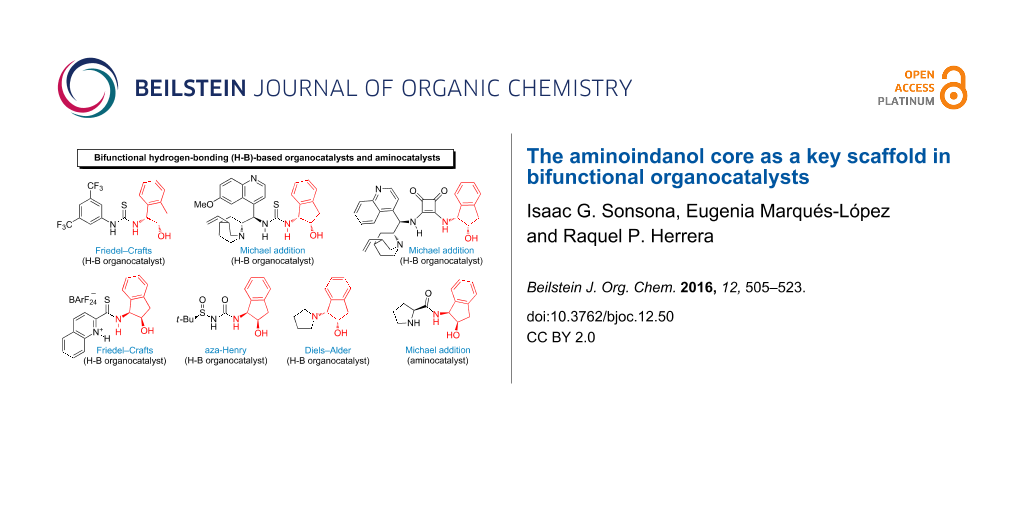
Abstract
The 1,2-aminoindanol scaffold has been found to be very efficient, enhancing the enantioselectivity when present in organocatalysts. This may be explained by its ability to induce a bifunctional activation of the substrates involved in the reaction. Thus, it is easy to find hydrogen-bonding organocatalysts ((thio)ureas, squaramides, quinolinium thioamide, etc.) in the literature containing this favored structural core. They have been successfully employed in reactions such as Friedel–Crafts alkylation, Michael addition, Diels–Alder and aza-Henry reactions. However, the 1,2-aminoindanol core incorporated into proline derivatives has been scarcely explored. Herein, the most representative and illustrative examples are compiled and this review will be mainly focused on the cases where the aminoindanol moiety confers bifunctionality to the organocatalysts.

Graphical Abstract
Introduction
The structural and chemical properties of the 1,2-aminoindanol scaffold 1 have transformed aminoindanol derivatives into versatile building blocks for the construction of catalysts and the efficient induction of chirality in asymmetric processes (Figure 1). Some examples of these properties are rigidity, disposition of the two stereogenic centers, ability of the hydroxy and amino groups to coordinate to some metals or to act as hydrogen-bond donors/acceptors, the different catalytic activity of these chemical groups and their possible derivatization. Thus, in the last decade, it has been widely employed in the field of asymmetric catalysis. Regarding the use of aminoindanol derivatives as ligands in organometallic catalytic complexes, the results have been outstanding. Examples are found in (a) the vanadium-catalyzed asymmetric oxidation of disulfides and sulfides, which are involved in the synthesis of ligands and pharmaceutical chiral synthetic precursors [1,2] and in (b) the transfer-hydrogenation reaction catalyzed by bifunctional chiral ruthenium complexes, employed in the synthesis of peptide mimics with an interesting trifluoroethylamine moiety [3-5]. However, it is in the field of asymmetric organocatalysis [6-8] where the aminoindanol core has gained more importance, being a recurrent structural motif in several organocatalytic species. Some examples are (a) the enantioselective reduction of ketones through the in situ formation of catalytically active oxazaborolidines using cis-1,2-aminoindanol derivatives [9,10] and (b) the synthesis of more active cooperative thiourea-urea-based organocatalysts, which employ the aminoindanol framework as structural linker between two hydrogen-bond-donor moieties [11]. The latter ones have exhibited efficient catalytic activity in the asymmetric Mannich reaction. In fact, the use of simple trans-(1R,2R)-aminoindanol (1c) as an efficient organocatalyst in the enantioselective synthesis of natural products as the TMC-954 core [12,13], has been recently reported. These examples show the high catalytic potential that this versatile motif exhibits [14].
![[1860-5397-12-50-1]](/bjoc/content/figures/1860-5397-12-50-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Different configurations of 1,2-aminoindanol 1a–d.
Figure 1: Different configurations of 1,2-aminoindanol 1a–d.
The concept of bifunctionality has been extensively explored in organocatalysis in the last decade [15,16]. The bifunctional organocatalyst contains two chemical groups that interact simultaneously with the substrates. This mode of activation increases the efficiency of the process, since the interactions favor a selective approach of the reactants. In the transition state, the chiral and rigid aminoindanol scaffold can be involved in different interactions with the substrates due to its capacity to interact through the hydroxy and amino groups. Although the aminoindanol scaffold appears in the structure of different catalysts (providing a suitable way to induce chirality), it is not always directly involved in the bifunctional activation of the substrates [17]. Herein, we show only those cases where the aminoindanol moiety confers bifunctionality to the organocatalysts, interacting with the reactants through both the hydroxy and amino groups.
Review
Bifunctional hydrogen-bonding-based organocatalysts
Most of the examples of bifunctional aminoindanol-containing organocatalysts present in literature correspond to catalysts acting through hydrogen bonding, such as thiourea, urea, squaramide, and thioamide frameworks. These have been efficiently employed in a few organocatalytic processes such as Friedel–Crafts alkylations, Michael additions, Diels–Alder reactions and aza-Henry reactions, as discussed below.
Friedel–Crafts-type alkylation reaction of indoles
To the best of our knowledge, the first example of an aminoindanol-containing bifunctional organocatalyst was reported by Ricci and co-workers in 2005 [18]. In this pioneering study, the authors used the easily prepared cis-(1R,2S)-aminoindanol-based thiourea derivative 4 to develop the first organocatalytic enantioselective Friedel–Crafts (F–C) alkylation of indoles, employing nitroalkenes as versatile electrophiles. In the presence of catalyst 4, the differently functionalized indole derivatives 2 reacted with aryl and alkyl nitroalkenes 3 in dichloromethane at low temperature. This afforded the optically active 2-indolyl-1-nitro compounds 5 (up to 88% yield and up to 89% ee, Scheme 1). These products were found to be valuable synthetic precursors of biologically active compounds such as tryptamines [19,20] and 1,2,3,4-tetrahydro-β-carbolines [21].
![[1860-5397-12-50-i1]](/bjoc/content/inline/1860-5397-12-50-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Asymmetric F–C alkylation catalyzed by thiourea 4.
Scheme 1: Asymmetric F–C alkylation catalyzed by thiourea 4.
In order to explain the sense of the asymmetric induction observed in the reaction, some experiments with structurally modified catalysts (4' and 4'') were carried out. The results obtained using indole (2a) and β-nitrostyrene (3a) supported the importance of the hydroxy group, since low yield and selectivity were observed when this group was trimethylsylilated (4') or was not present (4'') in the catalytic structure (Figure 2). Moreover, poor selectivity was also observed using N-methylindole, which supported a plausible catalyst–substrate interaction through the indolic proton.
![[1860-5397-12-50-2]](/bjoc/content/figures/1860-5397-12-50-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Results for the F–C reaction carried out with catalyst 4 and the structurally modified analogues, 4' and 4''.
Figure 2: Results for the F–C reaction carried out with catalyst 4 and the structurally modified analogues, 4'...
The authors proposed then a dual role of catalyst 4 in the activation of the substrates. Thus, in the transition state TS1 (Figure 3a), the substrates and catalyst would form a ternary complex where the thiourea moiety would activate the nitro group of the nitroalkene through hydrogen bonds. Simultaneously, the oxygen atom of the hydroxy group would interact with the NH of the indole by a weak hydrogen bond, driving the attack to the Si face of the nitroalkene in a stereocontrolled manner.
![[1860-5397-12-50-3]](/bjoc/content/figures/1860-5397-12-50-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: (a) Transition state TS1 originally proposed for the F–C reaction catalyzed by thiourea 4 [18]. (b) Transition state TS1’ proposed later, based on computational calculations [22].
Figure 3: (a) Transition state TS1 originally proposed for the F–C reaction catalyzed by thiourea 4 [18]. (b) Tra...
In a recent study of this F–C alkylation, Herrera’s group has provided computational evidence of the reaction pathway, which confirms the proposed bifunctional activation mode played by the thiourea catalyst 4 [22]. Remarkably, an interesting hydrogen-bonding interaction between the hydrogen atom of the hydroxy group and the nitro group was detected in this work (Figure 3b). This could explain the low reactivity (18% yield) and selectivity (39% ee) that the silyl ether-protected catalyst 4'' exhibited (Figure 2).
Encouraged by the development of more efficient organocatalytic systems, the same research group explored the influence of external acidic additives in this reaction. The authors envisioned that a cooperative effect between the chiral thiourea organocatalyst and a Brønsted acid (AH) could provide better results in terms of reactivity and enantioselectivity. Thus, in 2011, they published an article where it was proved that the synergic system between the thiourea ent-4 and mandelic acid led to the final products 5 with a significant increase of conversion and enantiomeric excess (Scheme 2) [23].
![[1860-5397-12-50-i2]](/bjoc/content/inline/1860-5397-12-50-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Asymmetric F–C alkylation catalyzed by thiourea ent-4 in the presence of D-mandelic acid as a Brønsted acid additive.
Scheme 2: Asymmetric F–C alkylation catalyzed by thiourea ent-4 in the presence of D-mandelic acid as a Brøns...
Experimental proofs exploring different catalysts and acids suggested that it is the thiourea which provides the sense of the enantioinduction. Therefore, the authors assumed the bifunctional transition state TS2, similar to the above mentioned TS1, where the external acid (AH) would only coordinate to the thiourea moiety enhancing its acidity and thus forming a more active catalytic species (Figure 4).
![[1860-5397-12-50-4]](/bjoc/content/figures/1860-5397-12-50-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Transition state TS2 proposed for the activation of the thiourea-based catalyst ent-4 by an external Brønsted acid.
Figure 4: Transition state TS2 proposed for the activation of the thiourea-based catalyst ent-4 by an externa...
Since the pioneering aminoindanol-containing organocatalyst 4, reported in 2005 [18], other research groups have studied the possibility of incorporating this scaffold into diverse organocatalysts.
In 2008, Seidel’s group published a new example of an asymmetric addition of indoles to nitroalkenes, employing a novel catalyst design [24]. The authors envisioned that a protonated 2-pyridyl substituent could increase the acidity of the thiourea group through an intramolecular N–H···S hydrogen-bonding interaction (analogous to the C–H···S that exists with the 3,5-bis-trifluoromethylphenyl moiety, commonly used in thiourea-based organocatalysts) [25]. Although this first approach did not provide a significant increase of the enantioselectivity, further modifications of the catalytic structure led to highly active catalysts. Indeed, the best results were obtained with the quinolinium thioamide 6, where the NH moiety adjacent to the pyridine ring of the analogous thiourea was “removed”. Likely, in this case, the intramolecular hydrogen-bonding interaction described above would yield a negligible stabilization due to the distance between N–H and S moieties. In contrast, it is suspected that both the thioamide N–H as well as the N–H on the quinolinium moiety are engaged in substrate binding, and thus, provide higher yields and selectivity in comparison with the catalyst 4 (up to 96% yield, up to 98% ee) (Scheme 3).
![[1860-5397-12-50-i3]](/bjoc/content/inline/1860-5397-12-50-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Friedel–Crafts alkylation of indoles catalyzed by the chiral thioamide 6.
Scheme 3: Friedel–Crafts alkylation of indoles catalyzed by the chiral thioamide 6.
The authors do not comment on whether the catalyst 6 acts in a bifunctional fashion or not, but it is reasonable to assume that the OH group is again involved in the transition state by a possible interaction with the indole derivatives 2. Indeed, as discussed below, other authors proposed the compound 6 as a plausible bifunctional catalyst. The Enders’ group used its enantiomer (ent-6) to develop a pioneering scalable one-pot multicatalytic method for the C2/C3-annulation of the indoles 2 (Scheme 4) [26]. In this work, an efficient enantioselective and sequential double Friedel–Crafts alkylation provided direct access to the tetracyclic seven-membered ring containing indoles 8. These pharmaceutically intriguing compounds exhibit anticancer [27] and antiproliferative activity [28].
![[1860-5397-12-50-i4]](/bjoc/content/inline/1860-5397-12-50-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Scalable tandem C2/C3-annulation of indoles, catalyzed by the thioamide ent-6.
Scheme 4: Scalable tandem C2/C3-annulation of indoles, catalyzed by the thioamide ent-6.
In the first catalytic cycle of the authors’ mechanistic hypothesis, the β-nitroalkene derivatives 7 are proposed to react with the indoles 2 in the presence of the organocatalyst ent-6 to afford the intermediates 9 with excellent enantioselectivity (Scheme 5). Furthermore, a bifunctional activation mode through the transition state TS3 was proposed. Herein, the NH from the thioamide and the protonated quinoline moiety would activate and fix the nitroalkene framework through hydrogen-bonding interactions. Simultaneously, the oxygen atom of the hydroxy group would orientate the attack of the indole by the Si face through the formation of a hydrogen bond with the indolic proton. In the second catalytic cycle, the intermediates 9 would react to give an intramolecular Friedel–Crafts alkylation. The alkyne moiety of 9 would be previously activated by a gold complex in the presence of p-toluenesulfonic acid hydrate as the additive. The final tetracyclic indoles 8 are released from the spirocyclic intermediates 11, following a ring-expansion and rearomatization/final protodeauration cascade process (Scheme 5) [26].
![[1860-5397-12-50-i5]](/bjoc/content/inline/1860-5397-12-50-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Plausible tandem process mechanism for the sequential, double Friedel–Crafts alkylation, which involves the hydrogen-bonding catalyst ent-6 and gold catalysis and leads to the tetracyclic indoles 8.
Scheme 5: Plausible tandem process mechanism for the sequential, double Friedel–Crafts alkylation, which invo...
In 2012, the same group reported an additional example of a one-pot multisequence reaction following a similar mode of activation. This method provided a route to access the enantiomerically enriched tetrahydrocarbazole scaffold-containing compounds 14 (Scheme 6 and Scheme 7) [29]. One of these valuable products is a synthetic precursor of the pharmacologically active compound 15, used to treat Alzheimer and other central nervous system diseases [30-34].
![[1860-5397-12-50-i6]](/bjoc/content/inline/1860-5397-12-50-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: One-pot multisequence process that allows the synthesis of interesting compounds 14. The pharmacologically active compound 15 can be obtained from the properly substituted product 14.
Scheme 6: One-pot multisequence process that allows the synthesis of interesting compounds 14. The pharmacolo...
![[1860-5397-12-50-i7]](/bjoc/content/inline/1860-5397-12-50-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Reaction pathway proposed for the preparation of the compounds 14.
Scheme 7: Reaction pathway proposed for the preparation of the compounds 14.
In the proposed reaction pathway, the nucleophilic addition of the indole derivatives 2 to the nitroalkene 13 progresses in a stereocontrolled manner due to the creation of a ternary complex with the chiral bifunctional thioamide ent-6 (TS4, Scheme 7). Herein, the catalyst activates both substrates simultaneously through hydrogen-bonding interactions between the thioamidic NH and the nitro group, and between the hydroxy group and the indolic proton. In the presence of AgSbF6, a soft Lewis acid, the stereogenic center-containing intermediates 16 are activated. This triggers an SN2-type attack/Ciamician–Plancher rearrangement [35]/rearomatization cascade process, affording the final products 14 (Scheme 7).
More recently, the same authors also provided an elegant and efficient solution to give direct access to cis-vicinal-substituted indane scaffolds through an organocatalyzed asymmetric domino-Michael addition/Henry reaction (Scheme 8) [36]. These heterocyclic products are important chiral building blocks for the synthesis of organocatalytic frameworks and ligands for chiral metal complexes, both with the potential ability to induce chirality. They also belong to a class of privileged pharmaceutical scaffolds and exhibit different biological activities, as it is the case of Crixivan [37,38], an HIV protease inhibitor which has been employed for AIDS treatment. The thermodynamic unfavorable cis conformation of these compounds represents a challenge for the development of suitable methods for their synthesis. Interestingly, the chiral thioamide ent-6, which contains a cis-vicinal-substituted indane motif, provided the best choice to this purpose. Therefore, in the presence of such a catalyst, the indole derivatives 2 reacted with 2-(2-nitrovinyl)benzaldehyde derivatives 20 to give the highly functionalized cis-1-hydroxy-2-nitroindane-based indole compounds 21 with excellent yield, high selectivity and good diastereomeric ratios (dr) (Scheme 8).
![[1860-5397-12-50-i8]](/bjoc/content/inline/1860-5397-12-50-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: The enantioselective synthesis of cis-vicinal-substituted indane scaffolds 21, catalyzed by ent-6.
Scheme 8: The enantioselective synthesis of cis-vicinal-substituted indane scaffolds 21, catalyzed by ent-6.
In the reaction pathway proposed to explain this domino reaction, both substrates are activated by the catalyst ent-6 through hydrogen-bonding interactions in a bifunctional manner. Thus, the ternary complex formed in the transition state TS5 leads to an enantioselective Friedel–Crafts-type Michael addition by the attack of indole 2 to the electrophilic prochiral center on the nitroalkene 20 in a stereocontrolled manner (Scheme 9). Afterwards, the hydrogen-bonding interactions are reorganized inside the complex, producing a bifunctional activation of both the nitro and the aldehyde groups through a cis-matched transition state TS6. It allows a kinetic controlled, enantioselective Henry reaction that leads to the final cis-product 21. The thermodynamically favorable trans-product 21 can be obtained through a tetramethylguanidine (TMG)-catalyzed epimerization process.
![[1860-5397-12-50-i9]](/bjoc/content/inline/1860-5397-12-50-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Asymmetric domino procedure (Michael addition/Henry cyclization), catalyzed by the thioamide ent-6 which involves a cis-matched transition state (TS6) that allows a kinetic control of the second reaction.
Scheme 9: Asymmetric domino procedure (Michael addition/Henry cyclization), catalyzed by the thioamide ent-6 ...
Other possible electrophiles have been contemplated in the Friedel–Crafts alkylation of indoles. For instance, Jørgensen’s group studied the use of α,β-unsaturated acyl phosphonates as suitable electrophiles for this kind of reaction, using several bifunctional aminoindanol-based organocatalytic scaffolds as catalysts (Scheme 10) [39]. Hence, the authors demonstrated that acyl phosphonates can be used as efficient hydrogen-bonding acceptors in their activation through hydrogen-bonding catalysis. The corresponding final esters or amides are obtained after proper treatment of the reaction mixture. During the screening of catalysts, the best enantioselectivity (74% ee) was obtained using ent-4 in the addition of indole (2a) to the acyl phosphonate 24a, in dichloromethane at room temperature, with subsequent addition of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and methanol to give the corresponding ester derivative 25a (Scheme 10a) [39]. The use of the analog squaramide 23 afforded the product with slightly lower selectivity (60% ee) and the Seidel’s thioamide 6 provided better activation (93% yield at −30 °C) of substrates but with an important loss of selectivity (20% ee). The removal of a hydrogen atom from the hydroxy group of the aminoindanol structure (such as in TIPS-ether catalysts ent-4''' and 22') and the loss of cis relationship between the hydroxy and amino groups (such as in catalyst 22) led to racemic mixtures (Scheme 10a). Under optimal conditions, the acyl phosphonates 24 reacted with indoles 2 in the presence of catalyst ent-4 providing the corresponding products 25 with high selectivity (up to 90% ee) (Scheme 10b) [39].
![[1860-5397-12-50-i10]](/bjoc/content/inline/1860-5397-12-50-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: The enantioselective addition of indoles 2 to α,β-unsaturated acyl phosphonates 24, a) screening of different catalysts and b) optimized conditions using catalyst ent-4.
Scheme 10: The enantioselective addition of indoles 2 to α,β-unsaturated acyl phosphonates 24, a) screening of...
Based on the experimental results, the authors proposed a bifunctional mode of activation (TS7), where the electrophile is fixed and activated by the thiourea framework through several hydrogen bonds. At the same time, the indole is oriented to attack the Re face of the Michael-type acceptor, by weak hydrogen-bonding interaction between the oxygen atom of the hydroxy group and the indolic proton (Figure 5).
![[1860-5397-12-50-5]](/bjoc/content/figures/1860-5397-12-50-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Proposed transition state TS7 for the Friedel–Crafts reaction of indole and α,β-unsaturated acyl phosphonates catalyzed by ent-4.
Figure 5: Proposed transition state TS7 for the Friedel–Crafts reaction of indole and α,β-unsaturated acyl ph...
Recently, the conjugated addition of indole derivatives to β,γ-unsaturated α-ketoesters was explored [40]. To this end, the catalytic activity of several chiral thioureas was studied, revealing the aminoindanol-based thiourea ent-4 as the most suitable catalyst for this process. The authors studied aliphatic derivatives because for this reaction these compounds had been much less explored than the aromatic ones. Thus, the different aliphatic β,γ-unsaturated α-ketoesters 26a–f reacted with the substituted indoles 2 in the presence of ent-4 to achieve the corresponding adducts 27 with good yields and enantioselectivities (up to 88% yield, up to 76% ee) (Scheme 11).
![[1860-5397-12-50-i11]](/bjoc/content/inline/1860-5397-12-50-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Study of aliphatic β,γ-unsaturated α-ketoesters 26 as substrates in the F–C alkylation of indoles catalyzed by ent-4.
Scheme 11: Study of aliphatic β,γ-unsaturated α-ketoesters 26 as substrates in the F–C alkylation of indoles c...
Although the absolute configuration was unknown at that point, the authors envisioned a plausible reaction pathway based on previously reported transition states (Figure 6). The catalyst ent-4 would activate and fix the electrophile through several hydrogen-bonding interactions with the NH groups of the thiourea. Simultaneously, the hydroxy group would be involved in the activation of the nucleophile, establishing a hydrogen bond with the indolic proton. This would conduct its attack over the Re face of the β,γ-unsaturated α-ketoesters, producing the addition in a stereocontrolled fashion. Some additional experimental proofs provided in the article supported this hypothesis [40].
![[1860-5397-12-50-6]](/bjoc/content/figures/1860-5397-12-50-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Possible transition states TS8 and TS9 in the asymmetric addition of indoles 2 to the β,γ-unsaturated α-ketoesters 26 catalyzed by ent-4.
Figure 6: Possible transition states TS8 and TS9 in the asymmetric addition of indoles 2 to the β,γ-unsaturat...
Michael addition to α,β-unsaturated compounds
Fernández, Lassaleta and co-workers provided an elegant, versatile and mild umpolung strategy, which leads to key synthetic precursors using the thiourea ent-4. In this study, an organocatalytic enantioselective addition of nucleophilic N,N-dialkylhydrazones to electron-deficient β,γ-unsaturated α-ketoesters was reported (Table 1) [41]. In the presence of catalyst ent-4, 1-methyleneaminopyrrolidine (28) reacted with the different β,γ-unsaturated α-ketoesters 26 in dichloromethane at low temperature to give the corresponding products 29, which are useful masked 1,4-dicarbonyl compounds with moderate to high yield and high selectivity, after moderate reaction times (Table 1).
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-50-i21.svg?max-width=637&scale=1.0)
The authors proposed the plausible transition state TS10, where the acidic hydrogen atoms of the thiourea could activate the β,γ-unsaturated α-ketoesters 26. Simultaneously, the hydrogen atom of the hydroxy group would coordinate and direct the hydrazone 28 to the Re face of the esters 26 in order to afford the absolute configuration found in the final products 29 of this process (Figure 7).
![[1860-5397-12-50-7]](/bjoc/content/figures/1860-5397-12-50-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Transition state TS10 proposed for the asymmetric addition of dialkylhydrazone 28 to the β,γ-unsaturated α-ketoesters 26 catalyzed by ent-4.
Figure 7: Transition state TS10 proposed for the asymmetric addition of dialkylhydrazone 28 to the β,γ-unsatu...
Another example of the bifunctional action of the indanol-based thiourea 4 was reported by Sibi’s group. There, 100 mol % of this compound was employed in the enantioselective conjugate addition of the hydroxylamine derivatives 31 to the enoates 30, affording the final products 32 with good yield (up to 98%) and high enantiomeric excess (up to 98% ee). This provided an efficient method that allows the preparation of biologically interesting β-amino acid derivatives (Table 2) [42].
Table 2: The enantioselective addition of the hydroxylamine derivatives 31 to the enoates 30 promoted by 4.
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-50-i22.svg?max-width=637&scale=1.0)
|
|||||
| Entry | 30 (R1, R2, R3) | 31 (R4) | Time (h) | Yield 32 (%) | ee 32 (%) |
|---|---|---|---|---|---|
| 1a | Me, H, Me | PhCH2 | 24 | 75 (32a) | 71 |
| 2a,b | Me, H, Me | PhCH2 | 168 | 63 (32a) | 71 |
| 3 | Me, H, Me | PhCH2 | 72 | 82 (32a) | 87 |
| 4a | Me, Br, Me | PhCH2 | 24 | 85 (32b) | 61 |
| 5a | Ph, H, Me | PhCH2 | 14 | 76 (32c) | 45 |
| 6a | Ph, Br, Me | PhCH2 | 12 | 72 (32d) | 31 |
| 7 | Me, H, Me | Ph2CH | 96 | 86 (32e) | 89 |
| 8 | Me, H, CO2Et | Ph2CH | 96 | 50 (32f) | 94 |
| 9 | Me, H, CO2Et | TBDMS | 96 | 42 (32g) | 90 |
| 10 | Me, H, Et | Ph2CH | 168 | 92 (32h) | 91 |
| 11 | Me, H, n-Pr | Ph2CH | 138 | 84 (32i) | 88 |
| 12 | Me, H, iPr | Ph2CH | 216 | 68 (32j) | 90 |
| 13 | Me, H, c-C6H11 | Ph2CH | 288 | 59 (32k) | 89 |
| 14b | Me, H, CH2OPMP | Ph2CH | 24 | 98 (32l) | 98 |
| 15a | Me, H, Ph | PhCH2 | 72 | 19 (32m) | 67 |
| 16 | Me, H, Me | TBDMS | 120 | 82 (32n) | 94 |
aReaction carried out at room temperature. b30 mol % of catalyst 4.
In this work, the authors compared the results achieved by means of 4 with other urea- and thiourea-based organocatalysts in order to understand the effect of the acidity, the structural rigidity, and the bifunctionality of the promoter. These reactions were performed in trifluorotoluene at room temperature with the Michael acceptor 30 (R1, R2, R3 = Me, H, Me) and O-benzylhydroxylamine (31, R4 = PhCH2), using a stoichiometric amount of the chiral activator and MS 4 Å as an additive. Some of the reported experiments supported the ability of the cis-2-aminoindanol structure to provide an adequate scaffold to induce chirality. In contrast, the catalysts ent-22 (with the trans-2-aminoindanol) or 4'' (with the aminoindane motif) and the flexible analogues 33–35, provided lower enantioselectivities or led to nearly racemic mixtures (Scheme 12). In the proposed transition state TS11, the α,β-unsaturated substrate is activated by an acidic thiourea template. Moreover, the hydroxylamine derivative is simultaneously oriented to attack the Si face of the Michel acceptor, through its interaction with the hydroxy group of the aminoindanol framework. In this case, a pyrazole moiety presents additional H-bond acceptor sites. These could play an important role in fixing the substrate to the catalyst and favoring a more rigid transition state (TS11) and thus leading to better selectivity (Scheme 12). The absolute configuration is only given for the compounds 32a–d, being S.
![[1860-5397-12-50-i12]](/bjoc/content/inline/1860-5397-12-50-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Different β-hydroxylamino-based catalysts tested in a Michael addition, and the transition state TS11 proposed for this reaction catalyzed by 4.
Scheme 12: Different β-hydroxylamino-based catalysts tested in a Michael addition, and the transition state TS...
Later, He and co-workers reported the use of several chiral multiple hydrogen-bond donating tertiary amine-based organocatalysts in the asymmetric addition of acetylacetone (36a) to the β-nitroalkenes 3. They found the thiourea 37 as a highly suitable catalytic structure to induce chirality in this process (Scheme 13) [43]. Under optimal conditions, this method provided highly enantioenriched γ-nitrocarbonyl compounds 38, which are versatile synthetic intermediates for the preparation of diverse chiral scaffolds.
![[1860-5397-12-50-i13]](/bjoc/content/inline/1860-5397-12-50-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Enantioselective addition of acetylacetone (36a) to nitroalkenes 3, catalyzed by 37 and the proposed transition state TS12.
Scheme 13: Enantioselective addition of acetylacetone (36a) to nitroalkenes 3, catalyzed by 37 and the propose...
Once again, a bifunctional activation mode as the origin of the asymmetric induction was proposed. In the plausible transition state TS12, acidic hydrogen atoms from both hydroxy and thiourea moieties would activate and fix the nitroalkene. Simultaneously, the tertiary amine of the cinchona framework would deprotonate the acidic proton of acetylacetone (36a), driving the attack of the nucleophile. The chiral environment present in the resulting ternary complex would confer the proper facial selectivity to afford the observed absolute configuration in the final products 38.
At the same time, Yuan and co-workers developed an interesting example of a scalable asymmetric Michael addition of 3-substituted oxindoles 39 to the protected 2-amino-1-nitroethenes 40, using the bifunctional tertiary amine aminoindanol-based organocatalyst 41 (Scheme 14) [44]. This catalytic study provides a straightforward synthetic route of the highly functionalized α,β-diamino-3,3’-disubstituted-oxindoles 42. These are key intermediates for the preparation of biologically and pharmacologically attractive compounds, such as (+)-alantrypinone [45], (−)-serantrypinone [46] and (−)-lapatin [47]. In the presence of the catalyst 41 (10 mol %), a broad scope of the oxindoles 39 reacted to give the quaternary stereocenters-containing products 42 with high diastereoselectivity (up to > 99:1 dr) and enantioselectivity (up to 90% ee).
![[1860-5397-12-50-i14]](/bjoc/content/inline/1860-5397-12-50-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Addition of 3-oxindoles 39 to 2-amino-1-nitroethenes 40, catalyzed by 41.
Scheme 14: Addition of 3-oxindoles 39 to 2-amino-1-nitroethenes 40, catalyzed by 41.
A bifunctional role played by the catalyst was again envisioned by the authors. In the transition state TS13 the tertiary amine group of the catalyst would activate the resulting enolized oxindole reagent 39 via deprotonation. Thus, 39 would be disposed to attack by its Re face to the Si face of the nitroethene derivative 40. Simultaneously, the latter would be fixed and activated by a hydrogen-bonding interaction with the hydroxy moiety of the catalyst, in its Z form, which is stabilized due to an intramolecular hydrogen bond (Scheme 14).
In 2012, Dong and co-workers studied the catalytic activity of several β-amino alcohol-based squaramide organocatalysts involved in the Michael addition of acetylacetone (36a) to β-nitrostyrene (3a) in dichloromethane at 15 °C (Scheme 15) [48]. Although high yields were obtained in all cases, the best enantioselectivity was provided by the bifunctional cis-aminoindanol-based squaramide 43. Under these conditions, several 1,3-dicarbonyl compounds 36 reacted with many different nitrostyrene derivatives 3 with very low catalytic charge (1 mol %), affording a broad scope of the enantiomerically enriched β-nitroalkyl products 38. A possible drawback of the method would be the low diastereoselectivity generally achieved for the nonsymmetrical 1,3-dicarbonyl compounds 36.
![[1860-5397-12-50-i15]](/bjoc/content/inline/1860-5397-12-50-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Michael addition of 1,3-dicarbonyl compounds 36 to the nitroalkenes 3 catalyzed by the squaramide 43.
Scheme 15: Michael addition of 1,3-dicarbonyl compounds 36 to the nitroalkenes 3 catalyzed by the squaramide 43...
In order to understand the role of the catalyst, the hydroxy group of the squaramide 43 was methylated (43'). Its catalytic activity was tested in the reaction of acetylacetone (36a) and β-nitrostyrene (3a), leading to very low enantiomeric excess (24% ee). This fact suggested the important role played by the hydroxy group in the activation and in the chiral induction of the process. The authors proposed the transition state TS14, where the NH groups and the OH group of the squaramide would coordinate to the nitroalkene 3 through hydrogen-bonding interactions with the nitro group. Simultaneously, the amine in the cinchona alkaloid would activate the 1,3-dicarbonyl compound 36 (Scheme 15). We would like to remark that although the authors indicated that the S enantiomer is obtained in their final products, they depicted the R configuration, as is drawn in the Scheme 15.
Aza-Henry reaction
Ellman’s group designed a set of pioneering (thio)urea scaffold-containing hydrogen-bonding organocatalysts with an N-sulfinyl moiety. As previously demonstrated, this chemical group increased the acidity of the catalyst and also served as a chiral controller [49-54]. Hence, in the presence of the catalyst 50 and diisopropylethylamine, a wide scope of the N-Boc-protected imines 48, including aliphatic ones, reacted with an excess of the nitroalkanes 49 at low temperature. This afforded the corresponding products 51 with high yield, diastereomeric ratio and excellent enantioselectivity (Scheme 16) [55].
![[1860-5397-12-50-i16]](/bjoc/content/inline/1860-5397-12-50-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Asymmetric aza-Henry reaction catalyzed by the aminoindanol-derived sulfinyl urea 50.
Scheme 16: Asymmetric aza-Henry reaction catalyzed by the aminoindanol-derived sulfinyl urea 50.
Some experimental results using the differently substituted aminoindane-derived sulfinyl ureas 50–50'' showed the important effect of the indanol framework in the diastereo- and enantio-selectivity of the process. The catalysts 50' (with the TBS-protected hydroxy group (TBS, tert-butyldimethylsilyl)) and 50'' (without the hydroxy group) exhibited poor enantioselectivity. These effects may suggest and support the bifunctional role played by the catalyst (Figure 8).
![[1860-5397-12-50-8]](/bjoc/content/figures/1860-5397-12-50-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Results for the aza-Henry reaction carried out with the structurally modified catalysts 50–50''.
Figure 8: Results for the aza-Henry reaction carried out with the structurally modified catalysts 50–50''.
Diels–Alder reaction
An important contribution in the construction of highly substituted carbocyclic compounds was disclosed by Tan’s group in 2009. In this work, the asymmetric Diels–Alder (D–A) reaction between the N-sulfonamide-3-hydroxy-2-pyridone-based dienes 52 and different dienophile substrates was developed using the bifunctional cis-2-trialkylaminoindanol organocatalyst ent-41 [56]. We show herein the reactivity of this family of dienes with several substituted maleimides 53, which in the presence of the above mentioned catalyst, afforded the highly substituted endo-adducts 54 with high yield and enantiomeric excess (Scheme 17). In this approach, the cis orientation of the hydroxy and cyclopentylamine groups of the catalyst was crucial to achieve high enantioselectivity.
![[1860-5397-12-50-i17]](/bjoc/content/inline/1860-5397-12-50-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Diels–Alder reaction catalyzed by the aminoindanol derivative ent-41.
Scheme 17: Diels–Alder reaction catalyzed by the aminoindanol derivative ent-41.
Aminocatalysis
Although aminoindanol-derived catalysts have been scarcely used in aminocatalysis, some relevant examples have been found in the literature, especially in the enantioselective addition of ketones to nitroalkene compounds. In this context, Alonso, Nájera and co-workers designed different alcohol-amino-derived prolinamide organocatalysts and in 2006 published an organocatalyzed direct asymmetric Michael addition of 3-pentanone (55a) to the nitrostyrenes 3 [57]. The corresponding syn-adducts 57 were obtained with excellent conversion, diastereomeric ratio and high enantiomeric excess when the cis-aminoindanol-based prolinamide 56, acting as bifunctional recyclable catalyst, was used (Scheme 18).
![[1860-5397-12-50-i18]](/bjoc/content/inline/1860-5397-12-50-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Asymmetric Michael addition of 3-pentanone (55a) to the nitroalkenes 3 through aminocatalysis.
Scheme 18: Asymmetric Michael addition of 3-pentanone (55a) to the nitroalkenes 3 through aminocatalysis.
Later, based on this previous work, the same research group extended the methodology to different ketones 55, rendering the syn-products 57 with excellent yield and high selectivity (Scheme 19) [58].
![[1860-5397-12-50-i19]](/bjoc/content/inline/1860-5397-12-50-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Substrate scope extension for the asymmetric Michael addition between the ketones 55 and the nitroalkenes 3 through aminocatalysis.
Scheme 19: Substrate scope extension for the asymmetric Michael addition between the ketones 55 and the nitroa...
In this case, the hydroxy group seems again to play an important role in the activation of the substrates, as well as in the selectivity of the process. The rigidity of the hydroxylamino moiety represents another important factor, where aminoindanol was the most appropriate scaffold for this asymmetric methodology among the catalysts tested. Based on the experimental results and computational calculations (DFT and B3LYP76-31G*), the authors proposed a reaction mechanism in which the catalyst 56 acts in a bifunctional way following the route depicted in Scheme 20. Thus, Michael addition of the enamine 58, formed from 3-pentanone (55a) and the catalyst 56, to the nitroalkene 3a takes place leading to the intermediate 59. The last step of the catalytic cycle involves the regeneration of the catalyst by hydrolysis, enabled by the small amount of water present in the solvent.
![[1860-5397-12-50-i20]](/bjoc/content/inline/1860-5397-12-50-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: A possible reaction pathway in the presence of the catalyst 56 and the plausible transition state TS15 proposed for this reaction.
Scheme 20: A possible reaction pathway in the presence of the catalyst 56 and the plausible transition state T...
The transition state TS15 based upon Seebach’s model [59] was envisioned as a plausible activation mode to explain the high asymmetric induction observed and the syn-diastereoselectivity exhibited by the catalyst 56. First, the activation of the ketone via enamine formation is produced. Furthermore, the acidic hydrogen atoms of the amide and the hydroxy groups present in the catalyst would activate and orientate the nitroalkene by hydrogen-bond formation. Thus, the attack of the formed enamine to the Re face of the nitroalkene is favored (Scheme 20). In this way, this example shows an efficient combination of covalent and non-covalent interactions in an interesting bifunctional activation mode.
Conclusion
The design, synthesis and application of catalysts acting in a bifunctional manner is a hot topic in the field of organocatalysis and thus widely investigated. Generally, this particular mode of activation allows the enhancement of both the reactivity and the selectivity of the processes, due to the generation of a more rigid transition state. Among the different ways of conferring this bifunctional character to the catalysts, the incorporation of the aminoindanol core into their structure has shown to be a very suitable method. In most of the examples gathered herein, this can be explained due to the presence of a hydroxy group in the catalyst that normally is able to interact with at least one of the substrates of the reaction, hence facilitating the approach of the reactants in a selective fashion. In many cases, this bifuntional role of the catalyst has been supported with experimental results and sometimes with computational calculations. This smart strategy has allowed the preparation of highly efficient organocatalysts, ranging from very simple structures to more complex ones. These are mainly hydrogen-bonding catalysts, but there is also an example of an aminoindanol-containing aminocatalyst. A broad variety of reactivities has been successfully covered, such as Friedel–Crafts alkylation, Michael addition, Diels–Alder and aza-Henry reactions. However, further exploration into the development of new bifunctional organocatalysts using aminoindanol or another appropriate scaffold and their application in different chemical processes still needs to be performed.
Acknowledgements
We thank the “Ministerio de Economía y Competitividad” (MINECO, Project CTQ2013-44367-C2-1-P), the University of Zaragoza (JIUZ-2014-CIE-07), the High Council of Scientific Investigation (CSIC) (PIE-201580I010) and the Government of Aragon DGA (Research Group E-104) for financial support of our research.
References
-
Weix, D. J.; Ellman, J. A. Org. Lett. 2003, 5, 1317–1320. doi:10.1021/ol034254b
Return to citation in text: [1] -
Lazar, A.; Sharma, P.; Singh, A. P. Microporous Mesoporous Mater. 2013, 170, 331–339. doi:10.1016/j.micromeso.2012.12.014
Return to citation in text: [1] -
Dai, X.; Cahard, D. Adv. Synth. Catal. 2014, 356, 1317–1328. doi:10.1002/adsc.201301115
Return to citation in text: [1] -
Sani, M.; Volonterio, A.; Zanda, M. ChemMedChem 2007, 2, 1693–1700. doi:10.1002/cmdc.200700156
Return to citation in text: [1] -
Piras, M.; Fleming, I. N.; Harrison, W. T. A.; Zanda, M. Synlett 2012, 23, 2899–2902. doi:10.1055/s-0032-1317557
Return to citation in text: [1] -
Berkessel, A.; Gröger, H. Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis; Wiley-VCH: Weinheim, 2005. doi:10.1002/3527604677
Return to citation in text: [1] -
Dalko, P. I., Ed. Enantioselective Organocatalysis: Reactions and Experimental Procedures; Wiley-VCH: Weinheim, 2007. doi:10.1002/9783527610945
Return to citation in text: [1] -
Dalko, P. I., Ed. Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions and Applications; Wiley-VCH: Weinheim, 2013. doi:10.1002/9783527658862
Return to citation in text: [1] -
Sibi, M. P.; Cook, G. R.; Liu, P. Tetrahedron Lett. 1999, 40, 2477–2480. doi:10.1016/S0040-4039(99)00281-6
Return to citation in text: [1] -
Turgut, Y.; Azizoglu, M.; Erdogan, A.; Arslan, N.; Hosgoren, H. Tetrahedron: Asymmetry 2013, 24, 853–859. doi:10.1016/j.tetasy.2013.05.016
Return to citation in text: [1] -
Probst, N.; Madarász, Á.; Valkonen, A.; Pápai, I.; Rissanen, K.; Neuvonen, A.; Pihko, P. M. Angew. Chem., Int. Ed. 2012, 51, 8495–8499. doi:10.1002/anie.201203852
Return to citation in text: [1] -
Kohno, J.; Koguchi, Y.; Nishio, M.; Nakao, K.; Kuroda, M.; Shimizu, R.; Ohnuki, T.; Komatsubara, S. J. Org. Chem. 2000, 65, 990–995. doi:10.1021/jo991375+
Return to citation in text: [1] -
Coste, A.; Couty, F.; Evano, G. C. R. Chim. 2008, 11, 1544–1573. doi:10.1016/j.crci.2008.06.003
See for a review.
Return to citation in text: [1] -
Coste, A.; Bayle, A.; Marrot, J.; Evano, G. Org. Lett. 2014, 16, 1306–1309. doi:10.1021/ol403675c
Return to citation in text: [1] -
Miyabe, H.; Takemoto, Y. Bull. Chem. Soc. Jpn. 2008, 81, 785–795. doi:10.1246/bcsj.81.785
Return to citation in text: [1] -
Connon, S. J. Chem. Commun. 2008, 2499–2510. doi:10.1039/B719249E
Return to citation in text: [1] -
Liu, X.; Lu, Y. Org. Biomol. Chem. 2010, 8, 4063–4065. doi:10.1039/c0ob00223b
Return to citation in text: [1] -
Herrera, R. P.; Sgarzani, V.; Bernardi, L.; Ricci, A. Angew. Chem., Int. Ed. 2005, 44, 6576–6579. doi:10.1002/anie.200500227
Return to citation in text: [1] [2] [3] -
Yevich, J. P.; Yocca, F. D. Curr. Med. Chem. 1997, 4, 295–312.
Return to citation in text: [1] -
Arendt, J.; Deacon, S. Chronobiol. Int. 1997, 14, 185–204. doi:10.3109/07420529709001155
Return to citation in text: [1] -
Laine, A. E.; Lood, C.; Koskinen, A. M. P. Molecules 2014, 19, 1544–1567. doi:10.3390/molecules19021544
Return to citation in text: [1] -
Roca-López, D.; Marqués-López, E.; Alcaine, A.; Merino, P.; Herrera, R. P. Org. Biomol. Chem. 2014, 12, 4503–4510. doi:10.1039/c4ob00348a
Return to citation in text: [1] [2] -
Marqués-López, E.; Alcaine, A.; Tejero, T.; Herrera, R. P. Eur. J. Org. Chem. 2011, 3700–3705. doi:10.1002/ejoc.201100506
Return to citation in text: [1] -
Ganesh, M.; Seidel, D. J. Am. Chem. Soc. 2008, 130, 16464–16465. doi:10.1021/ja8063292
Return to citation in text: [1] -
Wittkopp, A.; Schreiner, P. R. Chem. – Eur. J. 2003, 9, 407–414. doi:10.1002/chem.200390042
Return to citation in text: [1] -
Loh, C. C. J.; Badorrek, J.; Raabe, G.; Enders, D. Chem. – Eur. J. 2011, 17, 13409–13414. doi:10.1002/chem.201102793
Return to citation in text: [1] [2] -
Joseph, B.; Chapellier, V.; Mérour, J.-Y.; Léonce, S. Heterocycles 1998, 48, 1423–1430. doi:10.3987/COM-98-8178
Return to citation in text: [1] -
Joseph, B.; Alagille, D.; Mérour, J.-Y.; Léonce, S. Chem. Pharm. Bull. 2000, 48, 1872–1876. doi:10.1248/cpb.48.1872
Return to citation in text: [1] -
Loh, C. C. J.; Raabe, G.; Enders, D. Chem. – Eur. J. 2012, 18, 13250–13254. doi:10.1002/chem.201202908
Return to citation in text: [1] -
Glennon, R. A.; Lee, M.; Rangisetty, J. B.; Dukat, M.; Roth, B. L.; Savage, J. E.; MacBride, A.; Rauser, L.; Hufeisen, S.; Lee, D. K. H. J. Med. Chem. 2000, 43, 1011–1018. doi:10.1021/jm990550b
Return to citation in text: [1] -
Glennon, R. A.; Roth, B. L. Selective 5-HT6 Receptor Ligands. WO Patent WO2000/034242, June 15, 2000.
Return to citation in text: [1] -
Chang-Fong, J.; Rangisetty, J. B.; Dukat, M.; Setola, V.; Raffay, T.; Roth, B.; Glennon, R. A. Bioorg. Med. Chem. Lett. 2004, 14, 1961–1964. doi:10.1016/j.bmcl.2004.01.071
Return to citation in text: [1] -
Cole, D. C.; Lennox, W. J.; Stock, J. R.; Ellingboe, J. W.; Mazandarani, H.; Smith, D. L.; Zhang, G.; Tawa, G. J.; Schechter, L. E. Bioorg. Med. Chem. Lett. 2005, 15, 4780–4785. doi:10.1016/j.bmcl.2005.07.028
Return to citation in text: [1] -
Dukat, M.; Mosier, P. D.; Kolanos, R.; Roth, B. L.; Glennon, R. A. J. Med. Chem. 2008, 51, 603–611. doi:10.1021/jm070910s
Return to citation in text: [1] -
Ciamician, G.; Plancher, G. Ber. Dtsch. Chem. Ges. 1896, 29, 2475–2482. doi:10.1002/cber.18960290318
Return to citation in text: [1] -
Loh, C. C. J.; Atodiresei, I.; Enders, D. Chem. – Eur. J. 2013, 19, 10822–10826. doi:10.1002/chem.201302131
Return to citation in text: [1] -
Vacca, J. P.; Dorsey, B. D.; Schleif, W. A.; Levin, R. B.; McDaniel, S. L.; Darke, P. L.; Zugay, J.; Quintero, J. C.; Blahy, O. M.; Roth, E.; Sardana, V. V.; Schlabach, A. J.; Graham, P. I.; Condra, J. H.; Gotlib, L.; Holloway, M. K.; Lin, J.; Chen, I.-W.; Vastag, K.; Ostovic, D.; Anderson, P. S.; Emini, E. A.; Huff, J. R. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 4096–4100. doi:10.1073/pnas.91.9.4096
Return to citation in text: [1] -
Dorsey, B. D.; Levin, R. B.; McDaniel, S. L.; Vacca, J. P.; Guare, J. P.; Darke, P. L.; Zugay, J. A.; Emini, E. A.; Schleif, W. A.; Quintero, J. C.; Lin, J. H.; Chen, I.-W.; Holloway, M. K.; Fitzgerald, P. M. D.; Axel, M. G.; Ostovic, D.; Anderson, P. S.; Huff, J. R. J. Med. Chem. 1994, 37, 3443–3451. doi:10.1021/jm00047a001
Return to citation in text: [1] -
Jiang, H.; Paixão, M. W.; Monge, D.; Jørgensen, K. A. J. Am. Chem. Soc. 2010, 132, 2775–2783. doi:10.1021/ja9097803
Return to citation in text: [1] [2] [3] -
Juste-Navarro, V.; Marqués-López, E.; Herrera, R. P. Asian J. Org. Chem. 2015, 4, 884–889. doi:10.1002/ajoc.201500154
Return to citation in text: [1] [2] -
Herrera, R. P.; Monge, D.; Martín-Zamora, E.; Fernández, R.; Lassaletta, J. M. Org. Lett. 2007, 9, 3303–3306. doi:10.1021/ol071292c
Return to citation in text: [1] -
Sibi, M. P.; Itoh, K. J. Am. Chem. Soc. 2007, 129, 8064–8065. doi:10.1021/ja071739c
Return to citation in text: [1] -
Shi, X.; He, W.; Li, H.; Zhang, X.; Zhang, S. Tetrahedron Lett. 2011, 52, 3204–3207. doi:10.1016/j.tetlet.2011.04.043
Return to citation in text: [1] -
Liu, X.-L.; Wu, Z.-J.; Du, X.-L.; Zhang, X.-M.; Yuan, W.-C. J. Org. Chem. 2011, 76, 4008–4017. doi:10.1021/jo2004378
Return to citation in text: [1] -
Larsen, T. O.; Frydenvang, K.; Frisvad, J. C.; Christophersen, C. J. Nat. Prod. 1998, 61, 1154–1157. doi:10.1021/np980056v
Return to citation in text: [1] -
Ariza, M. R.; Larsen, T. O.; Petersen, B. O.; Duus, J. Ø.; Christophersen, C.; Barrero, A. F. J. Nat. Prod. 2001, 64, 1590–1592. doi:10.1021/np0101550
Return to citation in text: [1] -
Larsen, T. O.; Petersen, B. O.; Duus, J. Ø.; Sørensen, D.; Frisvad, J. C.; Hansen, M. E. J. Nat. Prod. 2005, 68, 871–874. doi:10.1021/np040248s
Return to citation in text: [1] -
Dong, Z.; Qiu, G.; Zhou, H.-B.; Dong, C. Tetrahedron: Asymmetry 2012, 23, 1550–1556. doi:10.1016/j.tetasy.2012.10.016
Return to citation in text: [1] -
Owens, T. D.; Hollander, F. J.; Oliver, A. G.; Ellman, J. A. J. Am. Chem. Soc. 2001, 123, 1539–1540. doi:10.1021/ja005635c
Return to citation in text: [1] -
Owens, T. D.; Souers, A. J.; Ellman, J. A. J. Org. Chem. 2003, 68, 3–10. doi:10.1021/jo020524c
Return to citation in text: [1] -
Schenkel, L. B.; Ellman, J. A. Org. Lett. 2003, 5, 545–548. doi:10.1021/ol027468m
Return to citation in text: [1] -
Pei, D.; Wang, Z.; Wei, S.; Zhang, Y.; Sun, J. Org. Lett. 2006, 8, 5913–5915. doi:10.1021/ol062633+
Return to citation in text: [1] -
Solà, J.; Revés, M.; Riera, A.; Verdaguer, X. Angew. Chem., Int. Ed. 2007, 46, 5020–5023. doi:10.1002/anie.200701040
Return to citation in text: [1] -
Tan, K. L.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2007, 46, 1315–1317. doi:10.1002/anie.200603354
Return to citation in text: [1] -
Robak, M. T.; Trincado, M.; Ellman, J. A. J. Am. Chem. Soc. 2007, 129, 15110–15111. doi:10.1021/ja075653v
Return to citation in text: [1] -
Soh, J. Y.-T.; Tan, C.-H. J. Am. Chem. Soc. 2009, 131, 6904–6905. doi:10.1021/ja900582a
Return to citation in text: [1] -
Almaşi, D.; Alonso, D. A.; Nájera, C. Tetrahedron: Asymmetry 2006, 17, 2064–2068. doi:10.1016/j.tetasy.2006.07.023
Return to citation in text: [1] -
Almaşi, D.; Alonso, D. A.; Gómez-Bengoa, E.; Nagel, Y.; Nájera, C. Eur. J. Org. Chem. 2007, 2328–2343. doi:10.1002/ejoc.200700031
Return to citation in text: [1] -
Blarer, S. J.; Seebach, D. Chem. Ber. 1983, 116, 2250–2260. doi:10.1002/cber.19831160616
Return to citation in text: [1]
| 37. | Vacca, J. P.; Dorsey, B. D.; Schleif, W. A.; Levin, R. B.; McDaniel, S. L.; Darke, P. L.; Zugay, J.; Quintero, J. C.; Blahy, O. M.; Roth, E.; Sardana, V. V.; Schlabach, A. J.; Graham, P. I.; Condra, J. H.; Gotlib, L.; Holloway, M. K.; Lin, J.; Chen, I.-W.; Vastag, K.; Ostovic, D.; Anderson, P. S.; Emini, E. A.; Huff, J. R. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 4096–4100. doi:10.1073/pnas.91.9.4096 |
| 38. | Dorsey, B. D.; Levin, R. B.; McDaniel, S. L.; Vacca, J. P.; Guare, J. P.; Darke, P. L.; Zugay, J. A.; Emini, E. A.; Schleif, W. A.; Quintero, J. C.; Lin, J. H.; Chen, I.-W.; Holloway, M. K.; Fitzgerald, P. M. D.; Axel, M. G.; Ostovic, D.; Anderson, P. S.; Huff, J. R. J. Med. Chem. 1994, 37, 3443–3451. doi:10.1021/jm00047a001 |
| 39. | Jiang, H.; Paixão, M. W.; Monge, D.; Jørgensen, K. A. J. Am. Chem. Soc. 2010, 132, 2775–2783. doi:10.1021/ja9097803 |
| 39. | Jiang, H.; Paixão, M. W.; Monge, D.; Jørgensen, K. A. J. Am. Chem. Soc. 2010, 132, 2775–2783. doi:10.1021/ja9097803 |
| 1. | Weix, D. J.; Ellman, J. A. Org. Lett. 2003, 5, 1317–1320. doi:10.1021/ol034254b |
| 2. | Lazar, A.; Sharma, P.; Singh, A. P. Microporous Mesoporous Mater. 2013, 170, 331–339. doi:10.1016/j.micromeso.2012.12.014 |
| 11. | Probst, N.; Madarász, Á.; Valkonen, A.; Pápai, I.; Rissanen, K.; Neuvonen, A.; Pihko, P. M. Angew. Chem., Int. Ed. 2012, 51, 8495–8499. doi:10.1002/anie.201203852 |
| 22. | Roca-López, D.; Marqués-López, E.; Alcaine, A.; Merino, P.; Herrera, R. P. Org. Biomol. Chem. 2014, 12, 4503–4510. doi:10.1039/c4ob00348a |
| 44. | Liu, X.-L.; Wu, Z.-J.; Du, X.-L.; Zhang, X.-M.; Yuan, W.-C. J. Org. Chem. 2011, 76, 4008–4017. doi:10.1021/jo2004378 |
| 9. | Sibi, M. P.; Cook, G. R.; Liu, P. Tetrahedron Lett. 1999, 40, 2477–2480. doi:10.1016/S0040-4039(99)00281-6 |
| 10. | Turgut, Y.; Azizoglu, M.; Erdogan, A.; Arslan, N.; Hosgoren, H. Tetrahedron: Asymmetry 2013, 24, 853–859. doi:10.1016/j.tetasy.2013.05.016 |
| 23. | Marqués-López, E.; Alcaine, A.; Tejero, T.; Herrera, R. P. Eur. J. Org. Chem. 2011, 3700–3705. doi:10.1002/ejoc.201100506 |
| 45. | Larsen, T. O.; Frydenvang, K.; Frisvad, J. C.; Christophersen, C. J. Nat. Prod. 1998, 61, 1154–1157. doi:10.1021/np980056v |
| 6. | Berkessel, A.; Gröger, H. Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis; Wiley-VCH: Weinheim, 2005. doi:10.1002/3527604677 |
| 7. | Dalko, P. I., Ed. Enantioselective Organocatalysis: Reactions and Experimental Procedures; Wiley-VCH: Weinheim, 2007. doi:10.1002/9783527610945 |
| 8. | Dalko, P. I., Ed. Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions and Applications; Wiley-VCH: Weinheim, 2013. doi:10.1002/9783527658862 |
| 18. | Herrera, R. P.; Sgarzani, V.; Bernardi, L.; Ricci, A. Angew. Chem., Int. Ed. 2005, 44, 6576–6579. doi:10.1002/anie.200500227 |
| 42. | Sibi, M. P.; Itoh, K. J. Am. Chem. Soc. 2007, 129, 8064–8065. doi:10.1021/ja071739c |
| 3. | Dai, X.; Cahard, D. Adv. Synth. Catal. 2014, 356, 1317–1328. doi:10.1002/adsc.201301115 |
| 4. | Sani, M.; Volonterio, A.; Zanda, M. ChemMedChem 2007, 2, 1693–1700. doi:10.1002/cmdc.200700156 |
| 5. | Piras, M.; Fleming, I. N.; Harrison, W. T. A.; Zanda, M. Synlett 2012, 23, 2899–2902. doi:10.1055/s-0032-1317557 |
| 22. | Roca-López, D.; Marqués-López, E.; Alcaine, A.; Merino, P.; Herrera, R. P. Org. Biomol. Chem. 2014, 12, 4503–4510. doi:10.1039/c4ob00348a |
| 43. | Shi, X.; He, W.; Li, H.; Zhang, X.; Zhang, S. Tetrahedron Lett. 2011, 52, 3204–3207. doi:10.1016/j.tetlet.2011.04.043 |
| 17. | Liu, X.; Lu, Y. Org. Biomol. Chem. 2010, 8, 4063–4065. doi:10.1039/c0ob00223b |
| 19. | Yevich, J. P.; Yocca, F. D. Curr. Med. Chem. 1997, 4, 295–312. |
| 20. | Arendt, J.; Deacon, S. Chronobiol. Int. 1997, 14, 185–204. doi:10.3109/07420529709001155 |
| 40. | Juste-Navarro, V.; Marqués-López, E.; Herrera, R. P. Asian J. Org. Chem. 2015, 4, 884–889. doi:10.1002/ajoc.201500154 |
| 15. | Miyabe, H.; Takemoto, Y. Bull. Chem. Soc. Jpn. 2008, 81, 785–795. doi:10.1246/bcsj.81.785 |
| 16. | Connon, S. J. Chem. Commun. 2008, 2499–2510. doi:10.1039/B719249E |
| 21. | Laine, A. E.; Lood, C.; Koskinen, A. M. P. Molecules 2014, 19, 1544–1567. doi:10.3390/molecules19021544 |
| 41. | Herrera, R. P.; Monge, D.; Martín-Zamora, E.; Fernández, R.; Lassaletta, J. M. Org. Lett. 2007, 9, 3303–3306. doi:10.1021/ol071292c |
| 14. | Coste, A.; Bayle, A.; Marrot, J.; Evano, G. Org. Lett. 2014, 16, 1306–1309. doi:10.1021/ol403675c |
| 39. | Jiang, H.; Paixão, M. W.; Monge, D.; Jørgensen, K. A. J. Am. Chem. Soc. 2010, 132, 2775–2783. doi:10.1021/ja9097803 |
| 12. | Kohno, J.; Koguchi, Y.; Nishio, M.; Nakao, K.; Kuroda, M.; Shimizu, R.; Ohnuki, T.; Komatsubara, S. J. Org. Chem. 2000, 65, 990–995. doi:10.1021/jo991375+ |
| 13. |
Coste, A.; Couty, F.; Evano, G. C. R. Chim. 2008, 11, 1544–1573. doi:10.1016/j.crci.2008.06.003
See for a review. |
| 18. | Herrera, R. P.; Sgarzani, V.; Bernardi, L.; Ricci, A. Angew. Chem., Int. Ed. 2005, 44, 6576–6579. doi:10.1002/anie.200500227 |
| 40. | Juste-Navarro, V.; Marqués-López, E.; Herrera, R. P. Asian J. Org. Chem. 2015, 4, 884–889. doi:10.1002/ajoc.201500154 |
| 25. | Wittkopp, A.; Schreiner, P. R. Chem. – Eur. J. 2003, 9, 407–414. doi:10.1002/chem.200390042 |
| 18. | Herrera, R. P.; Sgarzani, V.; Bernardi, L.; Ricci, A. Angew. Chem., Int. Ed. 2005, 44, 6576–6579. doi:10.1002/anie.200500227 |
| 46. | Ariza, M. R.; Larsen, T. O.; Petersen, B. O.; Duus, J. Ø.; Christophersen, C.; Barrero, A. F. J. Nat. Prod. 2001, 64, 1590–1592. doi:10.1021/np0101550 |
| 24. | Ganesh, M.; Seidel, D. J. Am. Chem. Soc. 2008, 130, 16464–16465. doi:10.1021/ja8063292 |
| 47. | Larsen, T. O.; Petersen, B. O.; Duus, J. Ø.; Sørensen, D.; Frisvad, J. C.; Hansen, M. E. J. Nat. Prod. 2005, 68, 871–874. doi:10.1021/np040248s |
| 48. | Dong, Z.; Qiu, G.; Zhou, H.-B.; Dong, C. Tetrahedron: Asymmetry 2012, 23, 1550–1556. doi:10.1016/j.tetasy.2012.10.016 |
| 35. | Ciamician, G.; Plancher, G. Ber. Dtsch. Chem. Ges. 1896, 29, 2475–2482. doi:10.1002/cber.18960290318 |
| 36. | Loh, C. C. J.; Atodiresei, I.; Enders, D. Chem. – Eur. J. 2013, 19, 10822–10826. doi:10.1002/chem.201302131 |
| 29. | Loh, C. C. J.; Raabe, G.; Enders, D. Chem. – Eur. J. 2012, 18, 13250–13254. doi:10.1002/chem.201202908 |
| 58. | Almaşi, D.; Alonso, D. A.; Gómez-Bengoa, E.; Nagel, Y.; Nájera, C. Eur. J. Org. Chem. 2007, 2328–2343. doi:10.1002/ejoc.200700031 |
| 30. | Glennon, R. A.; Lee, M.; Rangisetty, J. B.; Dukat, M.; Roth, B. L.; Savage, J. E.; MacBride, A.; Rauser, L.; Hufeisen, S.; Lee, D. K. H. J. Med. Chem. 2000, 43, 1011–1018. doi:10.1021/jm990550b |
| 31. | Glennon, R. A.; Roth, B. L. Selective 5-HT6 Receptor Ligands. WO Patent WO2000/034242, June 15, 2000. |
| 32. | Chang-Fong, J.; Rangisetty, J. B.; Dukat, M.; Setola, V.; Raffay, T.; Roth, B.; Glennon, R. A. Bioorg. Med. Chem. Lett. 2004, 14, 1961–1964. doi:10.1016/j.bmcl.2004.01.071 |
| 33. | Cole, D. C.; Lennox, W. J.; Stock, J. R.; Ellingboe, J. W.; Mazandarani, H.; Smith, D. L.; Zhang, G.; Tawa, G. J.; Schechter, L. E. Bioorg. Med. Chem. Lett. 2005, 15, 4780–4785. doi:10.1016/j.bmcl.2005.07.028 |
| 34. | Dukat, M.; Mosier, P. D.; Kolanos, R.; Roth, B. L.; Glennon, R. A. J. Med. Chem. 2008, 51, 603–611. doi:10.1021/jm070910s |
| 59. | Blarer, S. J.; Seebach, D. Chem. Ber. 1983, 116, 2250–2260. doi:10.1002/cber.19831160616 |
| 28. | Joseph, B.; Alagille, D.; Mérour, J.-Y.; Léonce, S. Chem. Pharm. Bull. 2000, 48, 1872–1876. doi:10.1248/cpb.48.1872 |
| 56. | Soh, J. Y.-T.; Tan, C.-H. J. Am. Chem. Soc. 2009, 131, 6904–6905. doi:10.1021/ja900582a |
| 26. | Loh, C. C. J.; Badorrek, J.; Raabe, G.; Enders, D. Chem. – Eur. J. 2011, 17, 13409–13414. doi:10.1002/chem.201102793 |
| 57. | Almaşi, D.; Alonso, D. A.; Nájera, C. Tetrahedron: Asymmetry 2006, 17, 2064–2068. doi:10.1016/j.tetasy.2006.07.023 |
| 26. | Loh, C. C. J.; Badorrek, J.; Raabe, G.; Enders, D. Chem. – Eur. J. 2011, 17, 13409–13414. doi:10.1002/chem.201102793 |
| 49. | Owens, T. D.; Hollander, F. J.; Oliver, A. G.; Ellman, J. A. J. Am. Chem. Soc. 2001, 123, 1539–1540. doi:10.1021/ja005635c |
| 50. | Owens, T. D.; Souers, A. J.; Ellman, J. A. J. Org. Chem. 2003, 68, 3–10. doi:10.1021/jo020524c |
| 51. | Schenkel, L. B.; Ellman, J. A. Org. Lett. 2003, 5, 545–548. doi:10.1021/ol027468m |
| 52. | Pei, D.; Wang, Z.; Wei, S.; Zhang, Y.; Sun, J. Org. Lett. 2006, 8, 5913–5915. doi:10.1021/ol062633+ |
| 53. | Solà, J.; Revés, M.; Riera, A.; Verdaguer, X. Angew. Chem., Int. Ed. 2007, 46, 5020–5023. doi:10.1002/anie.200701040 |
| 54. | Tan, K. L.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2007, 46, 1315–1317. doi:10.1002/anie.200603354 |
| 27. | Joseph, B.; Chapellier, V.; Mérour, J.-Y.; Léonce, S. Heterocycles 1998, 48, 1423–1430. doi:10.3987/COM-98-8178 |
| 55. | Robak, M. T.; Trincado, M.; Ellman, J. A. J. Am. Chem. Soc. 2007, 129, 15110–15111. doi:10.1021/ja075653v |
© 2016 G. Sonsona et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)