Abstract
Reactions of dihetaryl and aryl/hetaryl thioketones with 2-diazopropane, diazoethane, and (trimethylsilyl)diazomethane were studied at variable temperature. The experiments showed that reactions with 2-diazopropane carried out at –75 °C occur mainly via the initially formed, relatively stable 1,3,4-thiadiazolines as products of the [3 + 2]-cycloaddition of the diazo dipole onto the C=S bond. The latter decompose only at higher temperature (ca. −40 °C) to generate thiocarbonyl S-isopropanide. In the absence of the starting thioketone, the corresponding thiiranes and/or ethene derivatives, formed from them via spontaneous desulfurization, are the main products. In contrast, reactions with diazoethane occurred predominantly via initially formed diradicals, which in cascade processes gave sterically crowded 4,4,5,5-tetrahetaryl-1,3-dithiolanes as major products. Finally, the reaction of dihetaryl thioketones with (trimethylsilyl)diazomethane occur smoothly at −75 °C leading to the corresponding 4,4,5,5-tetrahetaryl-1,3-dithiolanes as the exclusive [3 + 2]-cycloadducts formed via a cascade of postulated diradicals. The presence of S or Se atoms in the hetaryl rings is of importance for stabilizing diradical intermediates. Remarkably, in no single case, the ‘head-to-head dimerization’ of aryl/hetaryl and dihetaryl substituted thiocarbonyl ylides was observed.

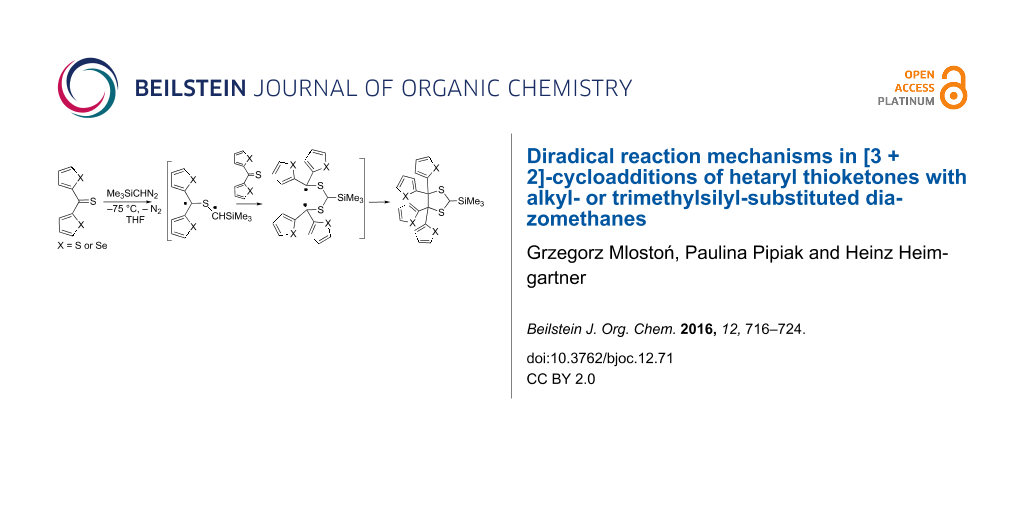
Graphical Abstract
Introduction
Cycloaddition reactions belong to the most important classes of organic reactions, and [3 + 2]- cycloadditions, also known as 1,3-dipolar cycloadditions or Huisgen reactions, offer a universal tool for the preparation of five-membered heterocycles with a variable number of heteroatoms in the ring [1,2]. In addition to their practical importance, discussions on the mechanism contribute significantly to the development of fundamental concepts in organic chemistry [3-7]. The first general concept of concerted [3 + 2]-cycloadditions was formulated by Huisgen [4]. However, some time later, Huisgen and co-workers reported stepwise [3 + 2]-cycloadditions via zwitterionic intermediates [8,9]. Large differences of energies of the frontier orbitals of dipole and dipolarophile, as well as sterically demanding groups at the terminus of the dipole, were pointed out as requirements for the initiation of the ‘zwitterionic pathway’. In addition to the experimental findings, new reports dealing with computational studies aimed at the demonstration of new zwitterionic [3 + 2]-cycloadditions were published [10-12]. Finally, a third concept for the interpretation of the mechanism of [3 + 2]-cycloadditions, formulated by Firestone, is based on the assumption that they occur via diradical intermediates [13-15].
Reactions of aromatic thioketones with diazomethane are well established. For example, in the case of thiobenzophenone (1a), the reaction performed at –65 ºC occurs without evolution of N2 and the in situ formed 2,2-diphenyl-1,3,4-thiadiazoline 2a can be subsequently used as a precursor of the reactive thiobenzophenone S-methanide (a thiocarbonyl ylide) 3a at ca. −45 °C, when the evolution of N2 takes place [16-20]. An analogous course of the reaction with diazomethane was observed in the case of thiofluorenone (1b, Scheme 1).
![[1860-5397-12-71-i1]](/bjoc/content/inline/1860-5397-12-71-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: ‘Head-to-head dimerization’ of diarylthioketone S-methanides 3a,b leading to 2,2,3,3-tetrasubstituted 1,4-dithianes 4a,b.
Scheme 1: ‘Head-to-head dimerization’ of diarylthioketone S-methanides 3a,b leading to 2,2,3,3-tetrasubstitut...
When the decomposition of 2a or 2b was performed in the presence of a suitable dipolarophile, the corresponding [3 + 2]-cycloadducts were formed, whereas in the absence of a dipolarophile, the ‘head-to-head dimerization’ leading to 2,2,3,3-tetraaryl-1,4-dithianes 4a,b is the exclusive reaction.
Heteroatoms such as S and Se are known to stabilize radical centers [21]. In our ongoing studies on thioketones and their applications in the cycloaddition chemistry, we described in a recent publication the unexpected course of the reaction of diazomethane with aryl/selenophen-2-yl thioketones of type 1c, leading to unusual dimers 5 of intermediate thiocarbonyl ylides of type 3c [22] (Scheme 2). In a competitive reaction, the latter react with the starting thioketone 1c to give 1,3-dithiolanes of type 6 which are, apparently, also formed via a diradical pathway, leading to the sterically crowded 4,4,5,5-tetrasubstituted isomers exclusively.
![[1860-5397-12-71-i2]](/bjoc/content/inline/1860-5397-12-71-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Diradical nature of the reactive intermediate 3c in the reaction of phenyl selenophen-2-yl thioketone 1c with diazomethane.
Scheme 2: Diradical nature of the reactive intermediate 3c in the reaction of phenyl selenophen-2-yl thioketo...
In other studies, performed with cycloaliphatic thioketones, e.g. with adamantane-2-thione or 2,2,4,4-tetramethyl-3-thioxocyclobutanone, the growing stability of the corresponding 1,3,4-thiadiazolines obtained in reactions with diazomethane, diazoethane, and 2-diazopropane was reported based on kinetic data [9,17-19]. The same tendency was observed in a series of reactions with aromatic thioketones: for example, the reaction of thiobenzophenone (1a) with diazoethane performed at −70 °C led to the formation of a relatively stable 2-methyl-1,3,4-thiadiazoline, which could be identified in the low temperature 1H NMR spectrum [20]. To the best of our knowledge, there are no reports on the course of reactions of either diaryl, aryl/hetaryl or dihetaryl thioketones with 2-diazopropane.
Prompted by these observations, we decided to examine reactions of aryl/hetaryl and dihetaryl thioketones 1 with some diazomethane derivatives, such as 2-diazopropane (7a), diazoethane (7b), and (trimethylsilyl)diazomethane (7c), and to compare their outcome with earlier reported reactions with diazomethane [22]. An important question was if in these reactions the corresponding 2-substituted 1,3,4-thiadiazolines of type 2 can be obtained at low temperature and subsequently used as precursors of new thiocarbonyl ylides. The latter may be potentially useful for the [3 + 2]-cycloaddition reactions with diverse dipolarophiles leading to five-membered S-heterocycles, such as di- and tetrahydrothiophenes, 1,3-oxathiolanes, 1,3-dithiolanes, etc.
Results and Discussion
The first experiment performed with thiobenzophenone (1a) and 2-diazopropane (7a) at ca. −75 °C in THF led to a change of the blue color of the solution without evolution of N2. The addition of the solution of 7a was continued until the color of the mixture changed to pale red. The mixture was warmed up, and around 0 °C the evolution of N2 was observed. The 1H NMR analysis of the crude product revealed the presence of only one singlet for two Me groups located at 1.62 ppm. After chromatographic work-up, the only product isolated as colorless crystals was identified as 2,2-dimethyl-3,3-diphenylthiirane (8a; Scheme 3, Table 1). The same result was obtained when the reaction was carried out at ca. −60 or −15 °C.
![[1860-5397-12-71-i3]](/bjoc/content/inline/1860-5397-12-71-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Formation of thiiranes 8 and/or 1,3-dithiolanes 10 in the reaction of aryl/aryl, aryl/hetaryl and dihetaryl thioketones 1 with 2-diazopropane (7a), diazoethane (7b), and (trimethylsilyl)diazomethane (7c) (Table 1).
Scheme 3: Formation of thiiranes 8 and/or 1,3-dithiolanes 10 in the reaction of aryl/aryl, aryl/hetaryl and d...
Table 1: Reactions of aryl/aryl, aryl/hetaryl, and hetaryl/hetaryl thioketones 1 with 2-diazopropane (7a), diazoethane (7b), and (trimethylsilyl)diazomethane (7c).
| Entry | Thioketone 1 | Diazoalkane 7 | T [°C] | Product 8/9 | Yields 8/9 [%]a | Product 10 (and/or 4) | Yield 10 (or 4) [%]a |
|---|---|---|---|---|---|---|---|
| 1 |
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-71-i5.svg?max-width=637&scale=1.0)
1a |
7a
R1 = R2 = Me |
−15
−60 −75 |
8a/9a |
86
80 (76b) 75 |
–
– – |
|
| 2 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-71-i6.svg?max-width=637&scale=1.0)
1b |
7a |
−15
−60 −75 |
8b/9b |
61 (41b)
85 76 |
10b |
14 (12d)
– – |
| 3 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-71-i7.svg?max-width=637&scale=1.0)
1c |
7a |
−15
−60 −75 |
8c/9c |
42
74 (57b) 63 |
10c |
61
13 (11d,e) 18 |
| 4 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-71-i8.svg?max-width=637&scale=1.0)
1d |
7a |
−15
−60 −75 |
8d/9d |
56
87 (75b + 2c) 97 |
10d |
58
36 (32d) 7 |
| 5 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-12-71-i9.svg?max-width=637&scale=1.0)
1e |
7a |
−15
−60 −75 |
8e/9e |
50
84 (61c) 68 |
10e |
49
20 (17d) 27 |
| 6 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-12-71-i10.svg?max-width=637&scale=1.0)
1f |
7a |
−15
−60 −75 |
8f/9f |
40 (17b + 7c)
85 96 |
10f |
67 (63d,e)
38 – |
| 7 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-12-71-i11.svg?max-width=637&scale=1.0)
1g |
7a |
−15
−60 −75 |
8g/9g |
38
51 (40c) 69 |
10g |
70
31 (28d,e) 36 |
| 8 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-12-71-i12.svg?max-width=637&scale=1.0)
1h |
7a |
−15
−60 −75 |
8h/9h |
36
88 (65c) 72 |
10h |
68
21 (17d,e) 21 |
| 9 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-12-71-i13.svg?max-width=637&scale=1.0)
1a |
7b
R1 = Me, R2 = H |
rt
−75 |
8i/9i |
traces (see, ref. [23])
93b + 3c (see, ref. [20]) |
10i |
87
(see, ref. [23]) – |
| 10 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-12-71-i14.svg?max-width=637&scale=1.0)
1b |
7b |
−15
−60 −75 |
8j/9j |
3
9 (6c) 14 |
10j |
82
73 (61d) 57 |
| 11 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-12-71-i15.svg?max-width=637&scale=1.0)
1d |
7b |
−15
−60 −75 |
8k/9k |
39
32 (25c) 69 |
10k |
85
81 (72d) 43 |
| 12 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-12-71-i16.svg?max-width=637&scale=1.0)
1e |
7b |
−15
−60 −75 |
8l/9l |
11
5 (3c) 12 |
10l |
76
86 (79d) 87 |
| 13 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-12-71-i17.svg?max-width=637&scale=1.0)
1f |
7b |
−15
−60 −75 |
8m/9m |
6
59 (48c) 62 |
10m |
79
35 (27d,e) 28 |
| 14 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-12-71-i18.svg?max-width=637&scale=1.0)
1c |
7b |
−15
−60 −75 |
8n/9n |
20
83 (71c) 65 |
10n |
75
36 (28d,e) 30 |
| 15 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-12-71-i19.svg?max-width=637&scale=1.0)
1a |
7c
R1 = Me3Si, R2 = H |
−75 | – |
10o
and 4c |
36 (33d)
39 (22f) |
|
| 16 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-12-71-i20.svg?max-width=637&scale=1.0)
1b |
7c | −75 | – | 4d |
53f
see ref. [26] |
|
| 17 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-12-71-i21.svg?max-width=637&scale=1.0)
1d |
7c | −75 | – | 10r | 95 (87d) | |
| 18 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-12-71-i22.svg?max-width=637&scale=1.0)
1e |
7c | −75 | – | 10s | 85 (76d) | |
aYields determined by 1H NMR with a weighed amount of 1,1,2,2-tetrachloroethane as a standard. bYield of isolated product 8. cYield of isolated product 9. dYield of isolated product 10. eIsolated as mixtures of isomeric products. fYield of isolated product 4.
Similar experiments were performed with thiofluorenone (1b) and 7a. Whereas at −75 °C and −60 °C the corresponding thiirane 8b and ethene 9b, formed via spontaneous desulfurization of 8b, were obtained as the sole products, in the experiment carried out at –15 ºC, the sterically crowded 1,3-dithiolane 10b was also detected as a minor product (14% yield, Table 1). In the latter case, thiirane 8b and ethene 9b were found in 24 and 37% yield, respectively. According to the literature, reaction of thiobenzophenone (1a) with diazoethane (7b) performed at –75 °C led to 5-methyl-2,2-diphenyl-1,3,4-thiadiazoline as the exclusive product [20]. After warming the reaction mixture to room temperature, the only products observed in the mixture were 2,2-diphenyl-3-methylthiirane (8i) and 1,1-diphenylpropene (9i) as the product of its desulfurization. On the other hand, the experiment with 1a and 7b performed at room temperature led to the 1,3-dithiolane 10i as the major product (87%) accompanied by small amounts of thiirane 8i and ethene 9i [23].
A different result was observed in the reaction of 7a with di(thiophen-2-yl) thioketone (1d). In that case, the reaction at −75 °C afforded also the expected thiirane 8d as the major product, accompanied by small amounts of the corresponding alkene 9d. However, in that case 4,4,5,5-tetrasubstituted 1,3-dithiolane 10d was also observed (7%). The amount of the latter product increased to 36% when the reaction was performed at −60 °C and to 58% at −15 °C. The corresponding experiments with 1c and 7b led to increased amounts of 1,3-dithiolane 10k, established as 43% at −75 °C, 81% at −60 °C, and 85% at −15 °C (Table 1). The same tendency, i.e., an increasing amount of 1,3-dithiolanes 10 in the case of the less-substituted diazoethane (7b) and at higher temperature, was observed for the reactions with thiofluorenone (1b), di(selenophen-2-yl)-, (phenyl)(thiophen-2-yl)-, (phenyl)(selenophen-2-yl)-, and other aryl hetaryl thioketones 1 (Table 1). As mentioned before, in some cases tri- and tetrasubstituted thiiranes partially underwent spontaneous extrusion of sulfur to form the corresponding tri- or tetrasubstituted ethene derivatives 9. In these cases, complete desulfurization was achieved by treatment of the reaction mixture with tris(diethylamino)phosphine, and the respective ethenes 9 were isolated as the final products.
It is worth mentioning that in the case of the non-symmetrical aryl/hetaryl thioketones 1c, 1f–h and 2-diazopropane (7a), the formed 1,3-dithiolanes 10 were isolated as mixtures of cis- and trans-isomers. In the 1H NMR spectra, they could be identified based on the presence of two and one signal, respectively, for the Me2C(2) group. The mixtures of 1,3-dithiolanes 10 obtained from the reactions of diazoethane (7b) with thioketones 1c,f consisted of only two instead of the expected three diastereoisomers. In all cases they belong to the same group of regioisomers. The regioselective formation of products 10 was proved by 13C NMR spectroscopy: the signals attributed to C(2) were found in narrow regions at 54.9–59.4 ppm for 10c, 10f–h and 43.1–45.6 ppm for 10m,n, respectively. In addition, the structure of the sterically crowded 4,4,5,5-tetrasubstituted aryl/hetaryl 1,3-dithiolanes 10 could be confirmed by the presence of only two signals for the three C(sp3)-atoms of the heterocycle, whereas in the isomeric 2,2,4,4-tetraaryl/hetaryl 1,3-dithiolanes, three signals for these atoms are expected (cf. [19]).
Trimethylsilyldiazomethane (7c) is widely applied as a practical and useful synthetic equivalent of the hazardous diazomethane [24,25]. In our earlier publications, its reactions with thiofluorenone (1b) and S-methyl (phosphonyl)dithioformate, leading to the expected 1,3,4-thiadiazoline derivatives, which are stable at −60 °C, were reported [26,27]. At higher temperature, in both reactions, dimers of the intermediate thiocarbonyl S-(trimethylsilyl)methanides were formed in the absence of a dipolarophile after evolution of N2. An analogous test experiment with 7c and thiobenzophenone (1a) was carried out in the course of the present study at −75 °C, and in this case slow decolorization of the reaction solution was observed. In contrast to the experiment with 1b, complete decolorization of the blue reaction solution was observed before the addition of the total, equimolar amount of 7c. After warming up and typical work-up procedure, the corresponding 1,3-dithiolane 10o and 1,4-dithiane 4c in a ratio of 2:1 were found as products identified in the 1H NMR spectrum of the crude reaction mixture. Thus, in this reaction dimerization of the intermediate thiocarbonyl ylide and its reaction with another molecule of 1a were competitive pathways. Finally, the reactions of 7c with symmetrically substituted dihetaryl thioketones 1d and 1e were performed at −75 °C, and in both cases, the sterically crowded 4,4,5,5-tetrahetaryl-1,3-dithiolanes 10r and 10s, respectively, were obtained as sole products.
The obtained results can be explained by the assumption that in the case of hetaryl thioketones 1 stepwise mechanisms via diradical intermediates govern the formation of the isolated 1,3-dithiolanes 10 (Scheme 3). Based on earlier studies, the stability of 1,3,4-thiadiazolines 2, which are considered as potential precursors of thiocarbonyl ylides 3, should be increased by the introduction of Me or Me3Si groups. Upon this assumption, all reactions performed at −75 °C should lead to the corresponding cycloadducts 2 with complete conversion of the starting thioketones 1. Only after warming up above −45/40 °C compounds 2 are expected to decompose yielding the reactive thiocarbonyl ylide 3. Under these conditions, the latter intermediates can undergo either 1,3-dipolar electrocyclization to give thiiranes 8 or dimerization leading to 1,4-dithianes 4 [20,26]. This reaction course resulting in the exclusive formation of thiiranes 8 was observed in the case of thiobenzophenone (1a) and thiofluorenone (1b) with 2-diazopropane (7a). However, the reaction of 1b with 7a carried out at −15 °C yielded also a small amount of 1,3-dithiolane 10b. This result can be interpreted by the partial decomposition of 2b in the presence of the non-converted thioketone 1b. The replacement of aromatic thioketones 1a,b by di(thiophen-2-yl) thioketone (1d) in the reaction with 7a resulted in the formation of comparable amounts of thiirane 8d and 1,3-dithiolane 10d, whereas at −75 °C 8d is again the major product. It is worth mentioning that the analogous experiment carried out at –60 ºC, i.e., below the expected decomposition temperature of 1,3,4-thiadiazoline 2d, led to substantial increase of the amount of 10d. A similar tendency was observed in other studied cases with hetaryl thioketones and 7a. The replacement of the latter by diazoethane (7b), leading to the less stable 1,3,4-thiadiazolines 2, resulted in a general increase of the corresponding 1,3-dithiolanes 10, which were formed in substantial amounts, even in experiments performed at −75 °C. However, the most striking results were observed in reactions performed with dihetaryl thioketones 1d and 1e with trimethylsilyldiazomethane (7c). In both experiments, the only products formed were the corresponding, sterically crowded 4,4,5,5,-tetrahetarylsubstituted 1,3-dithiolanes 10r and 10s. These results clearly demonstrate that 7c, similarly to diazomethane [22], reacts smoothly with dihetaryl thioketones 1 with no formation of the expected 1,3,4-thiadiazolines 2 and after release of N2 even at low temperature, the intermediate diradicals of type 3 attack the parent thioketones 1 yielding 1,3-dithiolanes 10 via stabilized 1,5-diradicals 12. In both cases no tendency for the formation of dimers of the intermediate ‘thiocarbonyl ylide’ was observed (Scheme 4).
![[1860-5397-12-71-i4]](/bjoc/content/inline/1860-5397-12-71-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed competitive mechanisms in the reactions of aryl/hetaryl and dihetaryl thioketones 1 with 2-diazopropane (7a), diazoethane (7b), and (trimethysilyl)diazomethane (7c).
Scheme 4: Proposed competitive mechanisms in the reactions of aryl/hetaryl and dihetaryl thioketones 1 with 2...
Based on these results, the reaction mechanism can be proposed as formulated in Scheme 4. It seems likely that the first step comprising the reaction of a hetaryl thioketone 1 with 7a, 7b, and 7c is not a concerted process, but the diradical or zwitterionic intermediate of type 11 is formed and, depending on the number and type of stabilizing substituents and the reaction temperature, they undergo two competitive reactions: elimination of N2 leading to thiocarbonyl ylide 3 or 1,5-ring closure to give 1,3,4-thiadiazoline 2. When the rapid elimination of N2 occurs in the presence of the non-converted thioketone 1, the stabilized 1,5-diradical 12 is formed as precursor of the sterically crowded 1,3-dithiolane 10. The alternatively formed 1,3,4-thiadiazolines 2a and 2b are expected to be stable at –75 ºC and eliminate N2 only at enhanced temperature generating thiocarbonyl ylide 3. As in that case the starting diaryl (or dihetaryl) thioketone 1 is completely consumed, the 1,3-dithiolane 10 cannot be formed. The results obtained with 7c demonstrate that its reaction with dihetaryl thioketones 1 occurs without formation of 1,3,4-thiadiazolines as intermediate [3 + 2]-cycloadducts. Elimination of N2 from the initially formed diradical of type 11 leads immediately to the new diradical species 3, which adds regioselectively to the C=S group of the non-converted thioketone 1.
This interpretation is consistent with the course of the reactions of aryl/hetaryl thioketones 1 with diazomethane, in which the formation of sterically crowded 1,3-dithiolanes side-by-side with 12-membered dimers of the thiocarbonyl ylide was observed [22]. In addition, it corresponds to the diradical mechanism postulated for the formal [3 + 2]-cycloaddition of aryl/hetaryl thioketones 1 with the in situ generated thiocarbonyl S-methanides [28]. The missing formation of dimers from thiocarbonyl ylides 3 derived from hetaryl thioketones can be explained by steric hindrance resulting from the presence of the Me or Me3Si groups in the S-methanide moiety. However, electronic effects resulting from the diradical nature of the intermediate thiocarbonyl ylides, can also play certain role. The same effect was observed in the case of diaryldiazomethanes used in reactions with aryl/hetaryl thioketones [29,30].
Experimental
General information: Melting points were determined in capillaries using a MEL-TEMP II apparatus (Aldrich) and are uncorrected. IR spectra were recorded with a FTIR NEXUS spectrophotometer as KBr pellets or as film; absorptions (ν) in cm−1. 1H and 13C NMR spectra were measured on a Bruker Avance III (1H at 600 and 13C at 150 MHz) instrument in CDCl3; chemical shifts (δ) are given in ppm, solvent signals as reference, coupling constants (J) in Hz. The multiplicity of the 13C signals was deduced from DEPT, supported by 1H,13C HMQC spectra. 1H NMR data are presented as follows: chemical shift, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant, integration. The mass spectra were recorded on a Finnigan MAT-95 (ESI). Elemental analyses were performed in the Microanalytical Laboratory of the Chemistry Faculty of the University of Łódź. Applied reagents such as 2-diazopropane (7a) and diazoethane (7b) were prepared by known methods according to the literature protocols [31,32]. Thiobenzophenone (1a), fluorene-9-thione (thiofluorenone, 1b), symmetrical dihetaryl thioketones 1d,e, and nonsymmetrical aryl/hetaryl thioketones 1c, 1f–h were obtained from the corresponding ketones using the known procedures [33]. Other reagents used in the present study were commercially available.
Reaction of thioketones 1a–h with 2-diazopropane (7a) and diazoethane (7b) – General procedure: Corresponding thioketones 1a–h (1 mmol) were dissolved in freshly distilled THF (2.5 mL) and the solution was cooled to the corresponding temperature (−15, −60, −75 °C; acetone/dry ice). Then, the mixture was treated with small portions of ethereal 2-diazopropane (7a) or diazoethane (7b) solution, until the intense color of the thioketone vanished. Then, the mixture was allowed to warm slowly to rt (ca. 2–4 h). After removal of the solvent under vacuum, the residue was subjected to 1H NMR analysis in CDCl3 solution with a weighed amount of 1,1,2,2-tetrachloroethane as a standard. Crude products were purified by CC (CHCl3/hexane 2:8).
In crude mixtures obtained from 2-diazopropane (7a) and thioketones 1c,e,g,h and from diazoethane (7b) and thioketones 1b–f, the presence of thiirane and the corresponding ethylene was evidenced based on the 1H NMR spectra. In these cases, no isolation of the thiirane was performed; after addition of tris(dimethylamino)phosphine desulfurization leading to the ethane derivative was carried out.
Reaction of thioketones 1d–e with (trimethylsilyl)diazomethane (7c) – General procedure: The corresponding thioketones 1d–e (1 mmol) were dissolved in freshly distilled THF (2.5 mL) and the solution was cooled to −75 °C (acetone/dry ice). Then, the mixture was treated with small portions of an ethereal solution of 7c (1 mmol). The mixture was kept in a cold bath, and the intense color of the thioketone vanished after 15 min. Subsequently, the mixture was allowed to slowly warm to rt (ca. 2–4 h). After removal of the solvent under vacuum, the residue was subjected to the 1H NMR analysis in CDCl3 solution with a weighed amount of 1,1,2,2-tetrachloroethane as an internal standard. Crude products were purified by CC (CHCl3/hexane 2:8).
2,2-Dimethyl-3,3-diphenylthiirane (8a): Yield: 182 mg (76%). White crystals; mp 66–67 °C (chromatographic purification); IR (KBr) ν: 2987 (w), 2921 (w), 1596 (w), 1490 (m), 1443 (m), 773 (m), 747 (m), 705 (s), 691 (m) cm−1; 1H NMR (600 MHz, CDCl3) δ 7.60–7.58 (m, 4H, Harom), 7.31–7.29 (m, 4H, Harom), 7.23–7.21 (m, 2H, Harom), 1.62 (s, 6H, CH3) ppm; 13C NMR (150 MHz, CDCl3) δ 142.4 (for 2 Carom), 129.4, 127.9, 126.8 (for 10 CHarom), 67.8, 52.9 (C-2, C-3), 27.9 (2 CH3) ppm; ESIMS m/z (%): 241 (100, [M + H]+); anal. calcd for C16H16S (240.36): C, 79.95; H, 6.71; S, 13.34; found: C, 79.69; H, 6.50; S, 13.40.
1,1-Di(thiophen-2-yl)-2-methylpropene (9d): Yield: 4 mg (2%). Yellow oil; IR (film) ν: 2927 (w), 2908 (w), 2850 (w), 1436 (m), 1369 (m), 1227 (m), 1016 (w), 827 (m), 693 (s) cm−1; 1H NMR (600 MHz, CDCl3) δ 7.29 (d, J = 5.4 Hz, 2H, Harom), 7.04–7.02 (m, 2H, Harom), 6.91 (d, J = 3.6 Hz, 2H, Harom), 2.03 (s, 6H, CH3) ppm; 13C NMR (150 MHz, CDCl3) δ 144.9, 137.1 (Carom, Carom-C=), 126.6, 126.4, 124.9 (for 6 CHarom), 122.9 (C=C(CH3)2), 23.3 (2 CH3) ppm; anal. calcd for C12H12S2 (220.35): C, 65.41; H, 5.49; S, 29.10; found: C, 65.31; H, 5.76; S, 28.81.
2,2-Dimethyl-4,5-diphenyl-4,5-di(thiophen-2-yl)-1,3-dithiolane (10f): Isolated as a mixture of cis-, trans-isomers (crude product ratio 65:35). Yield: 142 mg (63%). White crystals; mp 153–154 °C (MeOH/CHCl3); IR (KBr) ν: 3062 (w), 2919 (w), 1595 (w), 1490 (m), 1443 (m), 1231 (m), 1156 (m), 854 (w), 697 (s) cm−1; 1H NMR (600 MHz, CDCl3) δ 7.59–6.68 (m, 32H, Harom), 1.86 (s, 3H, CH3-cis), 1.70 (s, 6H, CH3-trans), 1.39 (s, 3H, CH3-cis) ppm; 13C NMR (150 MHz, CDCl3) δ 151.2, 149.8, 144.0, 142.9 (for 8 Carom), 131.3, 131.1, 130.5, 127.5, 127.1, 127.0, 126.6, 126.1, 125.4, 125.2, 125.0 (for 32 CHarom), 78.8, 78.4 (C-4 + C-5, for cis and trans), 55.4, 55.3 (C-2, cis and trans) 33.1 (CH3-cis), 32.9 (for 2 CH3-trans), 32.5 (CH3-cis) ppm; anal. calcd for C25H22S4 (450.70): C, 66.62; H, 4.92; S, 28.46; found: C, 66.46; H, 4.91; S, 28.33.
1,1-Di(thiophen-2-yl)-propene (9k): Yield: 52 mg (25%) – After desulfurization of tiirane 8k. Yellow oil; IR (film) ν: 3104 (w), 3071 (w), 2930 (w), 2908 (w), 2850 (w), 1438 (m), 1362 (w), 1249 (m), 1223 (m), 1036 (w), 850 (s), 836 (s), 818 (s), 695 (s) cm−1; 1H NMR (600 MHz, CDCl3) δ 7.23–7.22 (m, 1H, Harom), 7.01–7.00 (m, 1H, Harom), 6.96–6.94 (m, 1H, Harom), 6.90–6.89 (m, 1H, Harom), 6.80–6.79 (m, 1H, Harom), 6.71–6.70 (m, 1H, Harom), 6.18 (q, J = 7.2 Hz, 1H, =CH), 1.74 (d, J = 7.2 Hz, 3H, CH3) ppm; 13C NMR (150 MHz, CDCl3) δ 129.4, 139.8, 146.8 (2 Carom, Carom, C=), 127.6, 127.0, 126.6, 126.3, 125.5, 124.8, 123.4 (6 CHarom, =CH), 15.6 (CH3) ppm; anal. calcd for C11H10S2 (206.33): C, 64.04; H, 4.89; S, 31.08; found: C, 63.83; H, 4.98; S, 31.14.
2-Trimethylsilyl-4,4,5,5-tetra(thiophen-2-yl)-1,3-dithiolane (10r): Yield: 220 mg (87%). Yellow crystals: mp 152–154 °C (hexane/CH2Cl2); IR (KBr) ν: 2951 (w), 1618 (w), 1424 (m), 1250 (s), 1232 (m), 1122 (m), 1077 (m), 1050 (m), 842 (s), 752 (s), 699 (s), 632 (m) cm−1; 1H NMR (600 MHz, CDCl3) δ 7.23–7.22 (m, 2H, Harom), 7.20–7.19 (m, 2H, Harom), 6.96–6.95 (m, 2H, Harom), 6.89–6.87 (m, 2H, Harom), 6.83–6.81 (m, 4H, Harom), 3.89 (s, 1H, ((CH3)3Si)HC), 0.34 (s, 9H, (CH3)3Si) ppm; 13C NMR (150 MHz, CDCl3) δ 148.1, 146.1 (for 4 Carom), 130.2, 129.4, 127.0, 125.6, 125.5, 125.4 (for 12 CHarom), 74.9 (C-4, C-5), 36.5 (C-2), −1.53 ((CH3)3Si) ppm; anal. cald for C22H22S6Si (506.89): C, 52.13; H, 4.37; S, 37.96; found: C, 52.44; H, 4.55; S,37.71.
Supporting Information
| Supporting Information File 1: Experimental data for selected compounds 8–10, and the original 1H and 13C NMR spectra for all products. | ||
| Format: PDF | Size: 2.8 MB | Download |
References
-
Padwa, A., Ed. 1,3-Dipolar Cycloaddition Chemistry; John Wiley & Sons: Hoboken, NJ, U.S.A., 1984; Vol. 1, 2.
Return to citation in text: [1] -
Padwa, A.; Pearson, W. H., Eds. Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; John Wiley & Sons: Hoboken, NJ, U.S.A., 2002.
Return to citation in text: [1] -
Sustmann, R. Pure Appl. Chem. 1974, 40, 569–593. doi:10.1351/pac197440040569
Return to citation in text: [1] -
Huisgen, R. Angew. Chem., Int. Ed. 1963, 2, 565–598. doi:10.1002/anie.196305651
Return to citation in text: [1] [2] -
Huisgen, R. J. Org. Chem. 1976, 41, 403–419. doi:10.1021/jo00865a001
Return to citation in text: [1] -
Woodward, R. B.; Hoffmann, R. Angew. Chem., Int. Ed. Engl. 1969, 8, 781–853. doi:10.1002/anie.196907811
Return to citation in text: [1] -
Fukui, K.; Yonezawa, T.; Shingu, H. J. Chem. Phys. 1952, 20, 722–725. doi:10.1063/1.1700523
Return to citation in text: [1] -
Huisgen, R.; Mloston, G.; Giera, H.; Langhals, E. Tetrahedron 2002, 58, 507–519. doi:10.1016/S0040-4020(01)01147-4
Return to citation in text: [1] -
Huisgen, R.; Mloston, G.; Langhals, E. Helv. Chim. Acta 2001, 84, 1805–1820. doi:10.1002/1522-2675(20010613)84:6<1805::AID-HLCA1805>3.0.CO;2-T
Return to citation in text: [1] [2] -
Khlebnikov, A. F.; Konev, A. S.; Virtsev, A. A.; Yufit, D. S.; Mloston, G.; Heimgartner, H. Helv. Chim. Acta 2014, 97, 453–470. doi:10.1002/hlca.201300405
Return to citation in text: [1] -
Jasiński, R. J. Fluorine Chem. 2015, 176, 35–39. doi:10.1016/j.jfluchem.2015.04.020
Return to citation in text: [1] -
Siadati, S. A. Tetrahedron Lett. 2015, 56, 4857–4863. doi:10.1016/j.tetlet.2015.06.048
Return to citation in text: [1] -
Firestone, R. A. J. Org. Chem. 1968, 33, 2285–2290. doi:10.1021/jo01270a023
Return to citation in text: [1] -
Firestone, R. A. Tetrahedron 1977, 33, 3009–3039. doi:10.1016/0040-4020(77)80448-1
Return to citation in text: [1] -
Firestone, R. A. Int. J. Chem. Kinet. 2013, 45, 415–428. doi:10.1002/kin.20776
Return to citation in text: [1] -
Kalwinsch, I.; Li, X.; Gottstein, J.; Huisgen, R. J. Am. Chem. Soc. 1981, 103, 7032–7033. doi:10.1021/ja00413a073
Return to citation in text: [1] -
Huisgen, R.; Mloston, G.; Giera, H.; Langhals, E.; Polborn, K.; Sustmann, R. Eur. J. Org. Chem. 2005, 1519–1531. doi:10.1002/ejoc.200400765
Return to citation in text: [1] [2] -
Huisgen, R.; Li, X.; Mloston, G.; Knorr, R.; Stephenson, D. S. Tetrahedron 1999, 55, 12783–12796. doi:10.1016/S0040-4020(99)00790-5
Return to citation in text: [1] [2] -
Huisgen, R.; Li, X.; Mlostoń, G.; Fluka, C. Eur. J. Org. Chem. 2000, 1695–1702. doi:10.1002/(SICI)1099-0690(200005)2000:9<1695::AID-EJOC1695>3.0.CO;2-4
Return to citation in text: [1] [2] [3] -
Huisgen, R.; Kalwinsch, I.; Li, X.; Mloston, G. Eur. J. Org. Chem. 2000, 1685–1694. doi:10.1002/(SICI)1099-0690(200005)2000:9<1685::AID-EJOC1685>3.0.CO;2-6
Return to citation in text: [1] [2] [3] [4] [5] -
Oae, S. Organic Sulfur Chemistry: Structure and Mechanism; CRC Press, Inc.: Boca Raton, FL, U.S.A., 1992.
Return to citation in text: [1] -
Mlostoń, G.; Urbaniak, K.; Linden, A.; Heimgartner, H. Helv. Chim. Acta 2015, 98, 453–461. doi:10.1002/hlca.201500050
Return to citation in text: [1] [2] [3] [4] -
Schönberg, A.; Cernik, D.; Urban, W. Ber. Dtsch. Chem. Ges. 1931, 64, 2577–2581. doi:10.1002/cber.19310640941
Return to citation in text: [1] [2] [3] -
Shioiri, T.; Aoyama, T.; Snowden, T. e-EROS Encyclopedia of Reagents for Organic Synthesis, 2006. doi:10.1002/047084289X.rt298.pub2
Return to citation in text: [1] -
Fuchs, L. P., Ed. Handbook of Reagents for Organic Synthesis, Reagents for Silicon-Mediated Organic Synthesis; John Wiley & Sons: Hoboken, NJ, U.S.A., 2011; p 590.
Return to citation in text: [1] -
Mlostoń, G.; Urbaniak, K.; Linden, A.; Heimgartner, H. Pol. J. Chem. 2007, 81, 1849–1860.
Return to citation in text: [1] [2] [3] -
Mloston, G.; Urbaniak, K.; Linden, A.; Heimgartner, B. Heterocycles 2007, 73, 419–432. doi:10.3987/COM-07-S(U)12
Return to citation in text: [1] -
Mlostoń, G.; Pipiak, P.; Linden, A.; Heimgartner, H. Helv. Chim. Acta 2015, 98, 462–473. doi:10.1002/hlca.201500057
Return to citation in text: [1] -
Mlostoń, G.; Urbaniak, K.; Pawlak, A.; Heimgartner, H. Heterocycles 2016, in press. doi:10.3987/COM-15-S(T)8
Return to citation in text: [1] -
Mlostoń, G.; Hamera, R.; Heimgartner, H. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 2125–2133. doi:10.1080/10426507.2015.1071817
Return to citation in text: [1] -
Schweizer, E. E.; Kim, C. S. J. Org. Chem. 1971, 36, 4033–4041. doi:10.1021/jo00825a006
Return to citation in text: [1] -
Hudlicky, T.; Short, R. P. J. Org. Chem. 1982, 47, 1522–1527. doi:10.1021/jo00347a031
Return to citation in text: [1] -
Mlostoń, G.; Urbaniak, K.; Gębicki, K.; Grzelak, P.; Heimgartner, H. Heteroat. Chem. 2014, 25, 548–555. doi:10.1002/hc.21191
Return to citation in text: [1]
| 33. | Mlostoń, G.; Urbaniak, K.; Gębicki, K.; Grzelak, P.; Heimgartner, H. Heteroat. Chem. 2014, 25, 548–555. doi:10.1002/hc.21191 |
| 1. | Padwa, A., Ed. 1,3-Dipolar Cycloaddition Chemistry; John Wiley & Sons: Hoboken, NJ, U.S.A., 1984; Vol. 1, 2. |
| 2. | Padwa, A.; Pearson, W. H., Eds. Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; John Wiley & Sons: Hoboken, NJ, U.S.A., 2002. |
| 10. | Khlebnikov, A. F.; Konev, A. S.; Virtsev, A. A.; Yufit, D. S.; Mloston, G.; Heimgartner, H. Helv. Chim. Acta 2014, 97, 453–470. doi:10.1002/hlca.201300405 |
| 11. | Jasiński, R. J. Fluorine Chem. 2015, 176, 35–39. doi:10.1016/j.jfluchem.2015.04.020 |
| 12. | Siadati, S. A. Tetrahedron Lett. 2015, 56, 4857–4863. doi:10.1016/j.tetlet.2015.06.048 |
| 23. | Schönberg, A.; Cernik, D.; Urban, W. Ber. Dtsch. Chem. Ges. 1931, 64, 2577–2581. doi:10.1002/cber.19310640941 |
| 8. | Huisgen, R.; Mloston, G.; Giera, H.; Langhals, E. Tetrahedron 2002, 58, 507–519. doi:10.1016/S0040-4020(01)01147-4 |
| 9. | Huisgen, R.; Mloston, G.; Langhals, E. Helv. Chim. Acta 2001, 84, 1805–1820. doi:10.1002/1522-2675(20010613)84:6<1805::AID-HLCA1805>3.0.CO;2-T |
| 26. | Mlostoń, G.; Urbaniak, K.; Linden, A.; Heimgartner, H. Pol. J. Chem. 2007, 81, 1849–1860. |
| 4. | Huisgen, R. Angew. Chem., Int. Ed. 1963, 2, 565–598. doi:10.1002/anie.196305651 |
| 23. | Schönberg, A.; Cernik, D.; Urban, W. Ber. Dtsch. Chem. Ges. 1931, 64, 2577–2581. doi:10.1002/cber.19310640941 |
| 3. | Sustmann, R. Pure Appl. Chem. 1974, 40, 569–593. doi:10.1351/pac197440040569 |
| 4. | Huisgen, R. Angew. Chem., Int. Ed. 1963, 2, 565–598. doi:10.1002/anie.196305651 |
| 5. | Huisgen, R. J. Org. Chem. 1976, 41, 403–419. doi:10.1021/jo00865a001 |
| 6. | Woodward, R. B.; Hoffmann, R. Angew. Chem., Int. Ed. Engl. 1969, 8, 781–853. doi:10.1002/anie.196907811 |
| 7. | Fukui, K.; Yonezawa, T.; Shingu, H. J. Chem. Phys. 1952, 20, 722–725. doi:10.1063/1.1700523 |
| 20. | Huisgen, R.; Kalwinsch, I.; Li, X.; Mloston, G. Eur. J. Org. Chem. 2000, 1685–1694. doi:10.1002/(SICI)1099-0690(200005)2000:9<1685::AID-EJOC1685>3.0.CO;2-6 |
| 22. | Mlostoń, G.; Urbaniak, K.; Linden, A.; Heimgartner, H. Helv. Chim. Acta 2015, 98, 453–461. doi:10.1002/hlca.201500050 |
| 20. | Huisgen, R.; Kalwinsch, I.; Li, X.; Mloston, G. Eur. J. Org. Chem. 2000, 1685–1694. doi:10.1002/(SICI)1099-0690(200005)2000:9<1685::AID-EJOC1685>3.0.CO;2-6 |
| 21. | Oae, S. Organic Sulfur Chemistry: Structure and Mechanism; CRC Press, Inc.: Boca Raton, FL, U.S.A., 1992. |
| 22. | Mlostoń, G.; Urbaniak, K.; Linden, A.; Heimgartner, H. Helv. Chim. Acta 2015, 98, 453–461. doi:10.1002/hlca.201500050 |
| 16. | Kalwinsch, I.; Li, X.; Gottstein, J.; Huisgen, R. J. Am. Chem. Soc. 1981, 103, 7032–7033. doi:10.1021/ja00413a073 |
| 17. | Huisgen, R.; Mloston, G.; Giera, H.; Langhals, E.; Polborn, K.; Sustmann, R. Eur. J. Org. Chem. 2005, 1519–1531. doi:10.1002/ejoc.200400765 |
| 18. | Huisgen, R.; Li, X.; Mloston, G.; Knorr, R.; Stephenson, D. S. Tetrahedron 1999, 55, 12783–12796. doi:10.1016/S0040-4020(99)00790-5 |
| 19. | Huisgen, R.; Li, X.; Mlostoń, G.; Fluka, C. Eur. J. Org. Chem. 2000, 1695–1702. doi:10.1002/(SICI)1099-0690(200005)2000:9<1695::AID-EJOC1695>3.0.CO;2-4 |
| 20. | Huisgen, R.; Kalwinsch, I.; Li, X.; Mloston, G. Eur. J. Org. Chem. 2000, 1685–1694. doi:10.1002/(SICI)1099-0690(200005)2000:9<1685::AID-EJOC1685>3.0.CO;2-6 |
| 13. | Firestone, R. A. J. Org. Chem. 1968, 33, 2285–2290. doi:10.1021/jo01270a023 |
| 14. | Firestone, R. A. Tetrahedron 1977, 33, 3009–3039. doi:10.1016/0040-4020(77)80448-1 |
| 15. | Firestone, R. A. Int. J. Chem. Kinet. 2013, 45, 415–428. doi:10.1002/kin.20776 |
| 9. | Huisgen, R.; Mloston, G.; Langhals, E. Helv. Chim. Acta 2001, 84, 1805–1820. doi:10.1002/1522-2675(20010613)84:6<1805::AID-HLCA1805>3.0.CO;2-T |
| 17. | Huisgen, R.; Mloston, G.; Giera, H.; Langhals, E.; Polborn, K.; Sustmann, R. Eur. J. Org. Chem. 2005, 1519–1531. doi:10.1002/ejoc.200400765 |
| 18. | Huisgen, R.; Li, X.; Mloston, G.; Knorr, R.; Stephenson, D. S. Tetrahedron 1999, 55, 12783–12796. doi:10.1016/S0040-4020(99)00790-5 |
| 19. | Huisgen, R.; Li, X.; Mlostoń, G.; Fluka, C. Eur. J. Org. Chem. 2000, 1695–1702. doi:10.1002/(SICI)1099-0690(200005)2000:9<1695::AID-EJOC1695>3.0.CO;2-4 |
| 19. | Huisgen, R.; Li, X.; Mlostoń, G.; Fluka, C. Eur. J. Org. Chem. 2000, 1695–1702. doi:10.1002/(SICI)1099-0690(200005)2000:9<1695::AID-EJOC1695>3.0.CO;2-4 |
| 20. | Huisgen, R.; Kalwinsch, I.; Li, X.; Mloston, G. Eur. J. Org. Chem. 2000, 1685–1694. doi:10.1002/(SICI)1099-0690(200005)2000:9<1685::AID-EJOC1685>3.0.CO;2-6 |
| 23. | Schönberg, A.; Cernik, D.; Urban, W. Ber. Dtsch. Chem. Ges. 1931, 64, 2577–2581. doi:10.1002/cber.19310640941 |
| 29. | Mlostoń, G.; Urbaniak, K.; Pawlak, A.; Heimgartner, H. Heterocycles 2016, in press. doi:10.3987/COM-15-S(T)8 |
| 30. | Mlostoń, G.; Hamera, R.; Heimgartner, H. Phosphorus, Sulfur Silicon Relat. Elem. 2015, 190, 2125–2133. doi:10.1080/10426507.2015.1071817 |
| 31. | Schweizer, E. E.; Kim, C. S. J. Org. Chem. 1971, 36, 4033–4041. doi:10.1021/jo00825a006 |
| 32. | Hudlicky, T.; Short, R. P. J. Org. Chem. 1982, 47, 1522–1527. doi:10.1021/jo00347a031 |
| 22. | Mlostoń, G.; Urbaniak, K.; Linden, A.; Heimgartner, H. Helv. Chim. Acta 2015, 98, 453–461. doi:10.1002/hlca.201500050 |
| 28. | Mlostoń, G.; Pipiak, P.; Linden, A.; Heimgartner, H. Helv. Chim. Acta 2015, 98, 462–473. doi:10.1002/hlca.201500057 |
| 20. | Huisgen, R.; Kalwinsch, I.; Li, X.; Mloston, G. Eur. J. Org. Chem. 2000, 1685–1694. doi:10.1002/(SICI)1099-0690(200005)2000:9<1685::AID-EJOC1685>3.0.CO;2-6 |
| 26. | Mlostoń, G.; Urbaniak, K.; Linden, A.; Heimgartner, H. Pol. J. Chem. 2007, 81, 1849–1860. |
| 22. | Mlostoń, G.; Urbaniak, K.; Linden, A.; Heimgartner, H. Helv. Chim. Acta 2015, 98, 453–461. doi:10.1002/hlca.201500050 |
| 24. | Shioiri, T.; Aoyama, T.; Snowden, T. e-EROS Encyclopedia of Reagents for Organic Synthesis, 2006. doi:10.1002/047084289X.rt298.pub2 |
| 25. | Fuchs, L. P., Ed. Handbook of Reagents for Organic Synthesis, Reagents for Silicon-Mediated Organic Synthesis; John Wiley & Sons: Hoboken, NJ, U.S.A., 2011; p 590. |
| 26. | Mlostoń, G.; Urbaniak, K.; Linden, A.; Heimgartner, H. Pol. J. Chem. 2007, 81, 1849–1860. |
| 27. | Mloston, G.; Urbaniak, K.; Linden, A.; Heimgartner, B. Heterocycles 2007, 73, 419–432. doi:10.3987/COM-07-S(U)12 |
© 2016 Mlostoń et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)