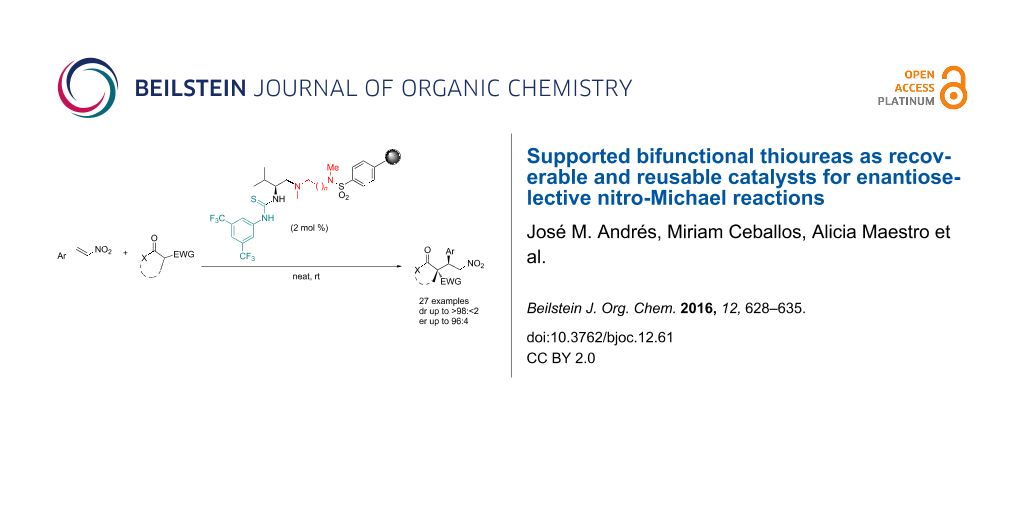
Abstract
The catalytic activity of different supported bifunctional thioureas on sulfonylpolystyrene resins has been studied in the nitro-Michael addition of different nucleophiles to trans-β-nitrostyrene derivatives. The activity of the catalysts depends on the length of the tether linking the chiral thiourea to the polymer. The best results were obtained with the thiourea derived from (L)-valine and 1,6-hexanediamine. The catalysts can be used in only 2 mol % loading, and reused for at least four cycles in neat conditions. The ball milling promoted additions also worked very well.

Graphical Abstract
Introduction
The use of chiral bifunctional thioureas that allow the simultaneous activation of a electrophile, by hydrogen bonding, and a nucleophile, by deprotonation, plays a major role in the stereoselective formation of C–C bonds in different transformations [1-5]. In these processes one of the major problems is related to the recovering of the catalysts. The support of the small molecules on different materials has been proposed as a solution, including their use in continuous flow processes [6-9]. The most popular supports include nanoparticles [10-12], inorganic solids [13,14], and different polystyrene derivatives [15-20].
Bifunctional thioureas were first supported on PEG [21], and later on different materials such as poly(methylhydrosiloxane) [16], polystyrene [18-22], and magnetic nanoparticles [12]. Cinchona-derived thioureas have been also prepared by co-polymerization of polyfunctionalized thiols with olefins [23].
Our interest in the search for novel bifunctional thioureas as organocatalysts [24-27] lead us to consider the preparation of different polymeric materials decorated with chiral bifunctional thioureas looking for a greener process [28], easier recovering and recyclability of the catalyst, and solvent-free reaction conditions. Along these lines, we have recently reported the bottom-up synthesis of polymeric thioureas [29], and the anchorage of (L)-valine-derived thiourea I [30] onto sulfonylpolystyrene resin leading to catalysts II–V (Figure 1), which differ in the length of the diamine linker or in the substitution pattern of the nitrogen in the sulfonamide. These materials, and the related unsupported thiourea VI, have been previously tested as excellent organocatalysts in the stereoselective aza-Henry reaction [31]. Now we describe the results obtained in different stereoselective nitro-Michael additions promoted by these materials.
![[1860-5397-12-61-1]](/bjoc/content/figures/1860-5397-12-61-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Parent and supported bifunctional thioureas used in this work.
Figure 1: Parent and supported bifunctional thioureas used in this work.
Results and Discussion
The ability of the supported catalysts (II–V) to promote the stereoselective nitro-Michael reaction was first tested in the reaction of trans-nitrostyrene (1a) with diethyl malonate (2a), leading to the enantioenriched addition product 4aa with a single stereocenter. In order to the creation of two tertiary-quaternary contiguous stereocenters (5aa) we also used ethyl 2-oxocyclopentanecarboxylate (3a) as nucleophile in neat conditions and in different solvents. For comparative purposes, the same reactions were studied in the presence of unsupported catalysts I and VI, and the results are summarized in Scheme 1 and Table 1.
![[1860-5397-12-61-i1]](/bjoc/content/inline/1860-5397-12-61-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reaction of nitrostyrene with diethyl malonate and 2-ethoxycarbonyl cyclopentanone.
Scheme 1: Reaction of nitrostyrene with diethyl malonate and 2-ethoxycarbonyl cyclopentanone.
Table 1: Screening of catalysts and optimization of the reaction conditions for the additions of diethyl malonate and ethyl 2-oxocyclopentanecarboxylate to β-nitrostyrene.
| Entrya | Catal. (mol %) | Solvent | T (°C) | t (h) | Product yieldb (%) | drc | erd |
|---|---|---|---|---|---|---|---|
| 1 | I (10) | neat | rt | 24 | 4aa (80) | – | 92:8 |
| 2 | II (10) | neat | rt | 120 | 4aa (77)e | – | 78:22 |
| 3 | III (10) | neat | rt | 120 | 4aa (63)f | – | 85:15 |
| 4 | IV (10) | neat | rt | 16 | 4aa (92) | – | 91:9 |
| 5 | IV (5) | neat | rt | 24 | 4aa (80) | – | 90:10 |
| 6 | V (5) | neat | rt | 16 | 4aa (92) | – | 89:11 |
| 7 | VI (5) | neat | rt | 16 | 4aa (100) | – | 94:6 |
| 8 | I (5) | neat | rt | 1 | 5aa (95) | 95:5 | 95:5 |
| 9 | II (10) | neat | rt | 4 | 5aa (100) | 89:11 | 90:10 |
| 10 | III /10) | neat | rt | 8 | 5aa (90) | 87:13 | 92:8 |
| 11 | IV (10) | neat | rt | 4 | 5aa (100) | 88:12 | 95:5 |
| 12 | IV (5) | neat | rt | 7 | 5aa (100) | 88:12 | 95:5 |
| 13 | V (5) | neat | rt | 0.5 | 5aa (98) | 89:11 | 95:5 |
| 14 | VI (5) | neat | rt | 2 | 5aa (100) | 89:11 | 95:5 |
| 15 | V (2) | neat | rt | 1 | 5aa (93) | 89:11 | 95:5 |
| 16 | V (2) | neat | 0 | 24 | 5aa (97) | 90:10 | 94:6 |
| 17g | V (2) | neat | rt | 8 | 5aa (86) | 88:12 | 94:6 |
| 18h | V (2) | neat | rt | 2.5 | 5aa (83) | 88:12 | 94:6 |
| 19 | V (2) | CH2Cl2 | rt | 1 | 5aa (74) | 90:10 | 93:7 |
| 20 | V (2) | PhMe | rt | 5 | 5aa (66) | 89:11 | 94:6 |
| 21 | V (2) | THF | rt | 1.5 | 5aa (55) | 89:11 | 94:6 |
| 22 | V (2) | MeCN | rt | 1.5 | 5aa (79) | 89:11 | 95:5 |
| 23 | V (2) | MeCN | 0 | 8 | 5aa (82) | 88:12 | 95:5 |
| 24g | V (2) | MeCN | rt | 8 | 5aa (86) | 89:11 | 92:8 |
aReaction performed with 2 equiv of nucleophile at room temperature. bYields refer to isolated compounds. cDetermined by 1H NMR in the reaction mixture. dDetermined by chiral HPLC. e13% of unreacted nitrostyrene was recovered. f15% of unreacted nitrostyrene was recovered. gReaction performed with 1.1 equiv of ketoester. hReaction performed with 1.5 equiv of ketoester.
Initially, the reactions were carried out in neat conditions at rt with twofold excess of nucleophile and 10 mol % of catalysts (entries 1–4 and 9–11 in Table 1). As a general trend, the reactions were faster, and much more stereoselective for ketoester 3a than for diethyl malonate 2a, and that the difference was specially remarkable when supported ethylenediamine-derived thioureas II and III were used as catalysts (compare entries 2 and 3 versus 9 and 10 in Table 1). It is noteworthy that the results obtained in the reaction catalyzed by supported thiourea IV were better than those observed in the addition catalyzed by the parent thiourea I (compare Table 1, entries 1 and 4).
The catalyst loading was decreased to 5 mol % for supported hexanediamine-derived catalyst IV, observing that the level of stereoselectivity was maintained for both reactions, although at expenses of slight increasing the reaction time (compare Table 1, entries 4 versus 5, and 11 versus 12). In these conditions, supported catalyst V, which differs from IV in the substitution pattern of the sulfonamide, was the best catalyst for the addition of both 2a and 3a to nitrostyrene, yielding products 4aa and 5aa, respectively, in much better yield maintaining the stereoselectivity in shorter reaction time (compare entries 5 versus 6 and 12 versus 13 in Table 1). An increase in both the yield and the enantioselectivity for the reaction of 1a with 2a (Table 1, entries 6 and 7), but no differences in the reaction of 1a with 3a (Table 1, entries 13 and 14) were observed when the unsupported thiourea VI, homologous to V, was used as organocatalyst.
Taking the supported thiourea V as the catalyst of choice, the effects of the catalyst loading, the temperature, the ratio of nucleophile, and the use of different solvents were studied for the reaction of 1a and 3a. Fortunately, the reduction of the amount of catalyst to 2 mol % did not influence the stereoselectivity, and only slightly decreased the yield to 93% (compare Table 1, entries 13 and 15). The reaction also proceeded at 0 °C, leading to the addition product 5aa in 97% and very good stereoselectivity, but in those conditions the reaction time increased to 24 h (Table 1, entry 16).
The reduction of the amount of the nucleophile to 1.1 equivalents (Table 1, entry 17) or 1.5 equivalents (Table 1, entry 18) had only a moderate effect on the yield of the reaction time, increasing it to 8 h and 2,5 h, respectively. The yield dropped to 55–74%, without change in the stereoselectivity, when the reaction was carried out in less polar solvents such as DCM, toluene, or THF (Table 1, entries 19–21), although both the yield and diastereo- and enantioselectivity were maintained when acetonitrile, a more polar solvent, was used (Table 1, entries 22–24).
We next consider the reaction of some 4-substituted nitrostyrenes (1b–d) with diethyl malonate (2a), and the addition of a range of β-functionalized nucleophiles 2b–g to 1a promoted by supported catalysts IV (5 mol %) and V (2 mol %), respectively (Scheme 2 and Table 2). The results obtained in the reactions of 4-chloro- (1b) and 4-fluoronitrostyrene (1c) were very similar than those obtained in the reaction with β-nitrostyrene (1a), maintaining the yield and the enantioselectivity, although 1c reacted slowly within 48 h (compare entries 1 and 2 in Table 2 versus entry 5 in Table 1), but the less reactive 4-methoxy derivative 1d only yielded the addition product in moderate 52% yield after 72 h of reaction (entry 3 in Table 2).
![[1860-5397-12-61-i2]](/bjoc/content/inline/1860-5397-12-61-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction of nitrostyrenes with malonates and β-diketones.
Scheme 2: Reaction of nitrostyrenes with malonates and β-diketones.
Table 2: Addition of malonates and β-diketones to nitrostyrenes catalyzed by IV and V.
| Entrya | Reagents | Catal. (mol %) | t (h) | Product yieldb (%) | drc | erd | Config. |
|---|---|---|---|---|---|---|---|
| 1 | 1b/2a | IV (5) | 6 | 4ba (76) | – | 83:17 | (S) |
| 2 | 1c/2a | IV (5) | 48 | 4ca (72) | – | 86:14 | (S) |
| 3 | 1d/2a | IV (5) | 72 | 4da (52) | – | 89:11 | (S) |
| 4 | 1a/2b | IV (5) | 36 | 4ab (64) | – | 92:8 | (R) |
| 5 | 1a/2d | IV (5) | 48 | 4ad (74) | – | 92:8 | (S) |
| 6 | 1a/2e | IV (5) | 5 | 4ae (88) | – | 89:11 | (S) |
| 7 | 1a/2f | IV (5) | 6 | 4af (70) | – | 93:7 | (R) |
| 8 | 1a/2g | IV (5) | 7 | 4ag (99) | 75:25 | 92:8 | (2R,3R) |
| 9 | 1a/2b | V (2) | 2 | 4ab (46) | – | 91:9 | (R) |
| 10 | 1a/2c | V (2) | 8 | 4ac (47) | – | 91:9 | (R) |
| 11 | 1a/2d | V (2) | 2 | 4ad (43) | – | 91:9 | (S) |
| 12 | 1a/2e | V (5) | 2 | 4ae (87) | – | 92:8 | (S) |
| 13 | 1a/2f | V (2) | 3 | 4af (65) | – | 93:7 | (R) |
| 14 | 1a/2g | V (2) | 6 | 4ag (81) | 74:26 | 94:6 | (2R,3R) |
aReaction performed with 2 equiv of nucleophile. bYields determined after chromatographic purifications. cDiastereomeric excess determined by 1H NMR of the crude reaction mixture. dEnantiomeric ratio determined by chiral HPLC analysis.
The reaction of different nucleophiles (2a–g) with 1a catalyzed by IV (5 mol %) also worked well in terms of enantioselectivity, maintaining the er near constant around or higher than 90:10, although the stereoselectivity for the reaction leading to 4ag, with two contiguous tertiary–quaternary stereocenters was only moderate (dr 75:25, entry 8 in Table 2). The main difference in those additions was related with the reaction time, because the less reactive malonates (2b and 2d) reacted slower than the acetylacetone (2e and 2f) or ethyl acetoacetate (2g) derivatives.
Similar results were obtained when the reaction was run in the presence of only 2 mol % of catalyst V (entries 9–14 in Table 2). The level of stereoselectivity was maintained, including the diastereoselectivity for the reaction of 1a with 2g (dr 74:26), but the yields for the reactions with 2-substituted malonates (2b–d) were only moderate (entries 9–11 in Table 2).
Next, we extend the study to the reaction of different β-functionalized cycloalkanones (3a–c), 2-acetylcyclopentanone (3d), and 2-acetyl-γ-lactone (3e) with nitrostyrene derivatives 1a–d (Scheme 3 and Table 3). The electronic nature of the nitroolefins only affects the yield and the reaction time, being longer as the donating effect of the substituent increases, but maintaining good diastereoselectivity and excellent enantioselectivity for all reactions (entries 2–4, Table 3).
![[1860-5397-12-61-i3]](/bjoc/content/inline/1860-5397-12-61-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of nitrostyrenes with β-keto esters and β-dicarbonyl compounds.
Scheme 3: Reaction of nitrostyrenes with β-keto esters and β-dicarbonyl compounds.
Table 3: Reaction of nitrostyrenes with β-substituted cycloalkanones catalyzed by V.
| Entrya | Reagents | Catal. (mol %) | t (h) | Product Yieldb (%) | drc | erd | Config. |
|---|---|---|---|---|---|---|---|
| 1 | 1a/3a | V (5) | 0.5 | 5aa (98) | 89:11 | 95:5 | (S,R) |
| 2 | 1b/3a | V (2) | 1 | 5ba (84) | 90:10 | 94:6 | (S,R) |
| 3 | 1c/3a | V (2) | 5 | 5ca (76) | 89:11 | 93:7 | (S,R) |
| 4 | 1d/3a | V (2) | 9 | 5da (74) | 89:11 | 95:5 | (S,R) |
| 5 | 1a/3a | V (2) | 1 | 5aa (93) | 89:11 | 95:5 | (S,R) |
| 6 | 1a/3b | V (2) | 96 | 5ab (63) | 88:12 | 96:4 | (S,R) |
| 7 | 1a/3b | V (5) | 10 | 5ab (62) | 88:12 | 96:4 | (S,R) |
| 8 | 1a/3c | V (2) | 24 | 5ac (73) | 82:18 | 96:4 | (S,R) |
| 9 | 1a/3d | V (2) | 2 | 5ad (92) | 83:17 | 93:7 | (R,R) |
| 10 | 1a/3e | V (2) | 1 | 5ae (89) | 70:30 | 92:8 | (R,R) |
| 11e | 1a/3a | V (5) | 1 | 5aa (81) | 88:12 | 94:6 | (S,R) |
| 12f | 1a/3a | V (5) | 1 | 5aa (76) | 88:12 | 95:5 | (S,R) |
| 13g | 1a/3a | V (5) | 2 | 5aa (82) | 88:12 | 94:6 | (S,R) |
aReaction performed with 2 equiv of nucleophile. bYields determined after chromatographic purifications. cDiastereomeric excess determined by 1H NMR of the crude reaction mixture. dEnantiomeric ratio determined by chiral HPLC analysis. eSecond cycle for entry 1 by using only 1.5 equivalents of nucleophile. fThird cycle for entry 1 by using only 1.5 equivalents of nucleophile. gFourth cycle for entry 1 by using only 1.5 equivalents of nucleophile.
With respect to the nucleophile, it is noteworthy that there seems to be a correlation: With growing size of the cycloalkanone an increase of the reaction time and a decrease of the yield can be observed. Cycloheptanone 3c, and specially cyclohexanone 3b reacted much more slowly than cyclopentanone derivative 3a (Table 3, entries 5, 6, and 8), although the reaction time could be reduced from 96 h to 10 h in the reaction of 1a with 3b by increasing the catalyst loading from 2 mol % to 5 mol % (Table 3, entry 7). It is also noteworthy that 2-acetyl-γ-lactone (3e) reacted very well with 1a, leading to 5ae in excellent yield and enantioselectivity, but only moderate diastereoselectivity (dr 70:30, Table 3, entry 10).
To test the recyclability of catalyst V we choose the reaction of 1a with 3a in neat conditions and 5 mol % of catalyst as model. Once the reaction had finished (TLC), the catalyst was recovered by filtration, and after washing with methanol and drying to constant weight, the catalyst was used in the next cycle (entries 1 and 11–13 in Table 3). Fortunately, catalyst V can be reused for at least four cycles yielding 5aa in high yields (76–82%), and maintaining both the diastereo- and enantioselectivity.
Finally, α-nitrocyclohexanone (6a) and ethyl α-nitropropionate (6b) were included in the screening process in order to compare the results obtained with more acidic nucleophiles with those obtained with β-dicarbonyl derivatives. The reactions were carried out under different conditions by varying the solvent, the temperature and using unsupported thioureas I and VI, and V as an example of a supported one (Scheme 4). The most significant difference between the reactions of nitroketone and nitroester refers to the stereoselectivity in both processes. The addition of nitroketone was totally diastereoselective and highly enantioselective (entries 1–12 in Table 4), whereas both the diastero- and enantioselectivities were moderate for the addition of nitroester (Table 4, entries 13–15). Additionally, supported catalyst V was able to promote more enantioselective transformations than unsupported catalyst I (compare Table 4, entry 13 versus 15). The decrease of the temperature slows down the reaction, increasing the reaction time from 1 h to 6 h for the reaction of 1a with 6b, catalyzed by I (Table 4, entries 13 and 14), and from 24 h to 48 h for the addition of 6a to 1a catalyzed for VI (Table 4, entries 7 and 8), but maintaining the diastereoselectivity and slightly increasing the enantioselectivity. The donor character of the substituent in the nitrostyrene derivative plays an important role in the process, increasing the reaction time, and decreasing the yield (compare Table 4, entries 5, 9, and 11).
![[1860-5397-12-61-i4]](/bjoc/content/inline/1860-5397-12-61-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of nitrostyrenes with α-nitrocyclohexanone and ethyl α-nitropropionate.
Scheme 4: Reaction of nitrostyrenes with α-nitrocyclohexanone and ethyl α-nitropropionate.
Table 4: Reactions of nitrostyrenes with α-nitrocyclohexanone and ethyl α-nitropropionate.
| Entrya | Reagents | Catal. (mol %) | t (h) | Solvent | Product yieldb (%) | drc | erd |
|---|---|---|---|---|---|---|---|
| 1 | 1a/6a | I (5) | 3 | DCM | 7aa (62) | >98:<2 | 84:16 |
| 2 | 1a/6a | I (5) | 1.5 | neat | 7aa (65) | >98:<2 | 90/10 |
| 3e | 1a/6a | I (5) | 1 | neat | 7aa (70) | >98:<2 | 90:10 |
| 4 | 1a/6a | V (5) | 48 | DCM | 7aa (60) | >98:<2 | 90:10 |
| 5 | 1a/6a | V (5) | 12 | MeCN | 7aa (77) | >98:<2 | 91:9 |
| 6e | 1a/6a | V (5) | 12 | neat | 7aa (85) | >98:<2 | 94:6 |
| 7 | 1a/6a | VI (5) | 24 | DCM | 7aa (64) | >98:<2 | 85:15 |
| 8f | 1a/6a | VI (5) | 48 | DCM | 7aa (52) | >98:<2 | 92:8 |
| 9 | 1b/6a | V (5) | 8 | MeCN | 7ba (91) | >98:<2 | 91:9 |
| 10e | 1b/6a | V (5) | 48 | neat | 7ba (48)g | >98:<2 | 90:10 |
| 11 | 1d/6a | V (5) | 24 | MeCN | 7da(73) | >98:<2 | 89:11 |
| 12e | 1d/6a | V (5) | 360 | neat | 7da(50)h | >98:<2 | 87:13 |
| 13 | 1a/6b | I (5) | 1 | neat | 7ab (75) | 74:26 | 69:31 |
| 14f | 1a/6b | I (5) | 6 | neat | 7ab (77) | 75:25 | 72:28 |
| 15 | 1a/6b | V (5) | 5 | neat | 7ab (90) | 76:24 | 74:26 |
aReaction performed with 1.5 equiv of nucleophile. bYields determined after chromatographic purifications. cDiastereomeric excess determined by 1H NMR of the crude reaction mixture (>98:<2 means that a single diastereomer was detected). dEnantiomeric ratio determined by chiral HPLC analysis. eBall mill conditions. fThe reaction was carried out at −20 °C. g37% of 2-nitrocyclohexanone was recovered unreacted. h30% of 2-nitrocyclohexanone was recovered unreacted.
The recent interest in alternative activation modes [32] for promoting C–C bond formations, led us to consider the catalytic addition of α-nitrocyclohexanone (6a) to nitrostyrene derivatives 1a, 1b, and 1d under ball milling conditions [33]. We chose these reactions for comparative purposes because they have been previously reported [34]. Although the experimental conditions have not been optimized, we observed that both the unsupported (I) and supported (V) catalysts worked very well in the reaction of 1a with 6a yielding the addition product 7aa in excellent yields and stereoselectivity (entries 3 and 6 in Table 4). On the contrary, the reactions of 6a with 1b and 1d are very slow and 37% and 30% of the nucleophile were recovered unchanged after 48 h and 360 h of reaction, respectively, although maintaining the stereoselectivity (Table 4, entries 10 and 12). The improvement of that process is under study.
Conclusion
In summary, supported bifunctional thioureas on sulfonylpolystyrene are able to promote highly stereoselective nitro-Michael reactions with different nucleophiles. The activity of the catalysts varies with the length of the tether between the polymer and the thiourea framework, and the best results were obtained by using catalysts IV and V, derived from 1,6-hexanediamine. The reactions work well by using only 2 mol % of catalyst loading in neat conditions, and the catalysts can easily be recovered and reused for four cycles. The results obtained with the described catalysts are similar to those previously reported by using bottom-up synthesized materials prepared by co-polymerization of monomeric thioureas as organocatalysts [29]. Catalyst V has been also used in the addition of α-nitrocyclohexanone (6a) to β-nitrostyrene (1a) under solvent-free conditions in a ball mill providing the addition product 7aa in excellent yield, total diastereoselectivity, and very good enantioselectitvity.
Experimental
General remarks
13C NMR (126 MHz) and 1H NMR (500 MHz) spectra were recorded in CDCl3 as the solvent. Chemical shifts for carbons are reported in ppm from TMS and are referenced to the carbon resonance of the solvent. Chemical shifts for protons are reported in ppm from TMS with the residual CHCl3 resonance as internal reference. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad), coupling constants in Hertz, and integration.
Flash chromatography was carried out using silica gel (230–240 mesh). TLC analysis was performed on glass-backed plates coated with silica gel 60 and an F254 indicator, and visualized by either UV irradiation or by staining with phosphomolybdic acid solution. Specific rotations were measured on a digital polarimeter using a 5 mL cell with a 1 dm path length, and a sodium lamp, and the concentration is given in g per 100 mL. Chiral HPLC analysis was performed by using Daicel Chiralcel OD or Chiralpak AD-H, analytical columns (250 × 4.6 mm) by using mixture of n-hexane/isopropanol as eluent. UV detection was monitored at 220 or at 254 nm. ESI mass spectra were obtained on an Agilent 5973 inert GC/MS system. Commercially available organic and inorganic compounds were used without further purification. Solvents were dried and stored over microwave–activated 4 Å molecular sieves. Supported thioureas II–V and unsupported thioureas I and VI were prepared according to reported procedures [30,31]. Racemic reference samples were prepared by using DABCO (5 mol %) following the same procedure as described below.
General procedure for the nitro-Michael reaction using homogeneous catalysts (I and VI). The reactions were carried out as previously described [29]. To a mixture of nitrostyrene (0.3 mmol) and catalyst (0.015 mmol, 0.05 equiv), the 1,3-dicarbonyl compound (0.6 mmol, 2 equiv) was added and the reaction mixture was stirred at rt in a Wheaton vial until consumption of the starting material (monitoring by TLC). The reaction mixture was purified by column chromatography to afford the Michael product. The anti- and syn-isomers of the Michael products were not separated by column chromatography. The diastereomeric ratio was determined by 1H NMR spectroscopy of the purified product.
General procedure for the nitro-Michael reaction using inmobilized catalysts (II, III, IV and V) [29]. To a mixture of β-nitrostyrene (0.3 mmol) and catalyst (0.015 mmol, 0.05 equiv), 1,3-dicarbonyl compound (0.6 mmol, 2 equiv) was added and the reaction mixture was stirred at rt in a Wheaton vial until consumption of the starting material (monitored by TLC). The catalyst was filtered off and washed with MeOH (3 × 1 mL). The solvent was removed under reduced pressure, and the residue was purified by column chromatography. The anti- and syn-isomers of the Michael products were not separated by column chromatography. The diastereomeric ratio was determined by 1H NMR spectroscopy of the purified product.
Recyclability of the supported thiourea catalysts in nitro-Michael reaction. The supported catalysts were recovered from the reaction mixtures by filtration, thoroughly washed with methanol, dried and reused in the next cycle.
General procedure for the Michael addition of 2-nitrocyclohexanone to β-nitrostyrene using immobilized catalyst VI
Method A, in organic solvent: The reactions were carried out as previously described [29]. To a stirred solution of 2-nitrocyclohexanone (43 mg, 0.3 mmol) and nitroalkene (0.45 mmol, 1.5 equiv) in an adequate solvent (0.4 mL), catalyst VI (15 mg, 0.015 mmol, 0.05 equiv) was added and the reaction mixture was stirred at room temperature in a Wheaton vial until the reaction was finished (TLC). The catalyst was filtered off and washed with DCM (3 × 1 mL). The solvent was removed under reduced pressure, the crude mixture subjected to flash chromatography to afford the Michael adduct. The diastereomeric ratio was determined by 1H NMR spectroscopy of the purified product. The enantiomeric excess was determined by chiral-phase HPLC analysis using mixtures of hexane/isopropanol as eluent.
Method B, under ball-milling conditions: Catalyst VI (15 mg, 0.015 mmol, 0.05 equiv), 2-nitrocyclohexanone (43 mg, 0.3 mmol) and nitroalkene (0.45 mmol, 1.5 equiv) were transferred to a clean, dry ball milling vessel (cylinder of 5 mL) loaded with two grinding balls with a 7 mm diameter. The vessel was placed in a Mixer Mill MM 200 and the mixture was milled at 5 Hz of vibrational frequency at room temperature until consumption of the starting material (monitored by TLC). The vessel and the balls were washed with CH2Cl2, the catalyst was filtered off and washed with CH2Cl2 and methanol. The resulting solution was concentrated in vacuo, and the product was purified by flash chromatography. The diastereomeric ratio was determined by 1H NMR spectroscopy of the purified product. The enantiomeric excess was determined by chiral-phase HPLC analysis using mixtures of hexane/isopropanol as eluent.
Supporting Information
| Supporting Information File 1: Physical and spectral data for all the compounds. Copies of 1H, 13C NMR spectra, and HPLC traces for all compounds synthesized. | ||
| Format: PDF | Size: 955.6 KB | Download |
References
-
Takemoto, Y. Org. Biomol. Chem. 2005, 3, 4299–4306. doi:10.1039/b511216h
Return to citation in text: [1] -
Taylor, M. S.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2006, 45, 1520–1543. doi:10.1002/anie.200503132
Return to citation in text: [1] -
Connon, S. J. Chem. Commun. 2008, 2499–2510. doi:10.1039/B719249E
Return to citation in text: [1] -
Connon, S. J. Synlett 2009, 354–376. doi:10.1055/s-0028-1087557
Return to citation in text: [1] -
Lu, L.-Q.; An, X.-L.; Chen, J.-R.; Xiao, W.-J. Synlett 2012, 490–508. doi:10.1055/s-0031-1290131
Return to citation in text: [1] -
Ayats, C.; Henseler, A. H.; Pericàs, M. A. ChemSusChem 2012, 5, 320–325. doi:10.1002/cssc.201100570
Return to citation in text: [1] -
Bortolini, O.; Caciolli, L.; Cavazzini, A.; Costa, V.; Greco, R.; Massi, A.; Pasti, L. Green Chem. 2012, 14, 992–1000. doi:10.1039/c2gc16673a
Return to citation in text: [1] -
Rodríguez-Escrich, C.; Pericàs, M. A. Eur. J. Org. Chem. 2015, 1173–1188. doi:10.1002/ejoc.201403042
Return to citation in text: [1] -
Kasaplar, P.; Ozkal, E.; Rodríguez-Escrich, C.; Pericàs, M. A. Green Chem. 2015, 17, 3122–3129. doi:10.1039/C5GC00496A
Return to citation in text: [1] -
Gawande, M. B.; Branco, P. S.; Varma, R. S. Chem. Soc. Rev. 2013, 42, 3371–3393. doi:10.1039/c3cs35480f
Return to citation in text: [1] -
Jiang, X.; Zhu, H.; Shi, X.; Zhong, Y.; Li, Y.; Wang, R. Adv. Synth. Catal. 2013, 355, 308–314. doi:10.1002/adsc.201201038
Return to citation in text: [1] -
Gleeson, O.; Davies, G.-L.; Peschiulli, A.; Tekoriute, R.; Gun’ko, Y. K.; Connon, S. J. Org. Biomol. Chem. 2011, 9, 7929–7940. doi:10.1039/c1ob06110k
Return to citation in text: [1] [2] -
Puglisi, A.; Annunziata, R.; Benaglia, M.; Cozzi, F.; Gervasini, A.; Bertacche, V.; Sala, M. C. Adv. Synth. Catal. 2009, 351, 219–229. doi:10.1002/adsc.200800635
Return to citation in text: [1] -
Yu, P.; He, J.; Guo, C. Chem. Commun. 2008, 2355–2357. doi:10.1039/b800640g
Return to citation in text: [1] -
Gruttadauria, M.; Giacalone, F.; Noto, R. Chem. Soc. Rev. 2008, 37, 1666–1688. doi:10.1039/b800704g
Return to citation in text: [1] -
Puglisi, A.; Benaglia, M.; Annunziata, R.; Siegel, J. S. ChemCatChem 2012, 4, 972–975. doi:10.1002/cctc.201200114
Return to citation in text: [1] [2] -
Fotaras, S.; Kokotos, C. G.; Kokotos, G. Org. Biomol. Chem. 2012, 10, 5613–5619. doi:10.1039/c2ob25693b
Return to citation in text: [1] -
Tuchman-Shukron, L.; Miller, S. J.; Portnoy, M. Chem. – Eur. J. 2012, 18, 2290–2296. doi:10.1002/chem.201102474
Return to citation in text: [1] [2] -
Li, J.; Yang, G.; Cui, Y. J. Appl. Polym. Sci. 2011, 121, 1506–1511. doi:10.1002/app.33676
Return to citation in text: [1] [2] -
Li, J.; Yang, G.; Qin, Y.; Yang, X.; Cui, Y. Tetrahedron: Asymmetry 2011, 22, 613–618. doi:10.1016/j.tetasy.2011.03.016
Return to citation in text: [1] [2] -
Miyabe, H.; Tuchida, S.; Yamauchi, M.; Takemoto, Y. Synthesis 2006, 3295–3300. doi:10.1055/s-2006-950196
Return to citation in text: [1] [2] -
Chuan, Y.; Chen, G.; Peng, Y. Tetrahedron Lett. 2009, 50, 3054–3058. doi:10.1016/j.tetlet.2009.04.011
Return to citation in text: [1] -
Fredriksen, K. A.; Kristensen, T. E.; Hansen, T. Beilstein J. Org. Chem. 2012, 8, 1126–1133. doi:10.3762/bjoc.8.125
Return to citation in text: [1] -
Manzano, R.; Andrés, J. M.; Muruzábal, M. D.; Pedrosa, R. Adv. Synth. Catal. 2010, 352, 3364–3372. doi:10.1002/adsc.201000612
Return to citation in text: [1] -
Manzano, R.; Andrés, J. M.; Pedrosa, R. Synlett 2011, 2203–2205. doi:10.1055/s-0030-1261139
Return to citation in text: [1] -
Manzano, R.; Andrés, J. M.; Álvarez, R.; Muruzábal, M. D.; Rodríguez de Lera, A.; Pedrosa, R. Chem. – Eur. J. 2011, 17, 5931–5938. doi:10.1002/chem.201100241
Return to citation in text: [1] -
Manzano, R.; Andrés, J. M.; Muruzábal, M.-D.; Pedrosa, R. J. Org. Chem. 2010, 75, 5417–5420. doi:10.1021/jo100792r
Return to citation in text: [1] -
Sheldon, R. A. Chem. Soc. Rev. 2012, 41, 1437–1451. doi:10.1039/C1CS15219J
Return to citation in text: [1] -
Andrés, J. M.; de La Cruz, N.; Valle, M.; Pedrosa, R. ChemPlusChem 2016, 81, 86–92. doi:10.1002/cplu.201500476
Return to citation in text: [1] [2] [3] [4] [5] -
Andrés, J. M.; Manzano, R.; Pedrosa, R. Chem. – Eur. J. 2008, 14, 5116–5119. doi:10.1002/chem.200800633
Return to citation in text: [1] [2] -
Pedrosa, R.; Andrés, J. M.; Ávila, D. P.; Ceballos, M.; Pindado, R. Green Chem. 2015, 17, 2217–2225. doi:10.1039/C4GC02474E
Return to citation in text: [1] [2] -
Baig, R. B. N.; Varma, R. S. Chem. Soc. Rev. 2012, 41, 1559–1584. doi:10.1039/C1CS15204A
See for a recent review.
Return to citation in text: [1] -
Stolle, A.; Szuppa, T.; Leonhardt, S. E. S.; Ondruschka, B. Chem. Soc. Rev. 2011, 40, 2317–2329. doi:10.1039/c0cs00195c
Return to citation in text: [1] -
Jörres, M.; Mersmann, S.; Raabe, G.; Bolm, C. Green Chem. 2013, 15, 612–616. doi:10.1039/c2gc36906k
Return to citation in text: [1]
| 29. | Andrés, J. M.; de La Cruz, N.; Valle, M.; Pedrosa, R. ChemPlusChem 2016, 81, 86–92. doi:10.1002/cplu.201500476 |
| 33. | Stolle, A.; Szuppa, T.; Leonhardt, S. E. S.; Ondruschka, B. Chem. Soc. Rev. 2011, 40, 2317–2329. doi:10.1039/c0cs00195c |
| 34. | Jörres, M.; Mersmann, S.; Raabe, G.; Bolm, C. Green Chem. 2013, 15, 612–616. doi:10.1039/c2gc36906k |
| 1. | Takemoto, Y. Org. Biomol. Chem. 2005, 3, 4299–4306. doi:10.1039/b511216h |
| 2. | Taylor, M. S.; Jacobsen, E. N. Angew. Chem., Int. Ed. 2006, 45, 1520–1543. doi:10.1002/anie.200503132 |
| 3. | Connon, S. J. Chem. Commun. 2008, 2499–2510. doi:10.1039/B719249E |
| 4. | Connon, S. J. Synlett 2009, 354–376. doi:10.1055/s-0028-1087557 |
| 5. | Lu, L.-Q.; An, X.-L.; Chen, J.-R.; Xiao, W.-J. Synlett 2012, 490–508. doi:10.1055/s-0031-1290131 |
| 15. | Gruttadauria, M.; Giacalone, F.; Noto, R. Chem. Soc. Rev. 2008, 37, 1666–1688. doi:10.1039/b800704g |
| 16. | Puglisi, A.; Benaglia, M.; Annunziata, R.; Siegel, J. S. ChemCatChem 2012, 4, 972–975. doi:10.1002/cctc.201200114 |
| 17. | Fotaras, S.; Kokotos, C. G.; Kokotos, G. Org. Biomol. Chem. 2012, 10, 5613–5619. doi:10.1039/c2ob25693b |
| 18. | Tuchman-Shukron, L.; Miller, S. J.; Portnoy, M. Chem. – Eur. J. 2012, 18, 2290–2296. doi:10.1002/chem.201102474 |
| 19. | Li, J.; Yang, G.; Cui, Y. J. Appl. Polym. Sci. 2011, 121, 1506–1511. doi:10.1002/app.33676 |
| 20. | Li, J.; Yang, G.; Qin, Y.; Yang, X.; Cui, Y. Tetrahedron: Asymmetry 2011, 22, 613–618. doi:10.1016/j.tetasy.2011.03.016 |
| 31. | Pedrosa, R.; Andrés, J. M.; Ávila, D. P.; Ceballos, M.; Pindado, R. Green Chem. 2015, 17, 2217–2225. doi:10.1039/C4GC02474E |
| 13. | Puglisi, A.; Annunziata, R.; Benaglia, M.; Cozzi, F.; Gervasini, A.; Bertacche, V.; Sala, M. C. Adv. Synth. Catal. 2009, 351, 219–229. doi:10.1002/adsc.200800635 |
| 14. | Yu, P.; He, J.; Guo, C. Chem. Commun. 2008, 2355–2357. doi:10.1039/b800640g |
| 32. |
Baig, R. B. N.; Varma, R. S. Chem. Soc. Rev. 2012, 41, 1559–1584. doi:10.1039/C1CS15204A
See for a recent review. |
| 10. | Gawande, M. B.; Branco, P. S.; Varma, R. S. Chem. Soc. Rev. 2013, 42, 3371–3393. doi:10.1039/c3cs35480f |
| 11. | Jiang, X.; Zhu, H.; Shi, X.; Zhong, Y.; Li, Y.; Wang, R. Adv. Synth. Catal. 2013, 355, 308–314. doi:10.1002/adsc.201201038 |
| 12. | Gleeson, O.; Davies, G.-L.; Peschiulli, A.; Tekoriute, R.; Gun’ko, Y. K.; Connon, S. J. Org. Biomol. Chem. 2011, 9, 7929–7940. doi:10.1039/c1ob06110k |
| 29. | Andrés, J. M.; de La Cruz, N.; Valle, M.; Pedrosa, R. ChemPlusChem 2016, 81, 86–92. doi:10.1002/cplu.201500476 |
| 6. | Ayats, C.; Henseler, A. H.; Pericàs, M. A. ChemSusChem 2012, 5, 320–325. doi:10.1002/cssc.201100570 |
| 7. | Bortolini, O.; Caciolli, L.; Cavazzini, A.; Costa, V.; Greco, R.; Massi, A.; Pasti, L. Green Chem. 2012, 14, 992–1000. doi:10.1039/c2gc16673a |
| 8. | Rodríguez-Escrich, C.; Pericàs, M. A. Eur. J. Org. Chem. 2015, 1173–1188. doi:10.1002/ejoc.201403042 |
| 9. | Kasaplar, P.; Ozkal, E.; Rodríguez-Escrich, C.; Pericàs, M. A. Green Chem. 2015, 17, 3122–3129. doi:10.1039/C5GC00496A |
| 30. | Andrés, J. M.; Manzano, R.; Pedrosa, R. Chem. – Eur. J. 2008, 14, 5116–5119. doi:10.1002/chem.200800633 |
| 12. | Gleeson, O.; Davies, G.-L.; Peschiulli, A.; Tekoriute, R.; Gun’ko, Y. K.; Connon, S. J. Org. Biomol. Chem. 2011, 9, 7929–7940. doi:10.1039/c1ob06110k |
| 24. | Manzano, R.; Andrés, J. M.; Muruzábal, M. D.; Pedrosa, R. Adv. Synth. Catal. 2010, 352, 3364–3372. doi:10.1002/adsc.201000612 |
| 25. | Manzano, R.; Andrés, J. M.; Pedrosa, R. Synlett 2011, 2203–2205. doi:10.1055/s-0030-1261139 |
| 26. | Manzano, R.; Andrés, J. M.; Álvarez, R.; Muruzábal, M. D.; Rodríguez de Lera, A.; Pedrosa, R. Chem. – Eur. J. 2011, 17, 5931–5938. doi:10.1002/chem.201100241 |
| 27. | Manzano, R.; Andrés, J. M.; Muruzábal, M.-D.; Pedrosa, R. J. Org. Chem. 2010, 75, 5417–5420. doi:10.1021/jo100792r |
| 29. | Andrés, J. M.; de La Cruz, N.; Valle, M.; Pedrosa, R. ChemPlusChem 2016, 81, 86–92. doi:10.1002/cplu.201500476 |
| 18. | Tuchman-Shukron, L.; Miller, S. J.; Portnoy, M. Chem. – Eur. J. 2012, 18, 2290–2296. doi:10.1002/chem.201102474 |
| 19. | Li, J.; Yang, G.; Cui, Y. J. Appl. Polym. Sci. 2011, 121, 1506–1511. doi:10.1002/app.33676 |
| 20. | Li, J.; Yang, G.; Qin, Y.; Yang, X.; Cui, Y. Tetrahedron: Asymmetry 2011, 22, 613–618. doi:10.1016/j.tetasy.2011.03.016 |
| 21. | Miyabe, H.; Tuchida, S.; Yamauchi, M.; Takemoto, Y. Synthesis 2006, 3295–3300. doi:10.1055/s-2006-950196 |
| 22. | Chuan, Y.; Chen, G.; Peng, Y. Tetrahedron Lett. 2009, 50, 3054–3058. doi:10.1016/j.tetlet.2009.04.011 |
| 29. | Andrés, J. M.; de La Cruz, N.; Valle, M.; Pedrosa, R. ChemPlusChem 2016, 81, 86–92. doi:10.1002/cplu.201500476 |
| 16. | Puglisi, A.; Benaglia, M.; Annunziata, R.; Siegel, J. S. ChemCatChem 2012, 4, 972–975. doi:10.1002/cctc.201200114 |
| 30. | Andrés, J. M.; Manzano, R.; Pedrosa, R. Chem. – Eur. J. 2008, 14, 5116–5119. doi:10.1002/chem.200800633 |
| 31. | Pedrosa, R.; Andrés, J. M.; Ávila, D. P.; Ceballos, M.; Pindado, R. Green Chem. 2015, 17, 2217–2225. doi:10.1039/C4GC02474E |
| 21. | Miyabe, H.; Tuchida, S.; Yamauchi, M.; Takemoto, Y. Synthesis 2006, 3295–3300. doi:10.1055/s-2006-950196 |
| 23. | Fredriksen, K. A.; Kristensen, T. E.; Hansen, T. Beilstein J. Org. Chem. 2012, 8, 1126–1133. doi:10.3762/bjoc.8.125 |
| 29. | Andrés, J. M.; de La Cruz, N.; Valle, M.; Pedrosa, R. ChemPlusChem 2016, 81, 86–92. doi:10.1002/cplu.201500476 |
© 2016 Andrés et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)