [47], which was subsequently TIPS-protected (39, Scheme 6).
![[1860-5397-12-36-i6]](/bjoc/content/inline/1860-5397-12-36-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Behavior of indolines after SnCl4-induced generation of allyl cations.
Scheme 6: Behavior of indolines after SnCl4-induced generation of allyl cations.
Hydroxyalkylation of 39 with β-cyclocitral (30) gave cyclization precursor 40 (68%). We were pleased to find that this time the SnCl4-induced cyclization afforded the desilylated tetracyclic indeno[1,2-f]indoline 41 in high yield (82%). We did not detect any regioisomer, probably because the TIPS group was still in place in the regioselecting step. In order to obtain a tricyclic cyclopentaindoline, the propargylic alcohol 43 (96%) was synthesized by Sonogashira coupling of N-TIPS-6-iodoindoline (39) and 42. Conversion of 43 to the (Z)-allylic alcohol 44 by modified (K2CO3) Lindlar hydrogenation (47%) followed. Treatment of 44 with SnCl4 in DCM afforded cyclopenta[f]indoline 45, albeit in the rather disappointing yield of 11%.
A reason for the much better yield of β-cyclocitral adduct 41 when compared to 45 may be a conformational restriction of the allyl cation, caused by the geminal methyl groups of 40. The intermediate is probably kept in the cisoid conformation required for the cyclization. Thus, switching from indole to indoline and changing the N-protecting group significantly improved the yield for the cyclocitral adduct, but this still does not appear to be sufficient for a strategy towards raputindole A (1).
Cyclization of propargylacetates. Key progress came when applying platinum and gold chemistry to propargylacetates 46 and 47 (Scheme 7), which were obtained by Sonogashira alkynylations of N-TIPS-6-iodoindoline (39), followed by acetylation (for experimental procedures, see Supporting Information File 1). Xuegong She and coworkers had obtained indanone derivatives from arylpropargylic esters in a Pt(II)-catalyzed reaction for which they propose a formal rearrangement of the acetoxy group, followed by cyclization [48]. When we treated 46 and 47 with PtI2 (10 mol %) in a CO atmosphere at elevated temperature (PhMe, 80 °C) we indeed obtained the tricyclic cyclopentanones 48 and 49, respectively. Yields were moderate in both cases (33% and 29%), but already better than in the case of the SnCl4-induced cyclization of (Z)-allylic alcohol 44 (Scheme 6). We only isolated the cyclopentanones, formed from the corresponding cyclopentenyl acetates. Presumably, the propargylacetate first undergoes a [3,3]-sigmatropic shift to the allenyl acetate, followed by a Pt(II)-catalyzed cyclization to the tricycle.
![[1860-5397-12-36-i7]](/bjoc/content/inline/1860-5397-12-36-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Pt(II) and Au(I)-catalyzed cyclizations of propargylacetates 46 and 47 afforded cyclopenta[f]indolinones 48 and 49, and tricyclic cyclopentenylacetates 50 and 51.
Scheme 7: Pt(II) and Au(I)-catalyzed cyclizations of propargylacetates 46 and 47 afforded cyclopenta[f]indoli...
When propargylacetates 46 and 47 were treated with catalytic amounts of Au(PPh3)Cl/AgBF4 (DCM, rt, dark), cyclopenta[f]indolines 50 and 51 were isolated in even better yields. It proved to be beneficial to filter the catalyst solution through a pad of Celite prior to addition of the starting material to remove AgCl. In that way we reached an 81% yield of tricycle 50. The acetoxy group again formally underwent a 1,3-shift to the benzylic position, which may also be based on a [3,3]-sigmatropic rearrangement. Alternatively, for a similar 1,3-shift, Nolan and coworkers proposed two sequential 1,2-shifts to occur after π-complexation of the triple bond by Au(I) [49]. Methanolysis (NaOMe/MeOH) of cyclopentenylacetate 50 afforded cyclopenta[f]indolinone 48 (96%).
Conclusion
It was our goal to explore how to efficiently assemble the cyclopenta[f]indole section present in the natural product raputindole A (1). As long as the cyclopentane ring would not be installed, it was indeed possible to work with 2,3-unsubstituted, N-Boc-protected indoles. In particular, the Mo/Au-catalyzed Meyer–Schuster rearrangement of propargylalcohols 7, 21, and 23 worked nicely, even in the presence of a triflyloxy group. However, all attempts of cyclization failed with the Boc-protected bisindoles. Two modifications changed things for the better regarding the cyclization: replacement of the Boc- by a non-coordinating TIPS-protecting group and the use of indolines instead of sensitive indoles. Cyclopentane anellation by SnCl4-induced cyclization of phenylvinylcarbinols became possible, at least for the sterically congested β-cyclocitral adduct 40 of N-TIPS-indoline. The less sterically demanding substrate 44 gave lower yields. Experiments employing Au(I) and Pt(II) catalysts point at how to continue, since tricycles 48, 49, and 50 were obtained in good yields. We will now investigate the synthesis and Au(I) and Pt(II)-catalyzed cyclizations of TIPS-protected bisindolines, following a modified retrosynthesis of raputindole A (1).
Supporting Information
| Supporting Information File 1: Experimental procedures and NMR spectra. | ||
| Format: PDF | Size: 5.3 MB | Download |
References
-
Vougogiannopoulou, K.; Fokialakis, N.; Aligiannis, N.; Cantrell, C.; Skaltsounis, A.-L. Org. Lett. 2010, 12, 1908–1911. doi:10.1021/ol100584w
Return to citation in text: [1] -
Ondeyka, J. G.; Helms, G. L.; Hensens, O. D.; Goetz, M. A.; Zink, D. L.; Tsipouras, A.; Shoop, W. L.; Slayton, L.; Dombrowski, A. W.; Polishook, J. D.; Ostlind, D. A.; Tsou, N. N.; Ball, R. G.; Singh, S. B. J. Am. Chem. Soc. 1997, 119, 8809–8816. doi:10.1021/ja971664k
Return to citation in text: [1] -
Belofsky, G. N.; Gloer, J. B.; Wicklow, D. T.; Dowd, P. F. Tetrahedron 1995, 51, 3959–3968. doi:10.1016/0040-4020(95)00138-X
Return to citation in text: [1] -
Xu, M.; Gessner, G.; Groth, I.; Lange, C.; Christner, A.; Bruhn, T.; Deng, Z.; Li, X.; Heinemann, S. H.; Grabley, S.; Bringmann, G.; Sattler, I.; Lin, W. Tetrahedron 2007, 63, 435–444. doi:10.1016/j.tet.2006.10.050
Return to citation in text: [1] -
Smetanina, O. F.; Kalinovsky, A. I.; Khudyakova, Y. V.; Pivkin, M. V.; Dmitrenok, P. S.; Fedorov, S. N.; Ji, H.; Kwak, J.-Y.; Kuznetsova, T. A. J. Nat. Prod. 2007, 70, 906–909. doi:10.1021/np060396d
Return to citation in text: [1] -
Gallagher, R. T.; Latch, G. C.; Keogh, R. G. Appl. Environ. Microbiol. 1980, 39, 272–273.
Return to citation in text: [1] -
Lauren, D. R.; Gallagher, R. T. J. Chromatogr. 1982, 248, 150–154. doi:10.1016/S0021-9673(00)83747-4
Return to citation in text: [1] -
de Jesus, A. E.; Steyn, P. S.; van Heerden, F. R.; Vleggaar, R. J. Chem. Soc., Perkin Trans. 1 1984, 697–701. doi:10.1039/p19840000697
Return to citation in text: [1] -
Wilkins, A. L.; Miles, C. O.; Ede, R. M.; Gallagher, R. T.; Munday, S. C. J. Agric. Food Chem. 1992, 40, 1307–1309. doi:10.1021/jf00020a002
Return to citation in text: [1] -
Herb, R.; Carroll, A. R.; Yoshida, W. Y.; Scheuer, P. J.; Paul, V. J. Tetrahedron 1990, 46, 3089–3092. doi:10.1016/S0040-4020(01)88399-X
Return to citation in text: [1] -
Capon, R. J.; Macleod, J. K.; Scammels, P. J. Tetrahedron 1986, 42, 6545–6550. doi:10.1016/S0040-4020(01)88117-5
Return to citation in text: [1] -
Muratake, H.; Mikawa, A.; Natsume, M. Tetrahedron Lett. 1992, 33, 4595–4598. doi:10.1016/S0040-4039(00)61322-9
Return to citation in text: [1] -
Macleod, J. K.; Monahan, L. C. Tetrahedron Lett. 1988, 29, 391–392. doi:10.1016/S0040-4039(00)80105-7
Return to citation in text: [1] -
Boger, D. L.; Zhang, M. J. Am. Chem. Soc. 1991, 113, 4230–4234. doi:10.1021/ja00011a026
Return to citation in text: [1] -
Muratake, H.; Natsume, M. Tetrahedron Lett. 1989, 30, 5771–5772. doi:10.1016/S0040-4039(00)76193-4
Return to citation in text: [1] -
Muratake, H.; Watanabe, M.; Goto, K.; Natsume, M. Tetrahedron 1990, 46, 4179–4192. doi:10.1016/S0040-4020(01)86755-7
Return to citation in text: [1] -
Muratake, H.; Seino, T.; Natsume, M. Tetrahedron Lett. 1993, 34, 4815–4818. doi:10.1016/S0040-4039(00)74096-2
Return to citation in text: [1] -
Muratake, H.; Mikawa, A.; Seino, T.; Natsume, M. Chem. Pharm. Bull. 1994, 42, 854–864. doi:10.1248/cpb.42.854
Return to citation in text: [1] -
Yasukouchi, T.; Kanematsu, K. Tetrahedron Lett. 1989, 30, 6559–6562. doi:10.1016/S0040-4039(01)89021-3
Return to citation in text: [1] -
Jackson, S. K.; Banfield, S. C.; Kerr, M. A. Org. Lett. 2005, 7, 1215–1218. doi:10.1021/ol047498k
Return to citation in text: [1] -
Jackson, S. K.; Kerr, M. A. J. Org. Chem. 2007, 72, 1405–1411. doi:10.1021/jo062350v
Return to citation in text: [1] -
Buszek, K. R.; Brown, N.; Luo, D. Org. Lett. 2009, 11, 201–204. doi:10.1021/ol802425m
Return to citation in text: [1] -
Chandrasoma, N.; Brown, N.; Brassfield, A.; Nerurkar, A.; Suarez, S.; Buszek, K. R. Tetrahedron Lett. 2013, 54, 913–917. doi:10.1016/j.tetlet.2012.11.125
Return to citation in text: [1] -
Brown, N.; Luo, D.; Decapo, J. A.; Buszek, K. R. Tetrahedron Lett. 2009, 50, 7113–7115. doi:10.1016/j.tetlet.2009.09.083
Return to citation in text: [1] -
Silva, L. F., Jr.; Craveiro, M. V. Org. Lett. 2008, 10, 5417–5420. doi:10.1021/ol8023105
Return to citation in text: [1] -
Silva, L. F., Jr.; Craveiro, M. V.; Tébéka, I. R. M. Tetrahedron 2010, 66, 3875–3895. doi:10.1016/j.tet.2010.03.089
Return to citation in text: [1] -
Saito, N.; Ichimaru, T.; Sato, Y. Org. Lett. 2012, 14, 1914–1917. doi:10.1021/ol300571b
Return to citation in text: [1] -
Tébéka, I. R. M.; Longato, G. B.; Craveiro, M. V.; de Carvalho, J. E.; Ruiz, A. L. T. G.; Silva, L. F., Jr. Chem. – Eur. J. 2012, 18, 16890–16901. doi:10.1002/chem.201202413
Return to citation in text: [1] -
Liu, W.; Lim, H. J.; RajanBabu, T. V. J. Am. Chem. Soc. 2012, 134, 5496–5499. doi:10.1021/ja3004733
Return to citation in text: [1] -
Magnus, P.; Mansley, T. E. Tetrahedron Lett. 1999, 40, 6909–6912. doi:10.1016/S0040-4039(99)01355-6
Return to citation in text: [1] -
Smith, A. B., III; Davulcu, A. H.; Kürti, L. Org. Lett. 2006, 8, 1669–1672. doi:10.1021/ol0602912
Return to citation in text: [1] -
Smith, A. B., III; Davulcu, A. H.; Cho, Y. S.; Ohmoto, K.; Kürti, L.; Ishiyama, H. J. Org. Chem. 2007, 72, 4596–4610. doi:10.1021/jo062422i
Return to citation in text: [1] -
Smith, A. B., III; Kürti, L.; Davulcu, A. H.; Cho, Y. S.; Ohmoto, K. J. Org. Chem. 2007, 72, 4611–4620. doi:10.1021/jo062423a
Return to citation in text: [1] -
Marsch, N.; Jones, P. G.; Lindel, T. Beilstein J. Org. Chem. 2015, 11, 1700–1706. doi:10.3762/bjoc.11.184
Return to citation in text: [1] [2] [3] -
Li, B. T. Y.; White, J. M.; Hutton, C. A. Aust. J. Chem. 2010, 63, 438–444. doi:10.1071/CH10033
Return to citation in text: [1] -
Camp, D.; Matthews, C. F.; Neville, S. T.; Rouns, M.; Scott, R. W.; Truong, Y. Org. Process Res. Dev. 2006, 10, 814–821. doi:10.1021/op0600761
Return to citation in text: [1] -
Egi, M.; Yamaguchi, Y.; Fujiwara, N.; Akai, S. Org. Lett. 2008, 10, 1867–1870. doi:10.1021/ol800596c
Return to citation in text: [1] -
Kern, J. C.; Terefenko, E.; Trybulski, E.; Berrodin, T. J.; Cohen, J.; Winneker, R. C.; Yudt, M. R.; Zang, Z.; Zhu, Y.; Zhang, P. Bioorg. Med. Chem. Lett. 2009, 19, 6666–6669. doi:10.1016/j.bmcl.2009.10.008
Return to citation in text: [1] -
Minatti, A.; Zheng, X.; Buchwald, S. L. J. Org. Chem. 2007, 72, 9253–9258. doi:10.1021/jo701741y
Return to citation in text: [1] -
Hirai, G.; Koizumi, Y.; Moharram, S. M.; Oguri, H.; Hirama, M. Org. Lett. 2002, 4, 1627–1630. doi:10.1021/ol025852d
Return to citation in text: [1] -
Ito, N.; Esaki, H.; Maesawa, T.; Imamiya, E.; Maegawa, T.; Sajiki, H. Bull. Chem. Soc. Jpn. 2008, 81, 278–286. doi:10.1246/bcsj.81.278
Return to citation in text: [1] -
Chancellor, D. R.; Davies, K. E.; De Moor, O.; Dorgan, C. R.; Johnson, P. D.; Lambert, A. G.; Lawrence, D.; Lecci, C.; Maillol, C.; Middleton, P. J.; Nugent, G.; Poignant, S. D.; Potter, A. C.; Price, P. D.; Pye, R. J.; Storer, R.; Tinsley, J. M.; van Well, R.; Vickers, R.; Vile, J.; Wilkes, F. J.; Wilson, F. X.; Wren, S. P.; Wynne, G. M. J. Med. Chem. 2011, 54, 3241–3250. doi:10.1021/jm200135z
Return to citation in text: [1] -
Hengartner, U.; Batcho, A. D.; Blount, J. F.; Leimgruber, W.; Larscheid, M. E.; Scott, J. W. J. Org. Chem. 1979, 44, 3748–3752. doi:10.1021/jo01336a002
Return to citation in text: [1] -
Okano, K.; Okuyama, K.; Fukuyama, T.; Tokuyama, H. Synlett 2008, 1977–1980. doi:10.1055/s-2008-1077980
Return to citation in text: [1] -
Horita, K.; Yoshioka, T.; Tanaka, T.; Oikawa, Y.; Yonemitsu, O. Tetrahedron 1986, 42, 3021–3028. doi:10.1016/S0040-4020(01)90593-9
Return to citation in text: [1] -
Alvarez-Manzaneda, E.; Chahboun, R.; Cabrera, E.; Alvarez, E.; Alvarez-Manzaneda, R.; Meneses, R.; Es-Samti, H.; Fernández, A. J. Org. Chem. 2009, 74, 3384–3388. doi:10.1021/jo900153y
Return to citation in text: [1] -
Anzalone, A. V.; Wang, T. Y.; Chen, Z.; Cornish, V. W. Angew. Chem., Int. Ed. 2013, 52, 650–654. doi:10.1002/anie.201205369
Return to citation in text: [1] -
Zheng, H.; Xie, X.; Yang, J.; Zhao, C.; Jing, P.; Fang, B.; She, X. Org. Biomol. Chem. 2011, 9, 7755–7762. doi:10.1039/c1ob06138k
Return to citation in text: [1] -
Marion, N.; Díez-González, S.; de Frémont, P.; Noble, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2006, 45, 3647–3650. doi:10.1002/anie.200600571
Return to citation in text: [1]
| 1. | Vougogiannopoulou, K.; Fokialakis, N.; Aligiannis, N.; Cantrell, C.; Skaltsounis, A.-L. Org. Lett. 2010, 12, 1908–1911. doi:10.1021/ol100584w |
| 11. | Capon, R. J.; Macleod, J. K.; Scammels, P. J. Tetrahedron 1986, 42, 6545–6550. doi:10.1016/S0040-4020(01)88117-5 |
| 38. | Kern, J. C.; Terefenko, E.; Trybulski, E.; Berrodin, T. J.; Cohen, J.; Winneker, R. C.; Yudt, M. R.; Zang, Z.; Zhu, Y.; Zhang, P. Bioorg. Med. Chem. Lett. 2009, 19, 6666–6669. doi:10.1016/j.bmcl.2009.10.008 |
| 7. | Lauren, D. R.; Gallagher, R. T. J. Chromatogr. 1982, 248, 150–154. doi:10.1016/S0021-9673(00)83747-4 |
| 8. | de Jesus, A. E.; Steyn, P. S.; van Heerden, F. R.; Vleggaar, R. J. Chem. Soc., Perkin Trans. 1 1984, 697–701. doi:10.1039/p19840000697 |
| 9. | Wilkins, A. L.; Miles, C. O.; Ede, R. M.; Gallagher, R. T.; Munday, S. C. J. Agric. Food Chem. 1992, 40, 1307–1309. doi:10.1021/jf00020a002 |
| 10. | Herb, R.; Carroll, A. R.; Yoshida, W. Y.; Scheuer, P. J.; Paul, V. J. Tetrahedron 1990, 46, 3089–3092. doi:10.1016/S0040-4020(01)88399-X |
| 39. | Minatti, A.; Zheng, X.; Buchwald, S. L. J. Org. Chem. 2007, 72, 9253–9258. doi:10.1021/jo701741y |
| 40. | Hirai, G.; Koizumi, Y.; Moharram, S. M.; Oguri, H.; Hirama, M. Org. Lett. 2002, 4, 1627–1630. doi:10.1021/ol025852d |
| 3. | Belofsky, G. N.; Gloer, J. B.; Wicklow, D. T.; Dowd, P. F. Tetrahedron 1995, 51, 3959–3968. doi:10.1016/0040-4020(95)00138-X |
| 4. | Xu, M.; Gessner, G.; Groth, I.; Lange, C.; Christner, A.; Bruhn, T.; Deng, Z.; Li, X.; Heinemann, S. H.; Grabley, S.; Bringmann, G.; Sattler, I.; Lin, W. Tetrahedron 2007, 63, 435–444. doi:10.1016/j.tet.2006.10.050 |
| 5. | Smetanina, O. F.; Kalinovsky, A. I.; Khudyakova, Y. V.; Pivkin, M. V.; Dmitrenok, P. S.; Fedorov, S. N.; Ji, H.; Kwak, J.-Y.; Kuznetsova, T. A. J. Nat. Prod. 2007, 70, 906–909. doi:10.1021/np060396d |
| 6. | Gallagher, R. T.; Latch, G. C.; Keogh, R. G. Appl. Environ. Microbiol. 1980, 39, 272–273. |
| 37. | Egi, M.; Yamaguchi, Y.; Fujiwara, N.; Akai, S. Org. Lett. 2008, 10, 1867–1870. doi:10.1021/ol800596c |
| 2. | Ondeyka, J. G.; Helms, G. L.; Hensens, O. D.; Goetz, M. A.; Zink, D. L.; Tsipouras, A.; Shoop, W. L.; Slayton, L.; Dombrowski, A. W.; Polishook, J. D.; Ostlind, D. A.; Tsou, N. N.; Ball, R. G.; Singh, S. B. J. Am. Chem. Soc. 1997, 119, 8809–8816. doi:10.1021/ja971664k |
| 34. | Marsch, N.; Jones, P. G.; Lindel, T. Beilstein J. Org. Chem. 2015, 11, 1700–1706. doi:10.3762/bjoc.11.184 |
| 34. | Marsch, N.; Jones, P. G.; Lindel, T. Beilstein J. Org. Chem. 2015, 11, 1700–1706. doi:10.3762/bjoc.11.184 |
| 35. | Li, B. T. Y.; White, J. M.; Hutton, C. A. Aust. J. Chem. 2010, 63, 438–444. doi:10.1071/CH10033 |
| 30. | Magnus, P.; Mansley, T. E. Tetrahedron Lett. 1999, 40, 6909–6912. doi:10.1016/S0040-4039(99)01355-6 |
| 31. | Smith, A. B., III; Davulcu, A. H.; Kürti, L. Org. Lett. 2006, 8, 1669–1672. doi:10.1021/ol0602912 |
| 32. | Smith, A. B., III; Davulcu, A. H.; Cho, Y. S.; Ohmoto, K.; Kürti, L.; Ishiyama, H. J. Org. Chem. 2007, 72, 4596–4610. doi:10.1021/jo062422i |
| 33. | Smith, A. B., III; Kürti, L.; Davulcu, A. H.; Cho, Y. S.; Ohmoto, K. J. Org. Chem. 2007, 72, 4611–4620. doi:10.1021/jo062423a |
| 36. | Camp, D.; Matthews, C. F.; Neville, S. T.; Rouns, M.; Scott, R. W.; Truong, Y. Org. Process Res. Dev. 2006, 10, 814–821. doi:10.1021/op0600761 |
| 13. | Macleod, J. K.; Monahan, L. C. Tetrahedron Lett. 1988, 29, 391–392. doi:10.1016/S0040-4039(00)80105-7 |
| 14. | Boger, D. L.; Zhang, M. J. Am. Chem. Soc. 1991, 113, 4230–4234. doi:10.1021/ja00011a026 |
| 15. | Muratake, H.; Natsume, M. Tetrahedron Lett. 1989, 30, 5771–5772. doi:10.1016/S0040-4039(00)76193-4 |
| 16. | Muratake, H.; Watanabe, M.; Goto, K.; Natsume, M. Tetrahedron 1990, 46, 4179–4192. doi:10.1016/S0040-4020(01)86755-7 |
| 17. | Muratake, H.; Seino, T.; Natsume, M. Tetrahedron Lett. 1993, 34, 4815–4818. doi:10.1016/S0040-4039(00)74096-2 |
| 18. | Muratake, H.; Mikawa, A.; Seino, T.; Natsume, M. Chem. Pharm. Bull. 1994, 42, 854–864. doi:10.1248/cpb.42.854 |
| 19. | Yasukouchi, T.; Kanematsu, K. Tetrahedron Lett. 1989, 30, 6559–6562. doi:10.1016/S0040-4039(01)89021-3 |
| 20. | Jackson, S. K.; Banfield, S. C.; Kerr, M. A. Org. Lett. 2005, 7, 1215–1218. doi:10.1021/ol047498k |
| 21. | Jackson, S. K.; Kerr, M. A. J. Org. Chem. 2007, 72, 1405–1411. doi:10.1021/jo062350v |
| 22. | Buszek, K. R.; Brown, N.; Luo, D. Org. Lett. 2009, 11, 201–204. doi:10.1021/ol802425m |
| 23. | Chandrasoma, N.; Brown, N.; Brassfield, A.; Nerurkar, A.; Suarez, S.; Buszek, K. R. Tetrahedron Lett. 2013, 54, 913–917. doi:10.1016/j.tetlet.2012.11.125 |
| 24. | Brown, N.; Luo, D.; Decapo, J. A.; Buszek, K. R. Tetrahedron Lett. 2009, 50, 7113–7115. doi:10.1016/j.tetlet.2009.09.083 |
| 25. | Silva, L. F., Jr.; Craveiro, M. V. Org. Lett. 2008, 10, 5417–5420. doi:10.1021/ol8023105 |
| 26. | Silva, L. F., Jr.; Craveiro, M. V.; Tébéka, I. R. M. Tetrahedron 2010, 66, 3875–3895. doi:10.1016/j.tet.2010.03.089 |
| 27. | Saito, N.; Ichimaru, T.; Sato, Y. Org. Lett. 2012, 14, 1914–1917. doi:10.1021/ol300571b |
| 28. | Tébéka, I. R. M.; Longato, G. B.; Craveiro, M. V.; de Carvalho, J. E.; Ruiz, A. L. T. G.; Silva, L. F., Jr. Chem. – Eur. J. 2012, 18, 16890–16901. doi:10.1002/chem.201202413 |
| 29. | Liu, W.; Lim, H. J.; RajanBabu, T. V. J. Am. Chem. Soc. 2012, 134, 5496–5499. doi:10.1021/ja3004733 |
| 12. | Muratake, H.; Mikawa, A.; Natsume, M. Tetrahedron Lett. 1992, 33, 4595–4598. doi:10.1016/S0040-4039(00)61322-9 |
| 34. | Marsch, N.; Jones, P. G.; Lindel, T. Beilstein J. Org. Chem. 2015, 11, 1700–1706. doi:10.3762/bjoc.11.184 |
| 43. | Hengartner, U.; Batcho, A. D.; Blount, J. F.; Leimgruber, W.; Larscheid, M. E.; Scott, J. W. J. Org. Chem. 1979, 44, 3748–3752. doi:10.1021/jo01336a002 |
| 41. | Ito, N.; Esaki, H.; Maesawa, T.; Imamiya, E.; Maegawa, T.; Sajiki, H. Bull. Chem. Soc. Jpn. 2008, 81, 278–286. doi:10.1246/bcsj.81.278 |
| 42. | Chancellor, D. R.; Davies, K. E.; De Moor, O.; Dorgan, C. R.; Johnson, P. D.; Lambert, A. G.; Lawrence, D.; Lecci, C.; Maillol, C.; Middleton, P. J.; Nugent, G.; Poignant, S. D.; Potter, A. C.; Price, P. D.; Pye, R. J.; Storer, R.; Tinsley, J. M.; van Well, R.; Vickers, R.; Vile, J.; Wilkes, F. J.; Wilson, F. X.; Wren, S. P.; Wynne, G. M. J. Med. Chem. 2011, 54, 3241–3250. doi:10.1021/jm200135z |
| 48. | Zheng, H.; Xie, X.; Yang, J.; Zhao, C.; Jing, P.; Fang, B.; She, X. Org. Biomol. Chem. 2011, 9, 7755–7762. doi:10.1039/c1ob06138k |
| 49. | Marion, N.; Díez-González, S.; de Frémont, P.; Noble, A. R.; Nolan, S. P. Angew. Chem., Int. Ed. 2006, 45, 3647–3650. doi:10.1002/anie.200600571 |
| 46. | Alvarez-Manzaneda, E.; Chahboun, R.; Cabrera, E.; Alvarez, E.; Alvarez-Manzaneda, R.; Meneses, R.; Es-Samti, H.; Fernández, A. J. Org. Chem. 2009, 74, 3384–3388. doi:10.1021/jo900153y |
| 47. | Anzalone, A. V.; Wang, T. Y.; Chen, Z.; Cornish, V. W. Angew. Chem., Int. Ed. 2013, 52, 650–654. doi:10.1002/anie.201205369 |
| 44. | Okano, K.; Okuyama, K.; Fukuyama, T.; Tokuyama, H. Synlett 2008, 1977–1980. doi:10.1055/s-2008-1077980 |
| 45. | Horita, K.; Yoshioka, T.; Tanaka, T.; Oikawa, Y.; Yonemitsu, O. Tetrahedron 1986, 42, 3021–3028. doi:10.1016/S0040-4020(01)90593-9 |
© 2016 Marsch et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)