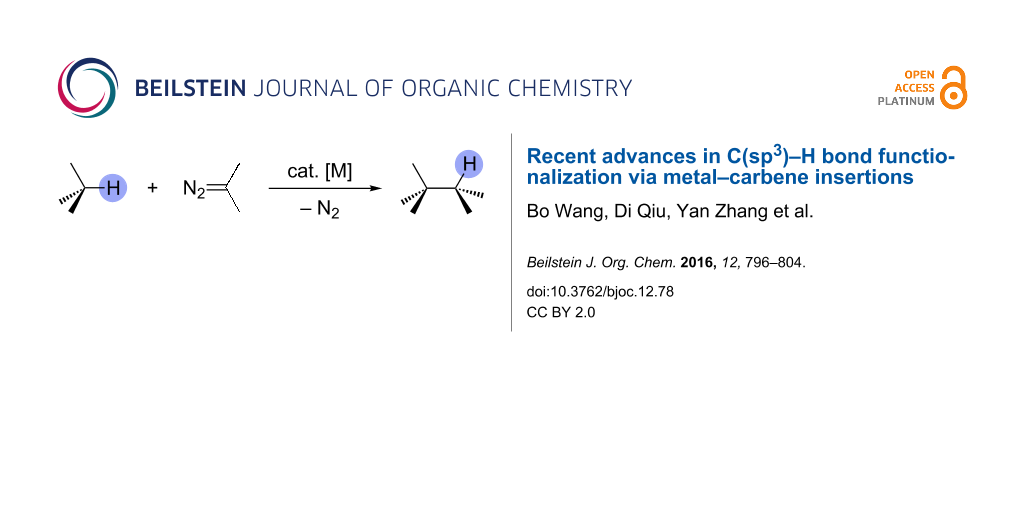
Abstract
The recent development of intermolecular C–H insertion in the application of C(sp3)–H bond functionalizations, especially for light alkanes, is reviewed. The challenging problem of regioselectivity in C–H bond insertions has been tackled by the use of sterically bulky metal catalysts, such as metal porphyrins and silver(I) complexes. In some cases, high regioselectivity and enantioselectivity have been achieved in the C–H bond insertion of small alkanes. This review highlights the most recent accomplishments in this field.

Graphical Abstract
Introduction
Direct functionalization of inactivated C–H bonds, especially C(sp3)–H bonds, have attracted significant attentions in recent years. The C(sp3)–H bond activation strategies based on radical reactions and transition metal catalysis have been explored, alongside the development of various directing groups for controlling the site-selectivity of the reaction. Regardless of the great efforts devoted to the field, the intermolecular C(sp3)–H bond activation of simple alkanes still remains a formidable challenge, obviously attributed to the inertness and ubiquitous nature of simple aliphatic C(sp3)–H bonds. In this context, catalytic metal–carbene C(sp3)–H bond insertion represents an alternative and unique approach for this purpose.
Metal–carbene insertion into a C(sp3)–H bond, well-recognized as one of the typical reactions of carbene species, have been studied extensively over the decades [1-8]. Mechanistically, the C(sp3)–H bond insertion reaction is considered to follow a concerted reaction pathway with a three-center two electron transition state (Scheme 1). Since late transition metals, typically Rh(II) complexes, are most commonly employed as the catalysts, the carbenic carbon of the metal–carbene species is positively charged in general, as shown by the resonance structure. Consequently, when the electron-deficient carbenic carbon approaches the C(sp3)–H bonds, the C–H bonds with high electron density will react preferentially [9]. However, the site-selectivity of C(sp3)–H bond insertion is also affected by steric factors. High regioselectivity of C(sp3)–H bond insertions has been observed in intramolecular reactions in most cases, in which the C(sp3)–H bond positioned 5 atoms away from the carbene center will normally react preferentially (1,5 C–H insertion). However, 1,3-, 1,4, and 1,6 C–H insertions are also possible, depending on the substrates and the catalysts. Although the site-selectivity of intramolecular metal–carbene C(sp3)–H bond insertion is affected by the combination of factors such as steric and electronic factors as well as catalysts, high site-selectivity is generally achievable, which makes this type of reaction a valuable tool for the construction of carbocycles from readily available starting materials [1-6].
![[1860-5397-12-78-i1]](/bjoc/content/inline/1860-5397-12-78-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Pathway for transition-metal-catalyzed carbene insertion into C(sp3)–H bonds.
Scheme 1: Pathway for transition-metal-catalyzed carbene insertion into C(sp3)–H bonds.
While most of the site-selective metal–carbene C(sp3)–H bond insertions are based on intramolecular reaction systems, or with relatively active C–H bonds, such as allylic, benzylic or the C–H bonds adjacent to the oxygen or nitrogen, the corresponding site-selective metal–carbene insertion into simple aliphatic C(sp3)–H bonds has also been challenged in intermolecular reaction systems and one has witnessed some exciting advances along this line. Thus, it would be an appropriate time to summarize the field in connection with direct C–H bond functionalization. Since catalytic metal–carbene C(sp3)–H bond insertions have been discussed in a series of excellent reviews [1-8], this short article will highlight the most recent developments in the field, with the emphasis on simple aliphatic C(sp3)–H bond insertions.
Review
Metal carbene C(sp3)–H bond insertions into relatively active C–H bonds
Compared to ordinary aliphatic C(sp3)–H bonds, the C(sp3)–H bonds located at allylic and benzylic sites and those at the α-position of oxygen or nitrogen, show high activity because of the stabilization of the partial positive charge developed in the transition state of the metal–carbene C–H bond insertion process. Such type of intramolecular metal–carbene C–H insertions shows high selectivities in many cases [10-20], and they have been successfully incorporated into the steps in natural product synthesis. Herein some selected recent examples are highlighted.
The C–H bond insertions at the α-positions of oxygen or nitrogen
Attributed to the stabilizing effect of oxygen and nitrogen toward the positive charge development at the neighboring positions, the metal–carbene C–H bond insertions at these positions are favored. Davies and co-workers have recently reported a highly site-selective and enantioselective C–H bond insertion of methyl ethers [21]. The use of 2,2,2-trichloroethyl aryldiazoacetates, in combination with sterically crowded chiral Rh(II) catalysts Rh2(R-BPCP)4, enhances the site-selectivity and the enantioselectivity of the reaction. Interestingly, the C–H bonds of a methyl group show high reactivity over the secondary C–H bonds, even the secondary benzylic C–H bonds (Scheme 2). Notably, for the site-selectivity of carbene insertion into primary, secondary and tertiary C–H bonds, the electronic and steric factors operate in the opposite directions. It is thus possible to tune or even revise the selectivity by judicious combination of reagents and catalysts.
![[1860-5397-12-78-i2]](/bjoc/content/inline/1860-5397-12-78-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Rh(II)-catalyzed site-selective and enantioselective C–H functionalization of methyl ether.
Scheme 2: Rh(II)-catalyzed site-selective and enantioselective C–H functionalization of methyl ether.
Rh(II)-catalyzed site-selective and enantioselective intramolecular carbene insertion into the C–H bond at the α-position of a tertiary amine have been previously established by Davies and co-workers [22-24]. Recently, this methodology has been used in the late-stage C–H functionalization of complex alkaloids and drug molecules (Scheme 3) [25].
![[1860-5397-12-78-i3]](/bjoc/content/inline/1860-5397-12-78-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Late-stage C–H functionalization with Rh(II)-catalyzed carbene C(sp3)–H insertion.
Scheme 3: Late-stage C–H functionalization with Rh(II)-catalyzed carbene C(sp3)–H insertion.
C–H bond insertions at the allylic and benzylic positions
Metal–carbene C–H insertion is also favored for allylic and benzylic sites. In 2014, Davies and co-workers reported an enantioselective C–H insertion catalyzed by chiral dirhodium catalysts. The reaction took place selectively at the primary C–H bond when chiral Rh(II) catalyst Rh2(R-BPCP)4 is employed. However, when chiral Rh(II) catalyst Rh2(R-DOSP)4 is used, secondary C–H bond insertion becomes predominant (Scheme 4) [26]. Subsequently, an extensive structure–selectivity relationship was carried out by Davies and Sigman [27]. The quantitative analysis of the substrate and the reagent shows that a non-bulky electron-rich carbene and a non-bulky catalyst prefer secondary or tertiary C–H bonds, while bulky electron-deficient carbene and bulky catalysts prefer primary C–H bonds (Scheme 5). Such quantitative understanding is very useful for the design of reagent/catalyst combination to achieve high selectivity of unactivated C(sp3)–H bond functionalization.
![[1860-5397-12-78-i4]](/bjoc/content/inline/1860-5397-12-78-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: The Rh(II)-catalyzed selective carbene insertion into benzylic C–H bonds.
Scheme 4: The Rh(II)-catalyzed selective carbene insertion into benzylic C–H bonds.
![[1860-5397-12-78-i5]](/bjoc/content/inline/1860-5397-12-78-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: The structure–selectivity relationship.
Scheme 5: The structure–selectivity relationship.
Metal carbene insertion into aliphatic C(sp3)–H bonds of light alkanes
Site-selective metal–carbene insertion into simple aliphatic C(sp3)–H bonds is generally more challenging simply because of the ubiquitous nature of C(sp3)–H bonds and the little difference between different C(sp3)–H bonds. The investigation in this field is focused on the development of novel transition metal catalysts through judicious combination of metal and ligands. In 1982, Callot and Metz reported Rh(III) porphyrins catalyzed intermolecular C–H insertion, which showed an extraordinary selectivity for 1° C–H bonds [28]. Other metal porphyrin complexes, such as Os [29], Fe, Cu, Ag [30], can also serve as catalysts for C–H insertions, although the C–H insertions with these catalysts are generally less efficient.
More recently, Che and co-workers developed a highly site-selective intermolecular insertion into primary C–H bonds catalyzed by robust and sterically encumbered [RhIII(2,4,6-Ph3tpp)Me]. Presumably attributed to the large steric hindrance of 2,4,6-Ph3tpp, the reaction for n-alkanes afforded a high 1°/2° ratio of up to 11.4:1 (Scheme 6) [31]. The results further demonstrate that steric bulkiness of the catalyst can override the electronic preference for secondary C(sp3)–H bond insertions.
![[1860-5397-12-78-i6]](/bjoc/content/inline/1860-5397-12-78-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Rh-porphyrin complexes for catalytic intermolecular C–H insertions.
Scheme 6: Rh-porphyrin complexes for catalytic intermolecular C–H insertions.
By employing chiral porphyrins D4-por*, the enantioselective intermolecular carbene insertion into C(sp3)–H bonds was achieved with moderate to high selectivities (Scheme 7). Besides, the reaction of diazo compounds with benzylic, allylic and alkane C(sp3)–H bonds afforded the insertion products in up to 80% yield and with up to 93% ee.
![[1860-5397-12-78-i7]](/bjoc/content/inline/1860-5397-12-78-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Asymmetric intermolecular C(sp3)–H insertion with chiral Rh-porphyrin catalyst.
Scheme 7: Asymmetric intermolecular C(sp3)–H insertion with chiral Rh-porphyrin catalyst.
Pérez and co-workers have explored different types of catalysts based on copper and silver in metal–carbene C(sp3)–H insertions. Tris(pyrazoly)borate complexes of copper (TpCu) [32-34] and silver (TpAg) [35-37] have been employed for C(sp3)–H insertion of alkanes (Figure 1). Compared to Rh(II) catalysts, these coinage metal-based catalysts are generally less efficient in carbene-transfer reactions, thus requiring a high catalyst loading in general. However, this drawback is compensated by much lower cost of these metals than rhodium.
![[1860-5397-12-78-1]](/bjoc/content/figures/1860-5397-12-78-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The structure of TpM catalysts.
Figure 1: The structure of TpM catalysts.
In 2008, Pérez, Díaz-Requejo and Etienne reported a perfluorinated F21-tris(pyrazoly)borate (F21-Tp) scorpionate ligand, which enhanced alkane C–H functionalization by carbene insertion with (F21-Tp)Cu and (F21-Tp)Ag catalysts (Figure 1) [38]. In particular, with silver catalyst remarkably low catalyst loading (ca. 0.5%), and high turnover number (up to 200), have been achieved when the reaction was carried out with hexane and EDA (Scheme 8).
![[1860-5397-12-78-i8]](/bjoc/content/inline/1860-5397-12-78-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Ag-Tpx-catalyzed intermolecular C–H insertion between EDA and alkanes.
Scheme 8: Ag-Tpx-catalyzed intermolecular C–H insertion between EDA and alkanes.
The high reactivity of silver-based catalysts encouraged further investigations on its applications in aliphatic C(sp3)–H insertions. The metal–carbene C–H bond insertion with methane, the ultimate simple alkane, thus becomes an attractive goal along this line. However, the major obstacle to achieve the methane C–H bond functionalization through metal–carbene C–H insertion is the low reactivity of the methane C–H bond as compared to most of the C–H bonds in organic compounds. This impedes the use of most organic solvents, while the gaseous nature of methane makes it impossible to form a homogenous reaction system without a solvent.
Pérez and co-workers addressed this problem by using supercritical carbon dioxide (scCO2) as the reaction medium. Methane can be dissolved in scCO2. Moreover, the electron-deficient nature of CO2 will prevent its reaction with the highly reactive metal–carbene species, which are electrophilic in nature. In 2011, Pérez, Etienne and Asensio achieved the TpxAg catalyzed C–H insertion of methane in supercritical CO2 (Scheme 9) [39]. The reaction between methane and EDA formed ethyl propionate in 19% yield (based on EDA) at 40 °C over 14 h in the presence of silver-perfluorinated catalysts (Fn-TpxAg). Other gaseous alkanes, including ethane, propane, butane and isobutene, have also been functionalized with the same catalytic system with EDA [40]. Notably, the site-selectivities of the C–H insertion of pentane in scCO2 media are essentially identical as compared to the corresponding reaction with pentane as the solvent.
![[1860-5397-12-78-i9]](/bjoc/content/inline/1860-5397-12-78-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Ag-Tpx-catalyzed C–H insertion of methane with EDA in scCO2.
Scheme 9: Ag-Tpx-catalyzed C–H insertion of methane with EDA in scCO2.
More recently, a new borate ligand, hydrotris((3,5-bis(trifluoromethyl)-4-bromo)pyrazol-1-yl)borate (Tp(CF3)2,Br), was introduced and similar C–H bond functionalization of methane was achieved by Tp(CF3)2,Br ML (M = Cu, Ag; L = MeCN or THF) (Figure 2) [41]. A detailed investigation on the TpML-type catalysts was also carried out [42]. Notably, the copper complex Tp(CF3)2,BrCu (NCMe) is soluble in methane/scCO2, thus forming a homogenous catalytic system.
![[1860-5397-12-78-2]](/bjoc/content/figures/1860-5397-12-78-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structure of TpM-type catalysts.
Figure 2: Structure of TpM-type catalysts.
With the new borate ligand, depending on the metal of the catalyst, different selectivities were observed when the reaction medium is changed from alkane to scCO2 (Scheme 10) [43]. When scCO2 was used as the solvent for the C–H insertion of hexane, a significant increase in the functionalization of the primary sites was observed in the presence of Tp(CF3)2,BrCu(MeCN); while hexane and scCO2 made little difference as compared to Tp(CF3)2,BrAg(thf) catalyzed reactions. These results may be related to the interaction of the ligand with carbon dioxide. A net electron density flux from the metal center to the ligand and to carbon dioxide was supported by experimental data and DFT studies. Such interaction increases the electrophilicity of the carbene moiety and thus lowers the activation energy for C–H bond insertion.
![[1860-5397-12-78-i10]](/bjoc/content/inline/1860-5397-12-78-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Comparison of site-selectivities of C–H insertion in different reaction media.
Scheme 10: Comparison of site-selectivities of C–H insertion in different reaction media.
Other ligands have also been explored for the metal–carbene C(sp3)–H insertion of simple alkanes. Mindiola and Caulton reported carbene C–H insertion catalyzed by a trinuclear cluster Ag3(μ2-3,5-(CF3)2PyrPy)3 (3,5-(CF3)2PyrPy = 3,5-bis(trifluoromethyl)-2,2’-pyridylpyrrolide) [44]. The C–H bond insertion of ethane, propane, butane and more heavier alkanes have been achieved except for methane (Scheme 11). The DFT studies suggested that formation of the silver carbene complex was the rate-determining step for alkane substrates such as ethane and propane. As for methane, the overall rate-determining step was carbene insertion into the C–H bond, attributed to the inertness of the methane C–H bond. As a result, the side reactions of the highly active metallocarbene intermediate override the C–H insertion.
![[1860-5397-12-78-i11]](/bjoc/content/inline/1860-5397-12-78-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: C(sp3)–H bond insertion catalyzed by trinuclear cluster Ag.
Scheme 11: C(sp3)–H bond insertion catalyzed by trinuclear cluster Ag.
Zinc catalysts, which were seldom used for metal–carbene C–H insertions, have been recently explored in the C–H insertion between EDA with alkanes [45]. A detailed computational study of the likely intermediate suggests that it is best described as a zinc-bound carbocation rather than a zinc carbene (Scheme 12). Notably, Zn is a cheap, earth-abundant 3d metal, thus making it attractive as catalyst for C–H bond functionalization.
![[1860-5397-12-78-i12]](/bjoc/content/inline/1860-5397-12-78-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Zn(II)-catalyzed C(sp3)–H bond insertion.
Scheme 12: Zn(II)-catalyzed C(sp3)–H bond insertion.
Conclusion
The intermolecular metal–carbene insertion represents a direct approach for the C–H bond functionalization of aliphatic C(sp3)–H bonds. While some progresses have been made in this area, significant challenge remains. From the selected examples summarized in this review it is obvious that the main issue is the balance of reactivity and selectivity. The factors governing the reactivity and site-selectivity of metal–carbene C–H insertion include: 1) the electrophilicity of the metal–carbene bond; 2) the nucleophilicity of the targeted C–H bond; 3) the steric bulk of the ligand around the metal; 4) the steric bulk of the substituents around the targeted C–H bond. While secondary and tertiary C(sp3)–H bonds are favored toward the electron-deficient carbenic carbon center in terms of electronic effects, they are unfavored in terms of steric hindrance. The primary C(sp3)–H bonds operate in the opposite way. Further studies in this area will be focused on the development of novel catalysts as well as the perceptive combination of catalyst/substrates in order to achieve highly selective C(sp3)–H bond insertions.
References
-
Davies, H. M. L.; Beckwith, R. E. J. Chem. Rev. 2003, 103, 2861–2904. doi:10.1021/cr0200217
Return to citation in text: [1] [2] [3] -
Doyle, M. P.; Duffy, R.; Ratnikov, M.; Zhou, L. Chem. Rev. 2010, 110, 704–724. doi:10.1021/cr900239n
Return to citation in text: [1] [2] [3] -
Taber, D. F.; Stiriba, S.-E. Chem. – Eur. J. 1998, 4, 990–992. doi:10.1002/(SICI)1521-3765(19980615)4:6<990::AID-CHEM990>3.0.CO;2-U
Return to citation in text: [1] [2] [3] -
Davies, H. M. L.; Denton, J. R. Chem. Soc. Rev. 2009, 38, 3061–3071. doi:10.1039/b901170f
Return to citation in text: [1] [2] [3] -
Egger, J.; Carreira, E. M. Nat. Prod. Rep. 2014, 31, 449–455. doi:10.1039/c3np70084d
Return to citation in text: [1] [2] [3] -
Lombard, F. J.; Coster, M. J. Org. Biomol. Chem. 2015, 13, 6419–6431. doi:10.1039/c5ob00311c
Return to citation in text: [1] [2] [3] -
Che, C.-M.; Lo, V. K.-Y.; Zhou, C.-Y.; Huang, J.-S. Chem. Soc. Rev. 2011, 40, 1950–1975. doi:10.1039/c0cs00142b
Return to citation in text: [1] [2] -
Caballero, A.; Díaz-Requejo, M. M.; Fructos, M. R.; Olmos, A.; Urbano, J.; Pérez, P. J. Dalton Trans. 2015, 44, 20295–20307. doi:10.1039/c5dt03450g
Return to citation in text: [1] [2] -
Wang, J.; Chen, B.; Bao, J. J. Org. Chem. 1998, 63, 1853–1862. doi:10.1021/jo971747j
Return to citation in text: [1] -
Suematsu, H.; Katsuki, T. J. Am. Chem. Soc. 2009, 131, 14218–14219. doi:10.1021/ja9065267
Return to citation in text: [1] -
Owens, C. P.; Varela-Álvarez, A.; Boyarskikh, V.; Musaev, D. G.; Davies, H. M. L.; Blakey, S. B. Chem. Sci. 2013, 4, 2590–2596. doi:10.1039/c3sc50886b
Return to citation in text: [1] -
Davies, H. M. L.; Jin, Q. Tetrahedron: Asymmetry 2003, 14, 941–949. doi:10.1016/s0957-4166(03)00154-x
Return to citation in text: [1] -
Bykowski, D.; Wu, K.-H.; Doyle, M. P. J. Am. Chem. Soc. 2006, 128, 16038–16039. doi:10.1021/ja066452e
Return to citation in text: [1] -
Hilt, G.; Galbiati, F. Org. Lett. 2006, 8, 2195–2198. doi:10.1021/ol060739z
Return to citation in text: [1] -
Nadeau, E.; Ventura, D. L.; Brekan, J. A.; Davies, H. M. L. J. Org. Chem. 2010, 75, 1927–1939. doi:10.1021/jo902644f
Return to citation in text: [1] -
Hansen, J. H.; Gregg, T. M.; Ovalles, S. R.; Lian, Y.; Autschbach, J.; Davies, H. M. L. J. Am. Chem. Soc. 2011, 133, 5076–5085. doi:10.1021/ja111408v
Return to citation in text: [1] -
Lian, Y.; Davies, H. M. L. J. Am. Chem. Soc. 2011, 133, 11940–11943. doi:10.1021/ja2051155
Return to citation in text: [1] -
Lian, Y.; Hardcastle, K. I.; Davies, H. M. L. Angew. Chem., Int. Ed. 2011, 50, 9370–9373. doi:10.1002/anie.201103568
Return to citation in text: [1] -
McMills, M. C.; Humes, R. J.; Pavlyuk, O. M. Tetrahedron Lett. 2012, 53, 849–851. doi:10.1016/j.tetlet.2011.12.019
Return to citation in text: [1] -
Wang, H.; Li, G.; Engle, K. M.; Yu, J.-Q.; Davies, H. M. L. J. Am. Chem. Soc. 2013, 135, 6774–6777. doi:10.1021/ja401731d
Return to citation in text: [1] -
Guptill, D. M.; Davies, H. M. L. J. Am. Chem. Soc. 2014, 136, 17718–17721. doi:10.1021/ja5107404
Return to citation in text: [1] -
Davies, H. M. L.; Venkataramani, C. Angew. Chem., Int. Ed. 2002, 41, 2197–2199. doi:10.1002/1521-3773(20020617)41:12<2197::AID-ANIE2197>3.0.CO;2-N
Return to citation in text: [1] -
Davies, H. M. L.; Jin, Q. Org. Lett. 2004, 6, 1769–1772. doi:10.1021/ol0495467
Return to citation in text: [1] -
Davies, H. M. L.; Ni, A. Chem. Commun. 2006, 3110–3112. doi:10.1039/B605047F
Return to citation in text: [1] -
He, J.; Hamann, L. G.; Davies, H. M. L.; Beckwith, R. E. J. Nat. Commun. 2015, 6, No. 5943. doi:10.1038/ncomms6943
Return to citation in text: [1] -
Qin, C.; Davies, H. M. L. J. Am. Chem. Soc. 2014, 136, 9792–9796. doi:10.1021/ja504797x
Return to citation in text: [1] -
Bess, E. N.; Guptill, D. M.; Davies, H. M. L.; Sigman, M. S. Chem. Sci. 2015, 6, 3057–3062. doi:10.1039/c5sc00357a
Return to citation in text: [1] -
Callot, H. J.; Metz, F. Tetrahedron Lett. 1982, 23, 4321–4324. doi:10.1016/s0040-4039(00)85590-2
Return to citation in text: [1] -
Li, Y.; Huang, J.-S.; Zhou, Z.-Y.; Che, C.-M. Chem. Commun. 2003, 1362–1363. doi:10.1039/b300441d
Return to citation in text: [1] -
Mbuvi, H. M.; Woo, L. K. Organometallics 2008, 27, 637–645. doi:10.1021/om7007502
Return to citation in text: [1] -
Thu, H.-Y.; Tong, G. S.-M.; Huang, J.-S.; Chan, S. L.-F.; Deng, Q.-H.; Che, C.-M. Angew. Chem., Int. Ed. 2008, 47, 9747–9751. doi:10.1002/anie.200803157
Return to citation in text: [1] -
Díaz-Requejo, M. M.; Belderrain, T. R.; Nicasio, M. C.; Trofimenko, S.; Pérez, P. J. J. Am. Chem. Soc. 2002, 124, 896–897. doi:10.1021/ja016798j
Return to citation in text: [1] -
Caballero, A.; Díaz-Requejo, M. M.; Belderrain, T. R.; Nicasio, M. C.; Trofimenko, S.; Pérez, P. J. J. Am. Chem. Soc. 2003, 125, 1446–1447. doi:10.1021/ja0291484
Return to citation in text: [1] -
Caballero, A.; Díaz-Requejo, M. M.; Belderrain, T. R.; Nicasio, M. C.; Trofimenko, S.; Pérez, P. J. Organometallics 2003, 22, 4145–4150. doi:10.1021/om034002e
Return to citation in text: [1] -
Dias, H. V. R.; Browning, R. G.; Richey, S. A.; Lovely, C. J. Organometallics 2004, 23, 1200–1202. doi:10.1021/om0499198
Return to citation in text: [1] -
Dias, H. V. R.; Browning, R. G.; Richey, S. A.; Lovely, C. J. Organometallics 2005, 24, 5784. doi:10.1021/om050832b
Return to citation in text: [1] -
Urbano, J.; Belderrain, T. R.; Nicasio, M. C.; Trofimenko, S.; Díaz-Requejo, M. M.; Pérez, P. J. Organometallics 2005, 24, 1528–1532. doi:10.1021/om050024k
Return to citation in text: [1] -
Despagnet-Ayoub, E.; Jacob, K.; Vendier, L.; Etienne, M.; Álvarez, E.; Caballero, A.; Díaz-Requejo, M. M.; Pérez, P. J. Organometallics 2008, 27, 4779–4787. doi:10.1021/om800531a
Return to citation in text: [1] -
Caballero, A.; Despagnet-Ayoub, E.; Díaz-Requejo, M. M.; Díaz-Rodríguez, A.; González-Núñez, M. E.; Mello, R.; Muñoz, B. K.; Ojo, W.-S.; Asensio, G.; Etienne, M.; Pérez, P. J. Science 2011, 332, 835–838. doi:10.1126/science.1204131
Return to citation in text: [1] -
Fuentes, M. A.; Olmos, A.; Muñoz, B. K.; Jacob, K.; González-Núñez, M. E.; Mello, R.; Asensio, G.; Caballero, A.; Etienne, M.; Pérez, P. J. Chem. – Eur. J. 2014, 20, 11013–11018. doi:10.1002/chem.201403217
Return to citation in text: [1] -
Gava, R.; Olmos, A.; Noverges, B.; Varea, T.; Álvarez, E.; Belderrain, T. R.; Caballero, A.; Asensio, G.; Pérez, P. J. ACS Catal. 2015, 5, 3726–3730. doi:10.1021/acscatal.5b00718
Return to citation in text: [1] -
Caballero, A.; Pérez, P. J. J. Organomet. Chem. 2015, 793, 108–113. doi:10.1016/j.jorganchem.2015.02.029
Return to citation in text: [1] -
Gava, R.; Olmos, A.; Noverges, B.; Varea, T.; Funes-Ardoiz, I.; Belderrain, T. R.; Caballero, A.; Maseras, F.; Asensio, G.; Pérez, P. J. ChemCatChem 2015, 7, 3254–3260. doi:10.1002/cctc.201500610
Return to citation in text: [1] -
Flores, J. A.; Komine, N.; Pal, K.; Pinter, B.; Pink, M.; Chen, C.-H.; Caulton, K. G.; Mindiola, D. J. ACS Catal. 2012, 2, 2066–2078. doi:10.1021/cs300256b
Return to citation in text: [1] -
Kulkarni, N. V.; Dash, C.; Jayaratna, N. B.; Ridlen, S. G.; Khani, S. K.; Das, A.; Kou, X.; Yousufuddin, M.; Cundari, T. R.; Dias, H. V. R. Inorg. Chem. 2015, 54, 11043–11045. doi:10.1021/acs.inorgchem.5b02134
Return to citation in text: [1]
| 40. | Fuentes, M. A.; Olmos, A.; Muñoz, B. K.; Jacob, K.; González-Núñez, M. E.; Mello, R.; Asensio, G.; Caballero, A.; Etienne, M.; Pérez, P. J. Chem. – Eur. J. 2014, 20, 11013–11018. doi:10.1002/chem.201403217 |
| 38. | Despagnet-Ayoub, E.; Jacob, K.; Vendier, L.; Etienne, M.; Álvarez, E.; Caballero, A.; Díaz-Requejo, M. M.; Pérez, P. J. Organometallics 2008, 27, 4779–4787. doi:10.1021/om800531a |
| 39. | Caballero, A.; Despagnet-Ayoub, E.; Díaz-Requejo, M. M.; Díaz-Rodríguez, A.; González-Núñez, M. E.; Mello, R.; Muñoz, B. K.; Ojo, W.-S.; Asensio, G.; Etienne, M.; Pérez, P. J. Science 2011, 332, 835–838. doi:10.1126/science.1204131 |
| 1. | Davies, H. M. L.; Beckwith, R. E. J. Chem. Rev. 2003, 103, 2861–2904. doi:10.1021/cr0200217 |
| 2. | Doyle, M. P.; Duffy, R.; Ratnikov, M.; Zhou, L. Chem. Rev. 2010, 110, 704–724. doi:10.1021/cr900239n |
| 3. | Taber, D. F.; Stiriba, S.-E. Chem. – Eur. J. 1998, 4, 990–992. doi:10.1002/(SICI)1521-3765(19980615)4:6<990::AID-CHEM990>3.0.CO;2-U |
| 4. | Davies, H. M. L.; Denton, J. R. Chem. Soc. Rev. 2009, 38, 3061–3071. doi:10.1039/b901170f |
| 5. | Egger, J.; Carreira, E. M. Nat. Prod. Rep. 2014, 31, 449–455. doi:10.1039/c3np70084d |
| 6. | Lombard, F. J.; Coster, M. J. Org. Biomol. Chem. 2015, 13, 6419–6431. doi:10.1039/c5ob00311c |
| 7. | Che, C.-M.; Lo, V. K.-Y.; Zhou, C.-Y.; Huang, J.-S. Chem. Soc. Rev. 2011, 40, 1950–1975. doi:10.1039/c0cs00142b |
| 8. | Caballero, A.; Díaz-Requejo, M. M.; Fructos, M. R.; Olmos, A.; Urbano, J.; Pérez, P. J. Dalton Trans. 2015, 44, 20295–20307. doi:10.1039/c5dt03450g |
| 10. | Suematsu, H.; Katsuki, T. J. Am. Chem. Soc. 2009, 131, 14218–14219. doi:10.1021/ja9065267 |
| 11. | Owens, C. P.; Varela-Álvarez, A.; Boyarskikh, V.; Musaev, D. G.; Davies, H. M. L.; Blakey, S. B. Chem. Sci. 2013, 4, 2590–2596. doi:10.1039/c3sc50886b |
| 12. | Davies, H. M. L.; Jin, Q. Tetrahedron: Asymmetry 2003, 14, 941–949. doi:10.1016/s0957-4166(03)00154-x |
| 13. | Bykowski, D.; Wu, K.-H.; Doyle, M. P. J. Am. Chem. Soc. 2006, 128, 16038–16039. doi:10.1021/ja066452e |
| 14. | Hilt, G.; Galbiati, F. Org. Lett. 2006, 8, 2195–2198. doi:10.1021/ol060739z |
| 15. | Nadeau, E.; Ventura, D. L.; Brekan, J. A.; Davies, H. M. L. J. Org. Chem. 2010, 75, 1927–1939. doi:10.1021/jo902644f |
| 16. | Hansen, J. H.; Gregg, T. M.; Ovalles, S. R.; Lian, Y.; Autschbach, J.; Davies, H. M. L. J. Am. Chem. Soc. 2011, 133, 5076–5085. doi:10.1021/ja111408v |
| 17. | Lian, Y.; Davies, H. M. L. J. Am. Chem. Soc. 2011, 133, 11940–11943. doi:10.1021/ja2051155 |
| 18. | Lian, Y.; Hardcastle, K. I.; Davies, H. M. L. Angew. Chem., Int. Ed. 2011, 50, 9370–9373. doi:10.1002/anie.201103568 |
| 19. | McMills, M. C.; Humes, R. J.; Pavlyuk, O. M. Tetrahedron Lett. 2012, 53, 849–851. doi:10.1016/j.tetlet.2011.12.019 |
| 20. | Wang, H.; Li, G.; Engle, K. M.; Yu, J.-Q.; Davies, H. M. L. J. Am. Chem. Soc. 2013, 135, 6774–6777. doi:10.1021/ja401731d |
| 32. | Díaz-Requejo, M. M.; Belderrain, T. R.; Nicasio, M. C.; Trofimenko, S.; Pérez, P. J. J. Am. Chem. Soc. 2002, 124, 896–897. doi:10.1021/ja016798j |
| 33. | Caballero, A.; Díaz-Requejo, M. M.; Belderrain, T. R.; Nicasio, M. C.; Trofimenko, S.; Pérez, P. J. J. Am. Chem. Soc. 2003, 125, 1446–1447. doi:10.1021/ja0291484 |
| 34. | Caballero, A.; Díaz-Requejo, M. M.; Belderrain, T. R.; Nicasio, M. C.; Trofimenko, S.; Pérez, P. J. Organometallics 2003, 22, 4145–4150. doi:10.1021/om034002e |
| 1. | Davies, H. M. L.; Beckwith, R. E. J. Chem. Rev. 2003, 103, 2861–2904. doi:10.1021/cr0200217 |
| 2. | Doyle, M. P.; Duffy, R.; Ratnikov, M.; Zhou, L. Chem. Rev. 2010, 110, 704–724. doi:10.1021/cr900239n |
| 3. | Taber, D. F.; Stiriba, S.-E. Chem. – Eur. J. 1998, 4, 990–992. doi:10.1002/(SICI)1521-3765(19980615)4:6<990::AID-CHEM990>3.0.CO;2-U |
| 4. | Davies, H. M. L.; Denton, J. R. Chem. Soc. Rev. 2009, 38, 3061–3071. doi:10.1039/b901170f |
| 5. | Egger, J.; Carreira, E. M. Nat. Prod. Rep. 2014, 31, 449–455. doi:10.1039/c3np70084d |
| 6. | Lombard, F. J.; Coster, M. J. Org. Biomol. Chem. 2015, 13, 6419–6431. doi:10.1039/c5ob00311c |
| 7. | Che, C.-M.; Lo, V. K.-Y.; Zhou, C.-Y.; Huang, J.-S. Chem. Soc. Rev. 2011, 40, 1950–1975. doi:10.1039/c0cs00142b |
| 8. | Caballero, A.; Díaz-Requejo, M. M.; Fructos, M. R.; Olmos, A.; Urbano, J.; Pérez, P. J. Dalton Trans. 2015, 44, 20295–20307. doi:10.1039/c5dt03450g |
| 35. | Dias, H. V. R.; Browning, R. G.; Richey, S. A.; Lovely, C. J. Organometallics 2004, 23, 1200–1202. doi:10.1021/om0499198 |
| 36. | Dias, H. V. R.; Browning, R. G.; Richey, S. A.; Lovely, C. J. Organometallics 2005, 24, 5784. doi:10.1021/om050832b |
| 37. | Urbano, J.; Belderrain, T. R.; Nicasio, M. C.; Trofimenko, S.; Díaz-Requejo, M. M.; Pérez, P. J. Organometallics 2005, 24, 1528–1532. doi:10.1021/om050024k |
| 1. | Davies, H. M. L.; Beckwith, R. E. J. Chem. Rev. 2003, 103, 2861–2904. doi:10.1021/cr0200217 |
| 2. | Doyle, M. P.; Duffy, R.; Ratnikov, M.; Zhou, L. Chem. Rev. 2010, 110, 704–724. doi:10.1021/cr900239n |
| 3. | Taber, D. F.; Stiriba, S.-E. Chem. – Eur. J. 1998, 4, 990–992. doi:10.1002/(SICI)1521-3765(19980615)4:6<990::AID-CHEM990>3.0.CO;2-U |
| 4. | Davies, H. M. L.; Denton, J. R. Chem. Soc. Rev. 2009, 38, 3061–3071. doi:10.1039/b901170f |
| 5. | Egger, J.; Carreira, E. M. Nat. Prod. Rep. 2014, 31, 449–455. doi:10.1039/c3np70084d |
| 6. | Lombard, F. J.; Coster, M. J. Org. Biomol. Chem. 2015, 13, 6419–6431. doi:10.1039/c5ob00311c |
| 30. | Mbuvi, H. M.; Woo, L. K. Organometallics 2008, 27, 637–645. doi:10.1021/om7007502 |
| 45. | Kulkarni, N. V.; Dash, C.; Jayaratna, N. B.; Ridlen, S. G.; Khani, S. K.; Das, A.; Kou, X.; Yousufuddin, M.; Cundari, T. R.; Dias, H. V. R. Inorg. Chem. 2015, 54, 11043–11045. doi:10.1021/acs.inorgchem.5b02134 |
| 9. | Wang, J.; Chen, B.; Bao, J. J. Org. Chem. 1998, 63, 1853–1862. doi:10.1021/jo971747j |
| 31. | Thu, H.-Y.; Tong, G. S.-M.; Huang, J.-S.; Chan, S. L.-F.; Deng, Q.-H.; Che, C.-M. Angew. Chem., Int. Ed. 2008, 47, 9747–9751. doi:10.1002/anie.200803157 |
| 26. | Qin, C.; Davies, H. M. L. J. Am. Chem. Soc. 2014, 136, 9792–9796. doi:10.1021/ja504797x |
| 28. | Callot, H. J.; Metz, F. Tetrahedron Lett. 1982, 23, 4321–4324. doi:10.1016/s0040-4039(00)85590-2 |
| 43. | Gava, R.; Olmos, A.; Noverges, B.; Varea, T.; Funes-Ardoiz, I.; Belderrain, T. R.; Caballero, A.; Maseras, F.; Asensio, G.; Pérez, P. J. ChemCatChem 2015, 7, 3254–3260. doi:10.1002/cctc.201500610 |
| 25. | He, J.; Hamann, L. G.; Davies, H. M. L.; Beckwith, R. E. J. Nat. Commun. 2015, 6, No. 5943. doi:10.1038/ncomms6943 |
| 29. | Li, Y.; Huang, J.-S.; Zhou, Z.-Y.; Che, C.-M. Chem. Commun. 2003, 1362–1363. doi:10.1039/b300441d |
| 44. | Flores, J. A.; Komine, N.; Pal, K.; Pinter, B.; Pink, M.; Chen, C.-H.; Caulton, K. G.; Mindiola, D. J. ACS Catal. 2012, 2, 2066–2078. doi:10.1021/cs300256b |
| 22. | Davies, H. M. L.; Venkataramani, C. Angew. Chem., Int. Ed. 2002, 41, 2197–2199. doi:10.1002/1521-3773(20020617)41:12<2197::AID-ANIE2197>3.0.CO;2-N |
| 23. | Davies, H. M. L.; Jin, Q. Org. Lett. 2004, 6, 1769–1772. doi:10.1021/ol0495467 |
| 24. | Davies, H. M. L.; Ni, A. Chem. Commun. 2006, 3110–3112. doi:10.1039/B605047F |
| 41. | Gava, R.; Olmos, A.; Noverges, B.; Varea, T.; Álvarez, E.; Belderrain, T. R.; Caballero, A.; Asensio, G.; Pérez, P. J. ACS Catal. 2015, 5, 3726–3730. doi:10.1021/acscatal.5b00718 |
| 21. | Guptill, D. M.; Davies, H. M. L. J. Am. Chem. Soc. 2014, 136, 17718–17721. doi:10.1021/ja5107404 |
| 27. | Bess, E. N.; Guptill, D. M.; Davies, H. M. L.; Sigman, M. S. Chem. Sci. 2015, 6, 3057–3062. doi:10.1039/c5sc00357a |
| 42. | Caballero, A.; Pérez, P. J. J. Organomet. Chem. 2015, 793, 108–113. doi:10.1016/j.jorganchem.2015.02.029 |
© 2016 Wang et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)