Abstract
Background: Derivatives of D-glucosamine and D-galactosamine represent an important family of the cell surface glycan components and their fluorinated analogs found use as metabolic inhibitors of complex glycan biosynthesis, or as probes for the study of protein–carbohydrate interactions. This work is focused on the synthesis of acetylated 3-deoxy-3-fluoro, 4-deoxy-4-fluoro and 3,4-dideoxy-3,4-difluoro analogs of D-glucosamine and D-galactosamine via 1,6-anhydrohexopyranose chemistry. Moreover, the cytotoxicity of the target compounds towards selected cancer cells is determined.
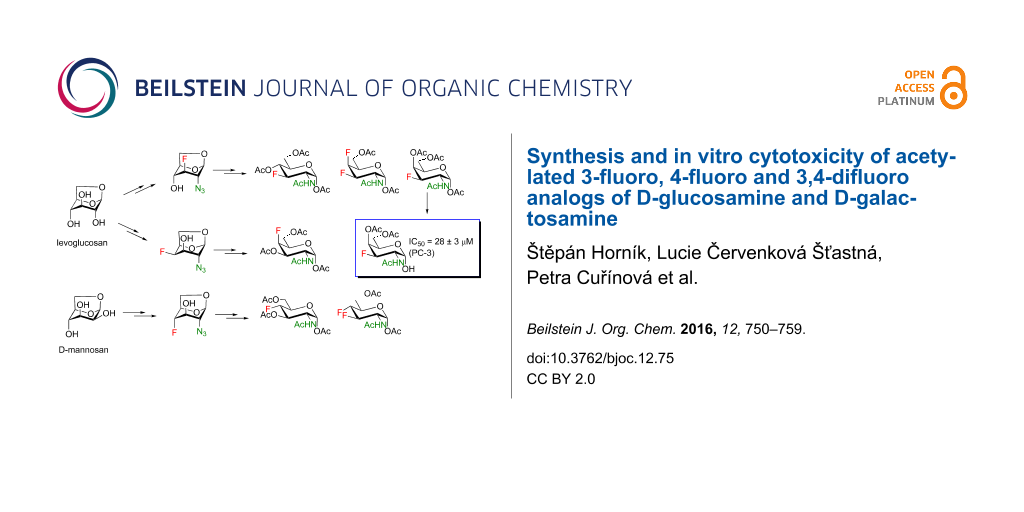
Results: Introduction of fluorine at C-3 was achieved by the reaction of 1,6-anhydro-2-azido-2-deoxy-4-O-benzyl-β-D-glucopyranose or its 4-fluoro analog with DAST. The retention of configuration in this reaction is discussed. Fluorine at C-4 was installed by the reaction of 1,6:2,3-dianhydro-β-D-talopyranose with DAST, or by fluoridolysis of 1,6:3,4-dianhydro-2-azido-β-D-galactopyranose with KHF2. The amino group was introduced and masked as an azide in the synthesis. The 1-O-deacetylated 3-fluoro and 4-fluoro analogs of acetylated D-galactosamine inhibited proliferation of the human prostate cancer cell line PC-3 more than cisplatin and 5-fluorouracil (IC50 28 ± 3 μM and 54 ± 5 μM, respectively).
Conclusion: A complete series of acetylated 3-fluoro, 4-fluoro and 3,4-difluoro analogs of D-glucosamine and D-galactosamine is now accessible by 1,6-anhydrohexopyranose chemistry. Intermediate fluorinated 1,6-anhydro-2-azido-hexopyranoses have potential as synthons in oligosaccharide assembly.

Graphical Abstract
Introduction
Derivatives of D-glucosamine (GlcN) and D-galactosamine (GalN) are essential amino sugar components of glycans in glycoproteins, glycolipids and proteoglycans. As such, they participate in functions performed by cell-surface glycans including cell adhesion and signaling [1]. Unnatural analogs of these amino sugars prepared by a selective replacement of a hydroxy group by fluorine have proved valuable tools to perturb glycan and glycosaminoglycan biosynthesis [2,3], to inhibit amino sugars processing enzymes [4,5], to probe interactions of amino sugars with their target enzymes and lectins [6,7], or to increase the hydrolytic stability of the glycosidic bond in amino sugar glycosides [8,9]. For example, acetylated 4-fluoro analogs of D-glucosamines 1–3, D-galactosamine 4, and 6-fluoro-D-galactosamine 9 (Figure 1) acted as metabolic inhibitors of the biosynthesis of cell-surface O- and N-linked glycans (including their lactosamine and sialyl LewisX terminal epitopes) [2,3,10,11], and glycosaminoglycans [3]. The resulting disruption of protein–(glycosamino)glycan interactions had important biomedical consequences such as reduced selectin-mediated tumor cell adhesion [12,13], suppressed selectin-mediated leukocyte migration [11,14,15], reduced angiogenesis [3], or inhibition of tumor growth by decreased galectin-mediated antitumor T cell apoptosis [16].
![[1860-5397-12-75-1]](/bjoc/content/figures/1860-5397-12-75-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of deoxofluorinated hexosamines.
Figure 1: Examples of deoxofluorinated hexosamines.
Some acetylated fluoro hexosamines also displayed antiproliferative properties in vitro [17]. For example, 3- and 4-fluoro-D-glucosamine analogs 5 [18] and 1 [19], and 4-fluoro-D-galactosamine analog 4 [19] (Figure 1), inhibited the growth of murine L1210 leukemia cells in micromolar range (IC50 27–35 μM). Compound 5 also inhibited the proliferation of the human pancreatic cancer cell line KP1-NL (IC50 30 μM) [20] and 4-fluoro-D-glucosamine analogs 1 and 2 were reported to inhibit the proliferation of the human prostate cancer cell line PC-3 (IC50 61 µm for 2) [2].
The remarkable ability of acetylated fluoro analogs of GlcNAc and GalNAc to perturb the (glycosamino)glycan biosynthesis and their antiproliferative properties aroused our interest in developing a methodology for the preparation of a complete series of acetylated 3-fluoro, 4-fluoro-, and 3,4-difluoro analogs 1, 4–8 (Figure 1) including previously unknown members of this class of hexosamine mimics: acetylated 3-fluoro-D-GalNAc 6, 3,4-difluoro-D-GlcNAc 7 and 3,4-difluoro-D-GalNAc 8, as well as 3-fluoro-D-GlcNAc 5, in which case the reported synthesis was troublesome and low-yielding [20]. To carry out the synthesis, flexible synthetic methods for the stereoselective introduction of fluorine at one or more designated positions of the hexosamine skeleton are necessary. The elaborated chemistry of 1,6-anhydrohexopyranose derivatives is suitable for this purpose [21-23]. Building on previous results from our [24] and other groups [25-28], we designed an approach based on stereoselective introduction of an azide as a masked amine group at C-2, and fluorine at C-3 and C-4 by nucleophilic displacement. Resulting 3-fluoro, 4-fluoro, and 3,4-difluoro analogs of 2-azido-1,6-anhydrohexopyranoses were then converted into the target fluoro analogs of D-glucosamine and D-galactosamine (Scheme 1). Dual protection of the anomeric and primary hydroxy groups in the form of the 1,6-anhydro bridge reduced the number of protecting groups, and the rigid bicyclic skeleton of 1,6-anhydrohexopyranoses enabled a high degree of regio- and stereocontrol necessary for the introduction of heteroatomic substituents at C-2, C-3, and C-4. The synthesis of the analogs 6–8 has not yet been reported to our knowledge, while the synthesis of 1, 4 and 5 represents an alternative to the published procedures [2,20,29]. Herein we also report on the cytotoxicity of prepared fluoro analogs in the human ovarian cancer A2780 and prostate cancer PC-3 cell lines. Preliminary results for the synthesis of compounds 5 and 6 were communicated earlier in a letter [30].
![[1860-5397-12-75-i1]](/bjoc/content/inline/1860-5397-12-75-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
Synthesis
The synthesis commenced with the introduction of an azide at the 2α-position of the 1,6-anhydropyranose skeleton by nucleophilic cleavage of a 2β,3β-epoxide or the displacement of the C-2β triflate ester using mainly a combination of published procedures (Scheme 2). 2-Azido alcohols 11, 12 and 15 were obtained from 1,6:2,3-dianhydro-4-O-benzyl-β-D-mannopyranose (10, one of the Černý epoxides available from D-glucal [31-33] or levoglucosan [34]). Regioselective azidolysis [21,35,36] of 10 yielded 2-azido alcohol 11 which was converted to 12 by benzoylation at O-3 and debenzylation at O-4. To prevent azide reduction, oxidative debenzylation [24] was applied instead of the more common hydrogenation. Debenzylation of 10 gave dianhydro derivative 13 [34,37] (available also directly from D-glucal [31,33] or from levoglucosan [38]) which was converted to 14 by Latrell–Dax inversion at C-4 [39]. O-Benzylation [40] of 14 followed by azidolysis [41] furnished 15. 2-Azido-3,4-epoxide 18 was prepared from readily available [42] 2,3-isopropylidene-D-mannosan (16) in five steps (Scheme 2). Tosylation of 16 [43], followed by hydrolysis of the benzylidene acetal [44] and oxirane ring closure [45] at C-4 delivered 1,6:3,4-dianhydro derivative 17. Formation of the triflate ester and azidolysis furnished 1,6:3,4-dianhydro-2-azido derivative 18. Lithium azide was found superior to sodium azide in conversion of the triflate to the azide 18, and the yield of 18 increased from 48% reported earlier [46] to 82%.
![[1860-5397-12-75-i2]](/bjoc/content/inline/1860-5397-12-75-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of starting 2-azido compounds. Reagents and conditions: (a) NaN3, NH4Cl, MeOC2H4OH, 79%; (b) i) BzCl, py; ii) NaBrO3, Na2S2O4, AcOEt, H2O, 47% over 2 steps; (c) Pd/C, H2, EtOH, 96%; (d) Tf2O, then (Bu)4NNO2, 69%; (e) (i) NaH, BnBr, THF, 79%; (ii) LiN3, NH4Cl, MeOC2H4OH, 100 °C, 81%; (f) (i) TsCl, py, (ii) AcOH, H2O; (iii) IRA 410 (OH−), MeOH, 78% over 3 steps; (g) (i) Tf2O, py, CH2Cl2; (ii) LiN3, DMF, 82% over 2 steps.
Scheme 2: Preparation of starting 2-azido compounds. Reagents and conditions: (a) NaN3, NH4Cl, MeOC2H4OH, 79%...
The synthesis of mono- and difluoro analogs of 2-azido-2-deoxy-1,6-anhydrohexopyranoses, which are key intermediates, is shown in Scheme 3. We first explored the reactions of azido alcohols 11, 12 and 15 (Scheme 3) with diethylaminosulfur trifluoride (DAST) to achieve the introduction of a nucleophilic fluorine atom. Reaction of 11 with DAST using a minor modification of the reported procedure [28] provided the D-gluco-configured 3-fluoro-derivative 19 (Scheme 3) with clean retention of configuration. To prepare the D-galacto-configured analog of 19, deoxofluorination of 15 by reaction with DAST in benzene at 75 °C (conditions used for fluorination of 11), or in dichloromethane at rt was attempted. Disappointingly, only unreacted 15 was recovered. A very slow formation of several unidentified fluorine-containing compounds (19F NMR) was observed on prolonged reaction times. Oxidative debenzylation of 19 gave fluorhydrin 20 which was subjected to epimerization at C-4 by Latrell–Dax inversion to furnish 2-azido-3-fluoro compound 21 with the desired D-galacto configuration (Scheme 3). The geminal fluorine–carbon coupling value 2JC4,F = 17.7 Hz in 21 is characteristic for a gauche relationship between the C3–F and C4–O bonds whereas 2JC4,F = 31.5 Hz in 20 is consistent with an antiperiplanar arrangement. The inversion at C-4 is also manifested by an increase in the 3JH4,H3/5 values (1.7 → 4.5 Hz) together with an increase of the 3JF,H4 coupling (13.9 → 26.4 Hz).
![[1860-5397-12-75-i3]](/bjoc/content/inline/1860-5397-12-75-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of mono and difluoro analogs of 2-azido-2-deoxy-1,6-anhydro-β-D-gluco- and galactopyranoses. Reagents and conditions: (a) DAST, benzene, 75–81 °C, 88%; (b) NaBrO3, Na2S2O4, EtOAc, H2O, 75%; (c) Tf2O, py, CH2Cl2; then (Bu)4NNO2, DMF, 67%; (d) DAST, benzene, 75–81 °C; or DAST, CH2Cl2, −25 °C to rt; (e) DAST, CH2Cl2, −25 °C to rt, 78%; (f) DAST, CH2Cl2, −25 °C to rt, 65%; (g) KHF2, C2H4(OH)2, 175 °C, 40–60% (26); (h) DAST, benzene, 75–81 °C, 46% (28), 12% (29); (i) DAST, CH2Cl2, −50 °C to rt, 52 %, (j) LiN3, NH4Cl, MeOC2H4OH, 100 °C, 74% (31).
Scheme 3: Preparation of mono and difluoro analogs of 2-azido-2-deoxy-1,6-anhydro-β-D-gluco- and galactopyran...
An attempt to introduce fluorine at C-4 on reaction of 12 with DAST furnished orthoester 25 instead of the desired 4-fluoro derivative. The structure of orthoester 25 was confirmed by single crystal X-ray analysis. The formation of 25 resulted from an intramolecular displacement of the unstable alkoxysulfur intermediate 23 with the oxygen of the benzoyl group and a subsequent reaction of the salt 24 with methanol upon quenching. A similar participation of a vicinal O-acetyl protecting group on reaction with DAST leading to the formation of an orthoacetate was reported [47]. Reaction of fluorohydrin 20 with DAST proceeded with inversion at C-4 giving 3,4-difluoro analog 22. The D-galacto configuration of 22 was manifested by the values of the vicinal coupling constants 3JH2,H3 = 1.6 Hz and 3JH3,H4 = 4.5 Hz, and the large value of 2JC5,F4 = 27.2 Hz confirmed the equatorial position of fluorine at C-4.
Cleavage of the oxirane ring in 18 on reaction with KHF2 in ethylene glycol [48,49] furnished the 4-fluoro derivative 26 (Scheme 3). The moderate yield of 26 (40–60%) can be attributed to the formation of the 4-O-(2-hydroxyethyl) derivative 27 arising from solvent participation, and to a partial decomposition (indicated by TLC) at high temperatures necessary for the reaction to proceed. The low values of the vicinal coupling constant 3JH3,H4 = 2.0 Hz and the large value of the geminal coupling constant 2JC3,F = 29.6 Hz evidenced a trans-diaxial relationship between the C-3 and C-4 substituents in 26. The reaction of fluorohydrin 26 with DAST in benzene under heating afforded, as the main product, 3,4-difluoro-D-gluco analog 28 (46%), and the rearranged 2,6-anhydro compound 29 (12%) as a side product. Products 28 and 29 can be separated by careful chromatography and their structures were verified by single crystal X-ray analysis.
Since fluorination of 12 with DAST failed to introduce fluorine at C-4β due to formation of orthoester 25, the reaction of 1,6:2,3-dianhydro-β-D-talopyranose 14 with DAST was utilized to give 1,6:2,3-dianhydro-4-deoxy-4-fluoro-β-D-talopyranose (30) with retention of the configuration at C-4 [39]. Azidolysis of the oxirane ring in the reaction with lithium azide furnished 2-azido derivative 31. Although nucleophilic cleavage of a three-membered ring annulated to the 1,6-anhydrohexopyranose skeleton usually occurs solely in trans-diaxial fashion [50], formation of the trans-diequatorial side-product 32 in ca. 8% was observed in NMR spectra. The desired D-galacto-configured 2-azido derivative 31 was separated from 32 by crystallization in 74% yield. The D-galacto configuration of 31 is evidenced by the vicinal coupling values 3JH4,H3/5 = 4.5 Hz and the long range coupling between equatorial protons H-1, H-3, and H-5 (4JH3,H1/5 = 1.4 Hz).
The reaction of the D-gluco-configured 3-hydroxy derivatives 11, and 26 with DAST, and the previously reported reactions of D-gluco-configured 3-hydroxy derivatives 33 [26], 34 [27], and 35 [25] (Scheme 4) with DAST are an important means of obtaining 3-deoxy-3-fluoro derivatives of D-gluco-configured aldohexopyranoses which are difficult to prepare otherwise. These reactions characteristically proceed with a clean retention of configuration which can be explained by an anchimeric assistance of the trans-diaxially positioned (with respect to C3–OH) polar groups at C-2 or C-4, or by an internal fluorine attack as in SNi substitution. A simple SN2 displacement leading to configurational inversion is probably suppressed by the steric effects of the axially positioned groups at C-2 and C-4, and repulsive effects of their aligned dipoles [51]. Compounds 11, and 33–35, possess a trans-diaxially positioned benzyloxy group at C-4 capable of participation through an oxiranium intermediate species (Scheme 4A) [52-54]. The formation of the rearranged difluoride 29 from alcohol 26 (Scheme 3) suggests an anchimeric assistance of the vicinal C-2 azido group. Although rare, azide participation was postulated before [52]. The main product 28 and rearranged difluoride 29 can arise from the same intermediate species 39 (Scheme 4B, pathways a and b, respectively). Compound 19 can, in principle, be also formed from 11 through azide participation (not shown). In such a case the reaction is unexpectedly sensitive to minor steric alterations of the substrate because the C-4 epimer 15 (Scheme 3) did not react. An internal fluorine attack from the β-face of the tetrahydropyran ring through a concerted (Scheme 4C) or contact ion-pair (Scheme 4D) SNi mechanism cannot be ruled out [55,56] because the bulky Et2NSF2O substituent at C-3 might force the substrate to adopt boat B3,O conformation [57] bringing the C-2, C-3 and C-4 substituents into a trans-equatorial arrangement unfavorable for anchimeric assistance.
![[1860-5397-12-75-i4]](/bjoc/content/inline/1860-5397-12-75-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Suggested mechanisms for deoxofluorination at C-3 of 1,6-anhydro-β-D-glucohexopyranose derivatives. A) participation of the O-benzyl group; B) participation of the azide group; C) internal fluorine attack, concerted mechanism; D) internal fluorine attack, contact ion pair mechanism.
Scheme 4: Suggested mechanisms for deoxofluorination at C-3 of 1,6-anhydro-β-D-glucohexopyranose derivatives....
With the D-gluco- and D-galacto-configured deoxofluoro derivatives of 2-azido-1,6-anhydrohexopyranoses in hand, we examined their conversion to the target acetylated hexosamine analogs. Initially, compound 19 was hydrogenated (Pd/C) and acetylated to afford acetamide 40 in modest yield (37%). Sulfuric acid-catalyzed [35] cleavage of the internal acetal with acetic anhydride gave a mixture containing 1,2-oxazoline 41 as the main product (Scheme 5). The structure of oxazoline 41 was confirmed by single crystal X-ray diffraction analysis which also confirmed the retention of configuration during the preceding fluorine introduction.
![[1860-5397-12-75-i5]](/bjoc/content/inline/1860-5397-12-75-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Formation of oxazoline 41 from 19.
Scheme 5: Formation of oxazoline 41 from 19.
To prevent oxazoline formation, the order of reactions was reversed, and triethylsilyl triflate (TESOTf)-catalyzed [58] acetolysis of the 1,6-anhydro bridge in 19 gave 42 (Table 1) as a mixture of anomers from which the α-anomer crystallized. TESOTf as a catalyst for acetolysis gave better results than sulfuric acid in terms of product purity. Hydrogenolysis of 42 on palladium in ethanol/HCl followed by acetylation of the amino group furnished the target acetylated 3-fluoro-D-GlcNAc 5 as a chromatographically separable mixture of anomers (Table 1). Addition of HCl was found necessary to effect a clean hydrogenolytic removal of the O-benzyl group in 42. The D-gluco configuration of 42 and 5 was reflected in the large values of 3JH3,H2/4 (7.6–10.2 Hz), and the chair inversion accompanying the cleavage of the 1,6-anhydro bridge (1C4 → 4C1) in a decrease of the geminal fluorine–carbon coupling value 2JC4,F (31.5 Hz in 40 → 18.4 Hz in 5 (α-anomer)) [59]. 1H and 13C NMR spectra of 5 reported in [20] are comparable with our data for the α-anomer (5α). A discrepancy in the values of specific optical rotation ![[Graphic 20]](/bjoc/content/inline/1860-5397-12-75-i26.svg?max-width=637&scale=1.18182) (reported +10°, +81° obtained by us) can be explained assuming that the value for the β-anomer (5β) was reported in [20]. Our synthesis of 5 is more practical (55% from 10 in 3 steps) than the earlier preparations which were reported troublesome owing to low yields (28% and 10%, respectively) of the key fluorination step [18,20].
(reported +10°, +81° obtained by us) can be explained assuming that the value for the β-anomer (5β) was reported in [20]. Our synthesis of 5 is more practical (55% from 10 in 3 steps) than the earlier preparations which were reported troublesome owing to low yields (28% and 10%, respectively) of the key fluorination step [18,20].
Table 1: Acetolysis of the fluorine-containing intermediates and hydrogenation.
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-75-i7.svg?max-width=637&scale=1.0)
|
|||||
| entry | starting compound | product of acetolysis | yield (%)a | target compound | yield (%)a |
|---|---|---|---|---|---|
| 1 |
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-75-i8.svg?max-width=637&scale=1.0)
19 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-75-i9.svg?max-width=637&scale=1.0)
42 |
96 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-75-i10.svg?max-width=637&scale=1.0)
5 |
82b |
| 2 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-12-75-i11.svg?max-width=637&scale=1.0)
21 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-12-75-i12.svg?max-width=637&scale=1.0)
43 |
83 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-12-75-i13.svg?max-width=637&scale=1.0)
6 |
74 |
| 3 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-12-75-i14.svg?max-width=637&scale=1.0)
26 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-12-75-i15.svg?max-width=637&scale=1.0)
|
78 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-12-75-i16.svg?max-width=637&scale=1.0)
1 |
86 |
| 16 | n.a. | ||||
| 4 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-12-75-i17.svg?max-width=637&scale=1.0)
31 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-12-75-i18.svg?max-width=637&scale=1.0)
46 |
94 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-12-75-i19.svg?max-width=637&scale=1.0)
4 |
78 |
| 5 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-12-75-i20.svg?max-width=637&scale=1.0)
28 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-12-75-i21.svg?max-width=637&scale=1.0)
47 |
c |
![[Graphic 16]](/bjoc/content/inline/1860-5397-12-75-i22.svg?max-width=637&scale=1.0)
7 |
44d |
| 6 |
![[Graphic 17]](/bjoc/content/inline/1860-5397-12-75-i23.svg?max-width=637&scale=1.0)
22 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-12-75-i24.svg?max-width=637&scale=1.0)
48 |
86 |
![[Graphic 19]](/bjoc/content/inline/1860-5397-12-75-i25.svg?max-width=637&scale=1.0)
8 |
70 |
aIsolated yield; bH2, Pd/C, EtOH, AcCl, then Ac2O, py was used for hydrogenation of 42; c47 was not isolated as pure compound, see text; doverall from 28; n.a. – not available.
For the remaining fluoro derivatives of 1,6-anhydro-2-azidohexopyranoses, TESOTf-catalyzed acetolysis and subsequent hydrogenation (Pd/C in a mixture of ethanol and acetic anhydride) was applied to obtain the target acetylated fluoro analogs (Table 1). Compound 21 was acetolyzed to 43 which was obtained as a mixture of anomers and characterized by NMR and finally hydrogenated to furnish acetylated 3-fluoro-GalNAc 6 isolated as a separable mixture of anomers. The D-galacto configuration of 6 is manifested by the lower 3JH3,H4 coupling value (3.4 Hz, α-anomer) in comparison with that of its C-4 epimer 5 (8.6 Hz, α-anomer). Acetolysis of the internal acetal in 26 proceeded more slowly and beside the desired product 44 (78% yield), 3-O-acetyl derivative 45, in which the 1,6-anhydro bridge remained intact, was also isolated. Hydrogenation of 44 furnished the known acetylated 4-fluoro-D-GlcNAc 1 [2]. The large values of the vicinal coupling constants 3JH3,H2/4 and 3JH4,H5 (8.9–11.1 Hz) confirmed the D-gluco configuration of 1 and the chair inversion when going from 26 to 1, which was also manifested by a decrease in the geminal coupling constant 2JC3,F (29.6 → 18.9 Hz, α-anomer). Acetolysis and subsequent hydrogenation of 31 provided the known peracetylated 4-deoxy-4-fluoro-D-galactosamine 4 [29]. The chair inversion of the tetrahydropyran ring associated with the cleavage of the 1,6-anhydro bridge is indicated by the increase in the coupling value 3JH2,H3 (1.4 → 11.6 Hz) and decrease in the coupling value 2JC5,F (27.4 → 18.4 Hz) when going from 31 to 4 (α-anomer). Acetolysis of 28 afforded diacetate 47 containing chromatographically inseparable impurities (NMR). They were removed in the next hydrogenation step to yield the peracetylated 3,4-difluoro analog of D-glucosamine 7 isolated as an α-anomer in 44% yield from 28. The D-gluco configuration of 7 is reflected in the large values of the vicinal coupling constants 3JH2,H3 = 10.5 Hz, 3JH3,H4 = 8.2 Hz, and 3JH4,H5 = 9.8 Hz. Acetolysis of 22 gave diacetate 48 as a separable mixture of anomers. Hydrogenation of the α-anomer furnished the peracetylated 3,4-difluoro analog of D-galactosamine 8. The D-galacto configuration of 8 was confirmed by an increase in the value of 3JH2,H3 (1.6 → 11.1 Hz), a decrease in the value of 2JC5,F4 (27.2 → 18.3 Hz), and an increase of 3JH3,F4 coupling (4.3 → 26.0 Hz) between 22 and 8.
To study the influence of 1-O-deacetylation on the cytotoxicity, the monofluorinated analogs 1, and 4–6 were subjected to anomeric deacetylation (Scheme 6). Compound 5 provided 1-O-deacetylated product 49 by treatment with BnNH2 in THF. Since acetylated 4-fluoro-D-GlcNAc 1 under these conditions did not react cleanly, we used piperidine-promoted [60] deacetylation to prepare 2 in 74% yield. Similarly, acetylated 4-fluoro-D-GalNAc 4 gave 50 in 60% yield. The attempted anomeric deacetylation of 3-fluoro-D-GalNAc 6 by treatment with piperidine followed by chromatography gave a fraction containing an inseparable side-product in addition to the expected deacetylated product 51. The side-product showed no fluorine resonance in 19F NMR and its molecular formula C17H28N2O7 assigned by LC–HRMS corresponded to a formal displacement of fluorine by piperidine, leading probably to compound 53. When pure 51 (prepared by another method, see below) was reacted with excess piperidine, high resolution ESIMS analysis of the reaction detected transient formation of an adduct ion corresponding to a supposed intermediate enal 52 (Scheme 6), while the adduct ion corresponding to 53 was the final product (see Supporting Information File 1). Presumably, piperidine as a relatively strong base effected dehydrofluorination of 51 to enal 52 which then added piperidine to give 53 as a byproduct (Scheme 6). To avoid the action of basic amines, a silica gel mediated anomeric deacetylation, recommended for 2-aminosugars [61], was tried. The reaction proceeded extremely slowly with our substrate 6 and the product 51 was obtained in only 40% yield after chromatography and recrystallization.
![[1860-5397-12-75-i6]](/bjoc/content/inline/1860-5397-12-75-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: 1-O-Deacetylation of monofluorinated hexosamines. Reagents and conditions: (a) BnNH2, THF, 62%; (b) C5H10NH, THF, 74%; (c) C5H10NH, THF, 60%; (d) silica gel, MeOH, 30 days, 40%.
Scheme 6: 1-O-Deacetylation of monofluorinated hexosamines. Reagents and conditions: (a) BnNH2, THF, 62%; (b)...
Cytotoxicity
Some acetylated fluorinated hexosamines (HexN), including peracetates of the α-methyl glycoside of 3-fluoro-D-ManNAc [18], 3-fluoro-D-GlcNAc 5 [18], 4-fluoro-D-GlcNAc 1 [19], 4-fluoro-D-GalNAc 4 [19], 4,4-difluoro-D-xylo-HexNAc [19], and 4,6-difluoro-D-GalNAc [19] were reported to exhibit antiproliferative properties against L1210 leukemia cells in micromolar concentrations (IC50 24–43 μM). It was found that O-deacetylated amino sugars were often inactive due to low lipophilicity and poor cellular uptake [19]. Compound 5 was also cytotoxic to the human pancreatic cancer cell line KP1-NL (IC50 = 30 μM) [20], and 1 and its 1-O-deacetylated derivative 2 inhibited cell proliferation of the human prostate cancer cell line PC-3 (IC50 61 µM for 2) [2]. Interestingly, while all 4-fluoro analogs 1–3 (Figure 1) reduced the expression of highly branched N-glycans in PC-3 cells, the 6-O-deacetylated analog 3 showed only negligible cytotoxicity [2] implying that the inhibition of proliferation and perturbation of N-glycan biosynthesis occur by different mechanisms.
Increased cytotoxicity as a result of 1-O-deacylation was noted for a variety of acylated (nonfluorinated) D-mannosamine and D-glucosamine derivatives [62,63]. Acylated hexosamine derivatives were subsequently studied as possible templates for the development of anticancer therapeutics [64,65]. While the ability of hexosamine derivatives and analogs to inhibit cell growth creates an avenue for their use in the development of anticancer drugs, it also limits their utility as agents to modify the cellular glycome [62]. The cytotoxic activity of peracetylated monofluoro analogs 1, and 4–6, their 1-O-deacetylated derivatives 2, and 49–51, difluoro analogs 7 and 8, and oxazoline 41 was therefore tested for 24 h on the human prostate cancer PC-3 cell line, and human ovarian cancer A2780 cell line using the MTT assay, and the obtained IC50 values were compared with those obtained for cisplatin and 5-fluorouracil.
All of the tested compounds induced only moderate to weak inhibition of A2780 cell proliferation (IC50 values ranging from 78 μM to 327 μM, Table 2) in comparison to cisplatin (IC50 12.9 μM). Anomeric deacetylation resulted in a higher activity in the case of 3-fluoro-D-GlcNAc (49, IC50 84 μM, Table 2, entry 6), and especially 4-fluoro-D-GlcNAc (2, IC50 78 μM, ca. 4-fold higher activity than that of the peracetylated 1, Table 2, entries 1 and 2), while 1-O-deacetylated 50 had a lower activity (IC50 142 μM, Table 2, entry 4) then the parent peracetate 4 (IC50 78 μM, Table 2, entry 2). All of the tested fluorinated D-glucosamine analogs also induced only weak inhibition on PC-3 cells (IC50 ranging from 134 μM to 337 μM, Table 2, entries 1, 2, 5, 6, and 9), and 1-O-deacetylation seemed to have a negative effect here (Table 2, entries 2 and 6). On the other hand, the 1-O-deacetylated form of 3-fluoro-D-galactosamine 51 (IC50 28 μM, Table 2, entry 8) and 4-fluoro-D-galactosamine 50 (IC50 54 μM, Table 2, entry 4) reduced proliferation of PC-3 cells more than cisplatin (IC50 182 μM, Table 2, entry 12) and 5-fluorouracil (IC50 69 μM, Table 2, entry 13). The effect of anomeric deacetylation was particularly pronounced in 3-fluoro-D-galactosamine 51 owing to inactivity of the corresponding peracetate 6. Finally, difluoro analogs 7 and 8, and oxazoline 41 exhibited weak (compound 7) to none (compounds 8 and 41) activity in the PC-cell line (Table 2, entries 9–11). Taken together, our results seem to corroborate in part the observations by Yarema at al. that the liberation of the anomeric hydroxy group in acylated hexosamines resulted in an enhancement of their cytotoxicity [62,63]. The effect of anomeric deacetylation was, however, found much more cell-line and substrate-specific for our fluoro analogs than it was for acylated natural hexosamines [62].
Table 2: IC50 mean values (μM) of selected fluorinated analogs after 24 h treatment using PC-3 and A2780 cancer cell lines.
| entry | compd. no. | abbreviationa | PC-3 | A2780 |
|---|---|---|---|---|
| 1 | 1 | 4F-Ac3GlcNAc | 235 ± 26 | 327 ± 48 |
| 2 | 2 | 4F-Ac2GlcNAc-1-OH | 337 ± 36 | 78 ± 9 |
| 3 | 4 | 4F-Ac3GalNAc | 101 ± 15 | 78 ± 9 |
| 4 | 50 | 4F-Ac2GalNAc-1-OH | 54 ± 5 | 142 ± 28 |
| 5 | 5 | 3F-Ac3GlcNAc | 134 ± 17 | 218 ± 29 |
| 6 | 49 | 3F-Ac2GlcNAc-1-OH | 308 ± 38 | 84 ± 6 |
| 7 | 6 | 3F-Ac3GalNAc | >500 | 249 ± 36 |
| 8 | 51 | 3F-Ac2GalNAc-1-OH | 28 ± 3 | 137 ± 24 |
| 9 | 7 | 3F,4F-Ac2GlcNAc | 199 ± 34 | 183 ± 26 |
| 10 | 8 | 3F,4F-Ac2GalNAc | >500 | 250 ± 41 |
| 11 | 41 | 3F-Ac2Glc-1,2-oxazoline | >1000 | 108 ± 14 |
| 12 | cisplatin | 182 ± 13 | 12.9 ± 1.5 | |
| 13 | 5-fluorouracil | 69 ± 6 | 52 ± 7 | |
aThese abbreviations are provided for quick orientation, anomeric deacetylation is indicated by '-1-OH'.
Conclusion
We have developed a synthetic route for the preparation of a series of 3- and 4-deoxofluorinated analogs of D-glucosamine and D-galactosamine. The choice of O-benzylated-1,6:2,3-dianhydro-β-D-mannopyranose 10 and 2,3-isopropylidene-D-mannosan 16 as starting material permits regio- and stereoselective introduction of both fluorine and nitrogen (as an azide) by nucleophilic substitution before acetolysis of the 1,6-anhydro bridge. The characteristic feature of the synthesis is the introduction of fluorine at C-3 by the reaction of D-gluco-configured 3-hydroxy derivatives with DAST with retention of configuration. The 1-O-deacetylated 3-fluoro and 4-fluoro analogs 51 and 50 of acetylated D-galactosamine were shown to be more cytotoxic in the PC-3 cell line than cisplatin and 5-fluorouracil. Most of the other fluoro analogs displayed moderate to low cytotoxicity. Fluoro analogs 6–8 are new compounds and their influence on the cell-surface glycan biosynthesis is currently being studied. We anticipate that 1,6-anhydro-2-azido-fluorohydrins 20, 21, 26 and 31 will find use as building blocks for the synthesis of fluorinated oligosaccharides and other glycoconjugates because they can be immediately employed as glycosyl acceptors or readily converted into glycosyl donors. Research in this way is now in progress in our laboratory.
Supporting Information
| Supporting Information File 1: Experimental procedures for compounds 1, 2, 4–8, 12, 18–22, 25, 26, 28, 29, 31, 40–42, 44, 45, and 48–51, HRMS results for reaction of 51 with piperidine, cell culture conditions and MTT assay, crystallographic data for compounds 25, 28, 29, and 41, and dose-response curves. | ||
| Format: PDF | Size: 905.4 KB | Download |
| Supporting Information File 2: NMR spectra for compounds 1, 2, 4–8, 12, 18–22, 25, 26, 28, 29, 31, 40–46, and 48–51. | ||
| Format: PDF | Size: 11.4 MB | Download |
References
-
Taylor, M. E.; Drickamer, K. Introduction to glycobiology, 3rd ed.; Oxford University Press: Oxford, United Kingdom, 2011.
Return to citation in text: [1] -
Nishimura, S.-I.; Hato, M.; Hyugaji, S.; Feng, F.; Amano, M. Angew. Chem., Int. Ed. 2012, 51, 3386–3390. doi:10.1002/anie.201108742
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Van Wijk, X. M.; Lawrence, R.; Thijssen, V. L.; van den Broek, S. A.; Troost, R.; van Scherpenzeel, M.; Naidu, N.; Oosterhof, A.; Griffioen, A. W.; Lefeber, D. J.; van Delft, F. L.; van Kuppevelt, T. H. FASEB J. 2015, 29, 2993–3002. doi:10.1096/fj.14-264226
Return to citation in text: [1] [2] [3] [4] -
Li, Y.; Zhou, Y.; Ma, Y.; Li, X. Carbohydr. Res. 2011, 346, 1714–1720. doi:10.1016/j.carres.2011.05.024
Return to citation in text: [1] -
Frantom, P. A.; Coward, J. K.; Blanchard, J. S. J. Am. Chem. Soc. 2010, 132, 6626–6627. doi:10.1021/ja101231a
Return to citation in text: [1] -
Pouilly, S.; Bourgeaux, V.; Piller, F.; Piller, V. ACS Chem. Biol. 2012, 7, 753–760. doi:10.1021/cb200511t
Return to citation in text: [1] -
Hartman, M. C. T.; Coward, J. K. J. Am. Chem. Soc. 2002, 124, 10036–10053. doi:10.1021/ja0127234
Return to citation in text: [1] -
Oberbillig, T.; Mersch, C.; Wagner, S.; Hoffmann-Röder, A. Chem. Commun. 2012, 48, 1487–1489. doi:10.1039/C1CC15139H
Return to citation in text: [1] -
Hoffmann-Röder, A.; Kaiser, A.; Wagner, S.; Gaidzik, N.; Kowalczyk, D.; Westerlind, U.; Gerlitzki, B.; Schmitt, E.; Kunz, H. Angew. Chem., Int. Ed. 2010, 49, 8498–8503. doi:10.1002/anie.201003810
Return to citation in text: [1] -
Barthel, S. R.; Antonopoulos, A.; Cedeno-Laurent, F.; Schaffer, L.; Hernandez, G.; Patil, S. A.; North, S. J.; Dell, A.; Matta, K. L.; Neelamegham, S.; Haslam, S. M.; Dimitroff, C. J. J. Biol. Chem. 2011, 286, 21717–21731. doi:10.1074/jbc.M110.194597
Return to citation in text: [1] -
Marathe, D. D.; Buffone, A., Jr.; Chandrasekaran, E. V.; Xue, J.; Locke, R. D.; Nasirikenari, M.; Lau, J. T. Y.; Matta, K. L.; Neelamegham, S. Blood 2010, 115, 1303–1312. doi:10.1182/blood-2009-07-231480
Return to citation in text: [1] [2] -
Woynarowska, B.; Skrincosky, D. M.; Haag, A.; Sharma, M.; Matta, K.; Bernacki, R. J. J. Biol. Chem. 1994, 269, 22797–22803.
http://www.jbc.org/content/269/36/22797.abstract
Return to citation in text: [1] -
Woynarowska, B.; Dimitroff, C. J.; Sharma, M.; Matta, K. L.; Bernacki, R. J. Glycoconjugate J. 1996, 13, 663–674. doi:10.1007/BF00731455
Return to citation in text: [1] -
Dimitroff, C. J.; Bernacki, R. J.; Sackstein, R. Blood 2003, 101, 602–610. doi:10.1182/blood-2002-06-1736
Return to citation in text: [1] -
Dimitroff, C. J.; Kupper, T. S.; Sackstein, R. J. Clin. Invest. 2003, 112, 1008–1018. doi:10.1172/JCI19220
Return to citation in text: [1] -
Cedeno-Laurent, F.; Opperman, M. J.; Barthel, S. R.; Hays, D.; Schatton, T.; Zhan, Q.; He, X.; Matta, K. L.; Supko, J. G.; Frank, M. H.; Murphy, G. F.; Dimitroff, C. J. J. Invest. Dermatol. 2012, 132, 410–420. doi:10.1038/jid.2011.335
Return to citation in text: [1] -
Goon, S.; Bertozzi, C. R. J. Carbohydr. Chem. 2002, 21, 943–977. doi:10.1081/CAR-120016493
Return to citation in text: [1] -
Sharma, M.; Bernacki, R. J.; Hillman, M. J.; Korytnyk, W. Carbohydr. Res. 1993, 240, 85–93. doi:10.1016/0008-6215(93)84174-5
Return to citation in text: [1] [2] [3] [4] -
Sharma, M.; Bernacki, R. J.; Paul, B.; Korytnyk, W. Carbohydr. Res. 1990, 198, 205–221. doi:10.1016/0008-6215(90)84293-4
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Wasonga, G.; Tatara, Y.; Kakizaki, I.; Huang, X. J. Carbohydr. Chem. 2013, 32, 392–409. doi:10.1080/07328303.2013.815196
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Arndt, S.; Hsieh-Wilson, L. C. Org. Lett. 2003, 5, 4179–4182. doi:10.1021/ol035606h
Return to citation in text: [1] [2] -
Černý, M.; Staněk, J., Jr. Adv. Carbohydr. Chem. Biochem. 1977, 34, 23–177. doi:10.1016/S0065-2318(08)60324-8
Return to citation in text: [1] -
Kulkarni, S. S.; Lee, J.-C.; Hung, S.-C. Curr. Org. Chem. 2004, 8, 475–509. doi:10.2174/1385272043485800
Return to citation in text: [1] -
Karban, J.; Sýkora, J.; Kroutil, J.; Císařová, I.; Padělková, Z.; Buděšínský, M. J. Org. Chem. 2010, 75, 3443–3446. doi:10.1021/jo1000912
Return to citation in text: [1] [2] -
Mtashobya, L.; Quiquempoix, L.; Linclau, B. J. Fluorine Chem. 2015, 171, 92–96. doi:10.1016/j.jfluchem.2014.08.023
Return to citation in text: [1] [2] -
Kobayashi, K.; Kondo, T. Macromolecules 1997, 30, 6531–6535. doi:10.1021/ma970691s
Return to citation in text: [1] [2] -
Sarda, P.; Escribano, F. C.; Alves, R. J.; Olesker, A.; Lukacs, G. J. Carbohydr. Chem. 1989, 8, 115–123. doi:10.1080/07328308908047996
Return to citation in text: [1] [2] -
Faghih, R.; Escribano, F. C.; Castillon, S.; Garcia, J.; Lukacs, G.; Olesker, A.; Thang, T. T. J. Org. Chem. 1986, 51, 4558–4564. doi:10.1021/jo00374a013
Return to citation in text: [1] [2] -
Berkin, A.; Szarek, W. A.; Kisilevsky, R. Carbohydr. Res. 2000, 326, 250–263. doi:10.1016/S0008-6215(00)00049-5
Return to citation in text: [1] [2] -
Karban, J.; Horník, Š.; Červenková Šťastná, L.; Sýkora, J. Synlett 2014, 25, 1253–1256. doi:10.1055/s-0033-1341187
Return to citation in text: [1] -
Hesek, D.; Lee, M.; Zhang, W.; Noll, B. C.; Mobashery, S. J. Am. Chem. Soc. 2009, 131, 5187–5193. doi:10.1021/ja808498m
Return to citation in text: [1] [2] -
Katavic, P. L.; Yong, K. W. L.; Herring, J. N.; Deseo, M. A.; Blanchfield, J. T.; Ferro, V.; Garson, M. J. Tetrahedron 2013, 69, 8074–8079. doi:10.1016/j.tet.2013.06.079
Return to citation in text: [1] -
Ganguli, A. R. S.; Coward, J. K. Tetrahedron: Asymmetry 2005, 16, 411–424. doi:10.1016/j.tetasy.2004.11.053
Return to citation in text: [1] [2] -
Trnka, T.; Černý, M. Collect. Czech. Chem. Commun. 1971, 36, 2216–2225. doi:10.1135/cccc19712216
Return to citation in text: [1] [2] -
Paulsen, H.; Stenzel, W. Chem. Ber. 1978, 111, 2348–2357. doi:10.1002/cber.19781110629
Return to citation in text: [1] [2] -
Karban, J.; Buděšínský, M.; Černý, M.; Trnka, T. Collect. Czech. Chem. Commun. 2001, 66, 799–819. doi:10.1135/cccc20010799
Return to citation in text: [1] -
Rehnberg, N.; Magnusson, G. J. Org. Chem. 1990, 55, 5467–5476. doi:10.1021/jo00307a017
Return to citation in text: [1] -
Doležalová, J.; Trnka, T.; Černý, M. Collect. Czech. Chem. Commun. 1982, 47, 2415–2422. doi:10.1135/cccc19822415
Return to citation in text: [1] -
Karban, J.; Císařová, I.; Strašák, T.; Červenková Šťastná, L.; Sýkora, J. Org. Biomol. Chem. 2012, 10, 394–403. doi:10.1039/C1OB06336G
Return to citation in text: [1] [2] -
Paulsen, H.; Kolář, Č.; Stenzel, W. Chem. Ber. 1978, 111, 2358–2369. doi:10.1002/cber.19781110630
Return to citation in text: [1] -
Oberdorfer, F.; Haeckel, R.; Lauer, G. Synthesis 1998, 201–206. doi:10.1055/s-1998-4484
Return to citation in text: [1] -
Zottola, M. A.; Alonso, R.; Vite, G. D.; Fraser-Reid, B. J. Org. Chem. 1989, 54, 6123–6125. doi:10.1021/jo00287a029
Return to citation in text: [1] -
Ogawa, S.; Nakamura, Y. Carbohydr. Res. 1992, 226, 79–89. doi:10.1016/0008-6215(92)84056-X
Return to citation in text: [1] -
Hann, R. M.; Hudson, C. S. J. Am. Chem. Soc. 1942, 64, 925–928. doi:10.1021/ja01256a053
Return to citation in text: [1] -
Staněk, J., Jr.; Černý, M. Synthesis 1972, 698–699. doi:10.1055/s-1972-21974
Return to citation in text: [1] -
Ogawa, S.; Aso, D. Carbohydr. Res. 1993, 250, 177–184. doi:10.1016/0008-6215(93)84164-2
Return to citation in text: [1] -
Wong, T. C.; Townsend, R. R.; Lee, Y. C. Carbohydr. Res. 1987, 170, 27–46. doi:10.1016/0008-6215(87)85003-6
Return to citation in text: [1] -
Bernet, B.; Vasella, A. Helv. Chim. Acta 2007, 90, 1874–1888. doi:10.1002/hlca.200790196
Return to citation in text: [1] -
Rönnols, J.; Manner, S.; Siegbahn, A.; Ellervik, U.; Widmalm, G. Org. Biomol. Chem. 2013, 11, 5465–5472. doi:10.1039/c3ob40991k
Return to citation in text: [1] -
Karban, J.; Kroutil, J. Adv. Carbohydr. Chem. Biochem. 2006, 60, 27–101. doi:10.1016/S0065-2318(06)60003-6
Return to citation in text: [1] -
Hale, K. J.; Hough, L.; Manaviazar, S.; Calabrese, A. Org. Lett. 2014, 16, 4838–4841. doi:10.1021/ol502193j
Return to citation in text: [1] -
Hanessian, S.; Saavedra, O. M.; Vilchis-Reyes, M. A.; Llaguno-Rueda, A. M. Med. Chem. Commun. 2014, 5, 1166–1171. doi:10.1039/C4MD00072B
Return to citation in text: [1] [2] -
Hartlieb, S.; Günzel, A.; Gerardy-Schahn, R.; Münster-Kühnel, A. K.; Kirschning, A.; Dräger, G. Carbohydr. Res. 2008, 343, 2075–2082. doi:10.1016/j.carres.2008.02.003
Return to citation in text: [1] -
Takahashi, Y.; Vasella, A. Helv. Chim. Acta 1992, 75, 1563–1571. doi:10.1002/hlca.19920750510
Return to citation in text: [1] -
Mori, Y.; Morishima, N. Chem. Pharm. Bull. 1991, 39, 1088–1090. doi:10.1248/cpb.39.1088
Return to citation in text: [1] -
Mori, Y.; Morishima, N. Bull. Chem. Soc. Jpn. 1994, 67, 236–241. doi:10.1246/bcsj.67.236
Return to citation in text: [1] -
Kroutil, J.; Buděšínský, M. Carbohydr. Res. 2007, 342, 147–153. doi:10.1016/j.carres.2006.11.028
Return to citation in text: [1] -
Zottola, M.; Rao, B. V.; Fraser-Reid, B. J. Chem. Soc., Chem. Commun. 1991, 969–970. doi:10.1039/c39910000969
Return to citation in text: [1] -
Wray, V. J. Chem. Soc., Perkin Trans. 2 1976, 1598–1605. doi:10.1039/p29760001598
Return to citation in text: [1] -
Rowell, R. M.; Feather, M. S. Carbohydr. Res. 1967, 4, 486–491. doi:10.1016/S0008-6215(00)81840-6
Return to citation in text: [1] -
Avalos, M.; Babiano, R.; Cintas, P.; Jiménez, J. L.; Palacios, J. C.; Valencia, C. Tetrahedron Lett. 1993, 34, 1359–1362. doi:10.1016/S0040-4039(00)91795-7
Return to citation in text: [1] -
Aich, U.; Campbell, C. T.; Elmouelhi, N.; Weier, C. A.; Sampathkumar, S.-G.; Choi, S. S.; Yarema, K. J. ACS Chem. Biol. 2008, 3, 230–240. doi:10.1021/cb7002708
Return to citation in text: [1] [2] [3] [4] -
Almaraz, R. T.; Aich, U.; Khanna, H. S.; Tan, E.; Bhattacharya, R.; Shah, S.; Yarema, K. J. Biotechnol. Bioeng. 2012, 109, 992–1006. doi:10.1002/bit.24363
Return to citation in text: [1] [2] -
Campbell, C. T.; Aich, U.; Weier, C. A.; Wang, J. J.; Choi, S. S.; Wen, M. M.; Maisel, K.; Sampathkumar, S.-G.; Yarema, K. J. J. Med. Chem. 2008, 51, 8135–8147. doi:10.1021/jm800873k
Return to citation in text: [1] -
Elmouelhi, N.; Aich, U.; Paruchuri, V. D. P.; Meledeo, M. A.; Campbell, C. T.; Wang, J. J.; Srinivas, R.; Khanna, H. S.; Yarema, K. J. J. Med. Chem. 2009, 52, 2515–2530. doi:10.1021/jm801661m
Return to citation in text: [1]
| 34. | Trnka, T.; Černý, M. Collect. Czech. Chem. Commun. 1971, 36, 2216–2225. doi:10.1135/cccc19712216 |
| 37. | Rehnberg, N.; Magnusson, G. J. Org. Chem. 1990, 55, 5467–5476. doi:10.1021/jo00307a017 |
| 31. | Hesek, D.; Lee, M.; Zhang, W.; Noll, B. C.; Mobashery, S. J. Am. Chem. Soc. 2009, 131, 5187–5193. doi:10.1021/ja808498m |
| 33. | Ganguli, A. R. S.; Coward, J. K. Tetrahedron: Asymmetry 2005, 16, 411–424. doi:10.1016/j.tetasy.2004.11.053 |
| 64. | Campbell, C. T.; Aich, U.; Weier, C. A.; Wang, J. J.; Choi, S. S.; Wen, M. M.; Maisel, K.; Sampathkumar, S.-G.; Yarema, K. J. J. Med. Chem. 2008, 51, 8135–8147. doi:10.1021/jm800873k |
| 65. | Elmouelhi, N.; Aich, U.; Paruchuri, V. D. P.; Meledeo, M. A.; Campbell, C. T.; Wang, J. J.; Srinivas, R.; Khanna, H. S.; Yarema, K. J. J. Med. Chem. 2009, 52, 2515–2530. doi:10.1021/jm801661m |
| 38. | Doležalová, J.; Trnka, T.; Černý, M. Collect. Czech. Chem. Commun. 1982, 47, 2415–2422. doi:10.1135/cccc19822415 |
| 62. | Aich, U.; Campbell, C. T.; Elmouelhi, N.; Weier, C. A.; Sampathkumar, S.-G.; Choi, S. S.; Yarema, K. J. ACS Chem. Biol. 2008, 3, 230–240. doi:10.1021/cb7002708 |
| 2. | Nishimura, S.-I.; Hato, M.; Hyugaji, S.; Feng, F.; Amano, M. Angew. Chem., Int. Ed. 2012, 51, 3386–3390. doi:10.1002/anie.201108742 |
| 62. | Aich, U.; Campbell, C. T.; Elmouelhi, N.; Weier, C. A.; Sampathkumar, S.-G.; Choi, S. S.; Yarema, K. J. ACS Chem. Biol. 2008, 3, 230–240. doi:10.1021/cb7002708 |
| 63. | Almaraz, R. T.; Aich, U.; Khanna, H. S.; Tan, E.; Bhattacharya, R.; Shah, S.; Yarema, K. J. Biotechnol. Bioeng. 2012, 109, 992–1006. doi:10.1002/bit.24363 |
| 2. | Nishimura, S.-I.; Hato, M.; Hyugaji, S.; Feng, F.; Amano, M. Angew. Chem., Int. Ed. 2012, 51, 3386–3390. doi:10.1002/anie.201108742 |
| 46. | Ogawa, S.; Aso, D. Carbohydr. Res. 1993, 250, 177–184. doi:10.1016/0008-6215(93)84164-2 |
| 43. | Ogawa, S.; Nakamura, Y. Carbohydr. Res. 1992, 226, 79–89. doi:10.1016/0008-6215(92)84056-X |
| 44. | Hann, R. M.; Hudson, C. S. J. Am. Chem. Soc. 1942, 64, 925–928. doi:10.1021/ja01256a053 |
| 41. | Oberdorfer, F.; Haeckel, R.; Lauer, G. Synthesis 1998, 201–206. doi:10.1055/s-1998-4484 |
| 42. | Zottola, M. A.; Alonso, R.; Vite, G. D.; Fraser-Reid, B. J. Org. Chem. 1989, 54, 6123–6125. doi:10.1021/jo00287a029 |
| 39. | Karban, J.; Císařová, I.; Strašák, T.; Červenková Šťastná, L.; Sýkora, J. Org. Biomol. Chem. 2012, 10, 394–403. doi:10.1039/C1OB06336G |
| 62. | Aich, U.; Campbell, C. T.; Elmouelhi, N.; Weier, C. A.; Sampathkumar, S.-G.; Choi, S. S.; Yarema, K. J. ACS Chem. Biol. 2008, 3, 230–240. doi:10.1021/cb7002708 |
| 63. | Almaraz, R. T.; Aich, U.; Khanna, H. S.; Tan, E.; Bhattacharya, R.; Shah, S.; Yarema, K. J. Biotechnol. Bioeng. 2012, 109, 992–1006. doi:10.1002/bit.24363 |
| 40. | Paulsen, H.; Kolář, Č.; Stenzel, W. Chem. Ber. 1978, 111, 2358–2369. doi:10.1002/cber.19781110630 |
| 62. | Aich, U.; Campbell, C. T.; Elmouelhi, N.; Weier, C. A.; Sampathkumar, S.-G.; Choi, S. S.; Yarema, K. J. ACS Chem. Biol. 2008, 3, 230–240. doi:10.1021/cb7002708 |
| 28. | Faghih, R.; Escribano, F. C.; Castillon, S.; Garcia, J.; Lukacs, G.; Olesker, A.; Thang, T. T. J. Org. Chem. 1986, 51, 4558–4564. doi:10.1021/jo00374a013 |
| 47. | Wong, T. C.; Townsend, R. R.; Lee, Y. C. Carbohydr. Res. 1987, 170, 27–46. doi:10.1016/0008-6215(87)85003-6 |
| 48. | Bernet, B.; Vasella, A. Helv. Chim. Acta 2007, 90, 1874–1888. doi:10.1002/hlca.200790196 |
| 49. | Rönnols, J.; Manner, S.; Siegbahn, A.; Ellervik, U.; Widmalm, G. Org. Biomol. Chem. 2013, 11, 5465–5472. doi:10.1039/c3ob40991k |
| 52. | Hanessian, S.; Saavedra, O. M.; Vilchis-Reyes, M. A.; Llaguno-Rueda, A. M. Med. Chem. Commun. 2014, 5, 1166–1171. doi:10.1039/C4MD00072B |
| 53. | Hartlieb, S.; Günzel, A.; Gerardy-Schahn, R.; Münster-Kühnel, A. K.; Kirschning, A.; Dräger, G. Carbohydr. Res. 2008, 343, 2075–2082. doi:10.1016/j.carres.2008.02.003 |
| 54. | Takahashi, Y.; Vasella, A. Helv. Chim. Acta 1992, 75, 1563–1571. doi:10.1002/hlca.19920750510 |
| 52. | Hanessian, S.; Saavedra, O. M.; Vilchis-Reyes, M. A.; Llaguno-Rueda, A. M. Med. Chem. Commun. 2014, 5, 1166–1171. doi:10.1039/C4MD00072B |
| 25. | Mtashobya, L.; Quiquempoix, L.; Linclau, B. J. Fluorine Chem. 2015, 171, 92–96. doi:10.1016/j.jfluchem.2014.08.023 |
| 51. | Hale, K. J.; Hough, L.; Manaviazar, S.; Calabrese, A. Org. Lett. 2014, 16, 4838–4841. doi:10.1021/ol502193j |
| 26. | Kobayashi, K.; Kondo, T. Macromolecules 1997, 30, 6531–6535. doi:10.1021/ma970691s |
| 27. | Sarda, P.; Escribano, F. C.; Alves, R. J.; Olesker, A.; Lukacs, G. J. Carbohydr. Chem. 1989, 8, 115–123. doi:10.1080/07328308908047996 |
| 39. | Karban, J.; Císařová, I.; Strašák, T.; Červenková Šťastná, L.; Sýkora, J. Org. Biomol. Chem. 2012, 10, 394–403. doi:10.1039/C1OB06336G |
| 50. | Karban, J.; Kroutil, J. Adv. Carbohydr. Chem. Biochem. 2006, 60, 27–101. doi:10.1016/S0065-2318(06)60003-6 |
| 57. | Kroutil, J.; Buděšínský, M. Carbohydr. Res. 2007, 342, 147–153. doi:10.1016/j.carres.2006.11.028 |
| 35. | Paulsen, H.; Stenzel, W. Chem. Ber. 1978, 111, 2348–2357. doi:10.1002/cber.19781110629 |
| 55. | Mori, Y.; Morishima, N. Chem. Pharm. Bull. 1991, 39, 1088–1090. doi:10.1248/cpb.39.1088 |
| 56. | Mori, Y.; Morishima, N. Bull. Chem. Soc. Jpn. 1994, 67, 236–241. doi:10.1246/bcsj.67.236 |
| 1. | Taylor, M. E.; Drickamer, K. Introduction to glycobiology, 3rd ed.; Oxford University Press: Oxford, United Kingdom, 2011. |
| 8. | Oberbillig, T.; Mersch, C.; Wagner, S.; Hoffmann-Röder, A. Chem. Commun. 2012, 48, 1487–1489. doi:10.1039/C1CC15139H |
| 9. | Hoffmann-Röder, A.; Kaiser, A.; Wagner, S.; Gaidzik, N.; Kowalczyk, D.; Westerlind, U.; Gerlitzki, B.; Schmitt, E.; Kunz, H. Angew. Chem., Int. Ed. 2010, 49, 8498–8503. doi:10.1002/anie.201003810 |
| 19. | Sharma, M.; Bernacki, R. J.; Paul, B.; Korytnyk, W. Carbohydr. Res. 1990, 198, 205–221. doi:10.1016/0008-6215(90)84293-4 |
| 29. | Berkin, A.; Szarek, W. A.; Kisilevsky, R. Carbohydr. Res. 2000, 326, 250–263. doi:10.1016/S0008-6215(00)00049-5 |
| 6. | Pouilly, S.; Bourgeaux, V.; Piller, F.; Piller, V. ACS Chem. Biol. 2012, 7, 753–760. doi:10.1021/cb200511t |
| 7. | Hartman, M. C. T.; Coward, J. K. J. Am. Chem. Soc. 2002, 124, 10036–10053. doi:10.1021/ja0127234 |
| 20. | Wasonga, G.; Tatara, Y.; Kakizaki, I.; Huang, X. J. Carbohydr. Chem. 2013, 32, 392–409. doi:10.1080/07328303.2013.815196 |
| 4. | Li, Y.; Zhou, Y.; Ma, Y.; Li, X. Carbohydr. Res. 2011, 346, 1714–1720. doi:10.1016/j.carres.2011.05.024 |
| 5. | Frantom, P. A.; Coward, J. K.; Blanchard, J. S. J. Am. Chem. Soc. 2010, 132, 6626–6627. doi:10.1021/ja101231a |
| 18. | Sharma, M.; Bernacki, R. J.; Hillman, M. J.; Korytnyk, W. Carbohydr. Res. 1993, 240, 85–93. doi:10.1016/0008-6215(93)84174-5 |
| 18. | Sharma, M.; Bernacki, R. J.; Hillman, M. J.; Korytnyk, W. Carbohydr. Res. 1993, 240, 85–93. doi:10.1016/0008-6215(93)84174-5 |
| 20. | Wasonga, G.; Tatara, Y.; Kakizaki, I.; Huang, X. J. Carbohydr. Chem. 2013, 32, 392–409. doi:10.1080/07328303.2013.815196 |
| 2. | Nishimura, S.-I.; Hato, M.; Hyugaji, S.; Feng, F.; Amano, M. Angew. Chem., Int. Ed. 2012, 51, 3386–3390. doi:10.1002/anie.201108742 |
| 3. | Van Wijk, X. M.; Lawrence, R.; Thijssen, V. L.; van den Broek, S. A.; Troost, R.; van Scherpenzeel, M.; Naidu, N.; Oosterhof, A.; Griffioen, A. W.; Lefeber, D. J.; van Delft, F. L.; van Kuppevelt, T. H. FASEB J. 2015, 29, 2993–3002. doi:10.1096/fj.14-264226 |
| 19. | Sharma, M.; Bernacki, R. J.; Paul, B.; Korytnyk, W. Carbohydr. Res. 1990, 198, 205–221. doi:10.1016/0008-6215(90)84293-4 |
| 2. | Nishimura, S.-I.; Hato, M.; Hyugaji, S.; Feng, F.; Amano, M. Angew. Chem., Int. Ed. 2012, 51, 3386–3390. doi:10.1002/anie.201108742 |
| 11. | Marathe, D. D.; Buffone, A., Jr.; Chandrasekaran, E. V.; Xue, J.; Locke, R. D.; Nasirikenari, M.; Lau, J. T. Y.; Matta, K. L.; Neelamegham, S. Blood 2010, 115, 1303–1312. doi:10.1182/blood-2009-07-231480 |
| 14. | Dimitroff, C. J.; Bernacki, R. J.; Sackstein, R. Blood 2003, 101, 602–610. doi:10.1182/blood-2002-06-1736 |
| 15. | Dimitroff, C. J.; Kupper, T. S.; Sackstein, R. J. Clin. Invest. 2003, 112, 1008–1018. doi:10.1172/JCI19220 |
| 16. | Cedeno-Laurent, F.; Opperman, M. J.; Barthel, S. R.; Hays, D.; Schatton, T.; Zhan, Q.; He, X.; Matta, K. L.; Supko, J. G.; Frank, M. H.; Murphy, G. F.; Dimitroff, C. J. J. Invest. Dermatol. 2012, 132, 410–420. doi:10.1038/jid.2011.335 |
| 20. | Wasonga, G.; Tatara, Y.; Kakizaki, I.; Huang, X. J. Carbohydr. Chem. 2013, 32, 392–409. doi:10.1080/07328303.2013.815196 |
| 12. |
Woynarowska, B.; Skrincosky, D. M.; Haag, A.; Sharma, M.; Matta, K.; Bernacki, R. J. J. Biol. Chem. 1994, 269, 22797–22803.
http://www.jbc.org/content/269/36/22797.abstract |
| 13. | Woynarowska, B.; Dimitroff, C. J.; Sharma, M.; Matta, K. L.; Bernacki, R. J. Glycoconjugate J. 1996, 13, 663–674. doi:10.1007/BF00731455 |
| 17. | Goon, S.; Bertozzi, C. R. J. Carbohydr. Chem. 2002, 21, 943–977. doi:10.1081/CAR-120016493 |
| 20. | Wasonga, G.; Tatara, Y.; Kakizaki, I.; Huang, X. J. Carbohydr. Chem. 2013, 32, 392–409. doi:10.1080/07328303.2013.815196 |
| 3. | Van Wijk, X. M.; Lawrence, R.; Thijssen, V. L.; van den Broek, S. A.; Troost, R.; van Scherpenzeel, M.; Naidu, N.; Oosterhof, A.; Griffioen, A. W.; Lefeber, D. J.; van Delft, F. L.; van Kuppevelt, T. H. FASEB J. 2015, 29, 2993–3002. doi:10.1096/fj.14-264226 |
| 58. | Zottola, M.; Rao, B. V.; Fraser-Reid, B. J. Chem. Soc., Chem. Commun. 1991, 969–970. doi:10.1039/c39910000969 |
| 2. | Nishimura, S.-I.; Hato, M.; Hyugaji, S.; Feng, F.; Amano, M. Angew. Chem., Int. Ed. 2012, 51, 3386–3390. doi:10.1002/anie.201108742 |
| 3. | Van Wijk, X. M.; Lawrence, R.; Thijssen, V. L.; van den Broek, S. A.; Troost, R.; van Scherpenzeel, M.; Naidu, N.; Oosterhof, A.; Griffioen, A. W.; Lefeber, D. J.; van Delft, F. L.; van Kuppevelt, T. H. FASEB J. 2015, 29, 2993–3002. doi:10.1096/fj.14-264226 |
| 10. | Barthel, S. R.; Antonopoulos, A.; Cedeno-Laurent, F.; Schaffer, L.; Hernandez, G.; Patil, S. A.; North, S. J.; Dell, A.; Matta, K. L.; Neelamegham, S.; Haslam, S. M.; Dimitroff, C. J. J. Biol. Chem. 2011, 286, 21717–21731. doi:10.1074/jbc.M110.194597 |
| 11. | Marathe, D. D.; Buffone, A., Jr.; Chandrasekaran, E. V.; Xue, J.; Locke, R. D.; Nasirikenari, M.; Lau, J. T. Y.; Matta, K. L.; Neelamegham, S. Blood 2010, 115, 1303–1312. doi:10.1182/blood-2009-07-231480 |
| 3. | Van Wijk, X. M.; Lawrence, R.; Thijssen, V. L.; van den Broek, S. A.; Troost, R.; van Scherpenzeel, M.; Naidu, N.; Oosterhof, A.; Griffioen, A. W.; Lefeber, D. J.; van Delft, F. L.; van Kuppevelt, T. H. FASEB J. 2015, 29, 2993–3002. doi:10.1096/fj.14-264226 |
| 59. | Wray, V. J. Chem. Soc., Perkin Trans. 2 1976, 1598–1605. doi:10.1039/p29760001598 |
| 21. | Arndt, S.; Hsieh-Wilson, L. C. Org. Lett. 2003, 5, 4179–4182. doi:10.1021/ol035606h |
| 22. | Černý, M.; Staněk, J., Jr. Adv. Carbohydr. Chem. Biochem. 1977, 34, 23–177. doi:10.1016/S0065-2318(08)60324-8 |
| 23. | Kulkarni, S. S.; Lee, J.-C.; Hung, S.-C. Curr. Org. Chem. 2004, 8, 475–509. doi:10.2174/1385272043485800 |
| 2. | Nishimura, S.-I.; Hato, M.; Hyugaji, S.; Feng, F.; Amano, M. Angew. Chem., Int. Ed. 2012, 51, 3386–3390. doi:10.1002/anie.201108742 |
| 20. | Wasonga, G.; Tatara, Y.; Kakizaki, I.; Huang, X. J. Carbohydr. Chem. 2013, 32, 392–409. doi:10.1080/07328303.2013.815196 |
| 18. | Sharma, M.; Bernacki, R. J.; Hillman, M. J.; Korytnyk, W. Carbohydr. Res. 1993, 240, 85–93. doi:10.1016/0008-6215(93)84174-5 |
| 18. | Sharma, M.; Bernacki, R. J.; Hillman, M. J.; Korytnyk, W. Carbohydr. Res. 1993, 240, 85–93. doi:10.1016/0008-6215(93)84174-5 |
| 60. | Rowell, R. M.; Feather, M. S. Carbohydr. Res. 1967, 4, 486–491. doi:10.1016/S0008-6215(00)81840-6 |
| 61. | Avalos, M.; Babiano, R.; Cintas, P.; Jiménez, J. L.; Palacios, J. C.; Valencia, C. Tetrahedron Lett. 1993, 34, 1359–1362. doi:10.1016/S0040-4039(00)91795-7 |
| 21. | Arndt, S.; Hsieh-Wilson, L. C. Org. Lett. 2003, 5, 4179–4182. doi:10.1021/ol035606h |
| 35. | Paulsen, H.; Stenzel, W. Chem. Ber. 1978, 111, 2348–2357. doi:10.1002/cber.19781110629 |
| 36. | Karban, J.; Buděšínský, M.; Černý, M.; Trnka, T. Collect. Czech. Chem. Commun. 2001, 66, 799–819. doi:10.1135/cccc20010799 |
| 24. | Karban, J.; Sýkora, J.; Kroutil, J.; Císařová, I.; Padělková, Z.; Buděšínský, M. J. Org. Chem. 2010, 75, 3443–3446. doi:10.1021/jo1000912 |
| 31. | Hesek, D.; Lee, M.; Zhang, W.; Noll, B. C.; Mobashery, S. J. Am. Chem. Soc. 2009, 131, 5187–5193. doi:10.1021/ja808498m |
| 32. | Katavic, P. L.; Yong, K. W. L.; Herring, J. N.; Deseo, M. A.; Blanchfield, J. T.; Ferro, V.; Garson, M. J. Tetrahedron 2013, 69, 8074–8079. doi:10.1016/j.tet.2013.06.079 |
| 33. | Ganguli, A. R. S.; Coward, J. K. Tetrahedron: Asymmetry 2005, 16, 411–424. doi:10.1016/j.tetasy.2004.11.053 |
| 19. | Sharma, M.; Bernacki, R. J.; Paul, B.; Korytnyk, W. Carbohydr. Res. 1990, 198, 205–221. doi:10.1016/0008-6215(90)84293-4 |
| 34. | Trnka, T.; Černý, M. Collect. Czech. Chem. Commun. 1971, 36, 2216–2225. doi:10.1135/cccc19712216 |
| 20. | Wasonga, G.; Tatara, Y.; Kakizaki, I.; Huang, X. J. Carbohydr. Chem. 2013, 32, 392–409. doi:10.1080/07328303.2013.815196 |
| 2. | Nishimura, S.-I.; Hato, M.; Hyugaji, S.; Feng, F.; Amano, M. Angew. Chem., Int. Ed. 2012, 51, 3386–3390. doi:10.1002/anie.201108742 |
| 20. | Wasonga, G.; Tatara, Y.; Kakizaki, I.; Huang, X. J. Carbohydr. Chem. 2013, 32, 392–409. doi:10.1080/07328303.2013.815196 |
| 29. | Berkin, A.; Szarek, W. A.; Kisilevsky, R. Carbohydr. Res. 2000, 326, 250–263. doi:10.1016/S0008-6215(00)00049-5 |
| 19. | Sharma, M.; Bernacki, R. J.; Paul, B.; Korytnyk, W. Carbohydr. Res. 1990, 198, 205–221. doi:10.1016/0008-6215(90)84293-4 |
| 30. | Karban, J.; Horník, Š.; Červenková Šťastná, L.; Sýkora, J. Synlett 2014, 25, 1253–1256. doi:10.1055/s-0033-1341187 |
| 19. | Sharma, M.; Bernacki, R. J.; Paul, B.; Korytnyk, W. Carbohydr. Res. 1990, 198, 205–221. doi:10.1016/0008-6215(90)84293-4 |
| 24. | Karban, J.; Sýkora, J.; Kroutil, J.; Císařová, I.; Padělková, Z.; Buděšínský, M. J. Org. Chem. 2010, 75, 3443–3446. doi:10.1021/jo1000912 |
| 19. | Sharma, M.; Bernacki, R. J.; Paul, B.; Korytnyk, W. Carbohydr. Res. 1990, 198, 205–221. doi:10.1016/0008-6215(90)84293-4 |
| 25. | Mtashobya, L.; Quiquempoix, L.; Linclau, B. J. Fluorine Chem. 2015, 171, 92–96. doi:10.1016/j.jfluchem.2014.08.023 |
| 26. | Kobayashi, K.; Kondo, T. Macromolecules 1997, 30, 6531–6535. doi:10.1021/ma970691s |
| 27. | Sarda, P.; Escribano, F. C.; Alves, R. J.; Olesker, A.; Lukacs, G. J. Carbohydr. Chem. 1989, 8, 115–123. doi:10.1080/07328308908047996 |
| 28. | Faghih, R.; Escribano, F. C.; Castillon, S.; Garcia, J.; Lukacs, G.; Olesker, A.; Thang, T. T. J. Org. Chem. 1986, 51, 4558–4564. doi:10.1021/jo00374a013 |
| 19. | Sharma, M.; Bernacki, R. J.; Paul, B.; Korytnyk, W. Carbohydr. Res. 1990, 198, 205–221. doi:10.1016/0008-6215(90)84293-4 |
© 2016 Horník et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)