Abstract
A one-pot transformation, which involves the reaction of ketones with aldehydes in the presence of metal halides to furnish tetrahydro-2H-pyran-2,4-diols in a highly diastereoselective manner, is investigated thoroughly by experiments and computations. The reaction was also successfully implemented on a flow micro reactor system.

Graphical Abstract
Introduction
Since its discovery in the late nineteenth century the aldol reaction has become one of the most powerful tools in the field of carbon–carbon bond formation [1-5]. It is widely used in the formation of many natural products [6-11], stereoselective syntheses [12-16], and tandem reactions [17-19]. While the latter processes usually comprise only one aldol reaction, tandem reaction sequences containing two consecutive aldol steps are mostly limited to the trimerization of enolates [20-22].
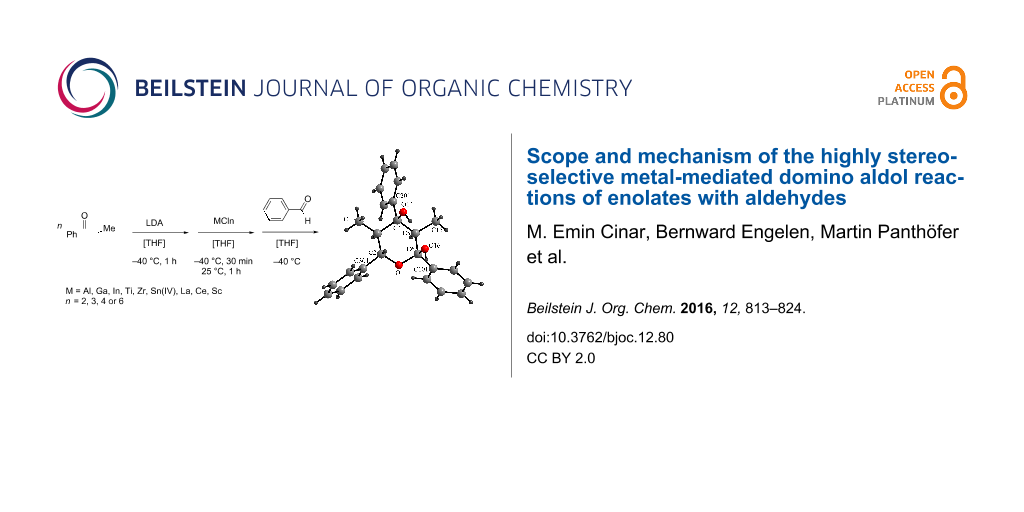
Metal enolates (Ti [23], Zr [24], Si [25], and Sn [26]) and boron enolates [27] have adopted a considerable significance because of their high potential to control the stereochemical outcome of the bond formation [28-30]. However, the other group III metal enolates have been almost completely omitted over the years [31]. We have already reported a domino aldol–aldol–hemiacetal process that furnishes racemic tetrahydro-2H-pyran-2,4-diols in a highly stereoselective manner (Scheme 1) [30-37].
![[1860-5397-12-80-i1]](/bjoc/content/inline/1860-5397-12-80-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of racemic tetrahydro-2H-pyran-2,4-diols rac-5 from enolates 2 and aldehydes 3.
Scheme 1: Synthesis of racemic tetrahydro-2H-pyran-2,4-diols rac-5 from enolates 2 and aldehydes 3.
In this paper, the domino aldol–aldol–hemiacetal reaction involving several metals (Al, Ga, In, Ti, Zr, Sn), and various aldehydes and ketones is studied experimentally and computationally.
Results
Metal effect
The experiments were performed to screen suitable metal fragments for their ability to promote the domino aldol reaction by studying the reaction between propiophenone (1a) and benzaldehyde (3a: Ar = Ph) (Scheme 2). The enolate was generated from propiophenone by deprotonation with lithium diisopropylamide (LDA) at −40 °C in tetrahydrofuran (THF) and was subsequently reacted with 0.33 equivalents of MCl3 or 0.25 equivalents of MCl4, respectively. The resulting metal enolate was then treated with a stoichiometric amount of benzaldehyde (3a) and stirred for 2 h at 0 °C, room temperature or 67 °C. The hemiacetal 5a was obtained in varying yields along with some amount of the monoaldol 6a [38,39] (obtained as a mixture of two diastereomers; syn/anti ≈ 1:1) and condensation product 6ac (Scheme 2, Table 1).
![[1860-5397-12-80-i2]](/bjoc/content/inline/1860-5397-12-80-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of rac-5a–j and monoaldol products 6a–i and 6ac–ic as obtained from propiophenone (1a) in the presence of metal chloride and substituted arylaldehydes.
Scheme 2: Synthesis of rac-5a–j and monoaldol products 6a–i and 6ac–ic as obtained from propiophenone (1a) in...
Table 1: Effect of metals on the domino aldol reaction of 1a and 3a at different temperatures (reaction time: 2 h) on the yield of 5a and 6a.
| Entry | Metal | Yield of 5a and 6a at various temperatures | |||||
|---|---|---|---|---|---|---|---|
| at 0 °C | at 25 °C | at 67 °C | |||||
| 5a (%) | 6a (%) | 5a (%) | 6a (%) | 5a (%) | 6a (%) | ||
| 1 | Al | 18 | 19 | 64a | 8 | 8 | − |
| 2 | Ga | 17 (+20)b | 22 | 70 | 19 | 67 | − |
| 3 | In | 76 | 3 | 85a | 13 | 60 | 2 |
| 4 | Tib | 19 | 26 | 38 | 14 | 50a | 5 |
| 5 | Zr | 61 | 10 | 76c | 23 | 21 | − |
| 6 | Sn(IV) | 7 | 20 | 36 (+29)b,c | 22 | 17 | 5 |
Aside of the ions mentioned in Table 1 the metal-mediated domino aldol reaction was also probed with LaCl3, La(OTf)3, CeCl3, Sc(OTf)3, BF3 and SnCl2 resulting in failure. While SnCl2 afforded 6a in 90% yield, LaCl3, La(OTf)3, CeCl3, and Sc(OTf)3 furnished 6a in 25, 52, 10 and 55% yield, respectively. In almost all cases the mono aldolate 6a was the main product at lower temperatures, e.g., at 0 °C, along with the domino aldol product 5a obtained in the range of 7–26% yield.
Higher yields of 5a were obtained at 0 °C in the presence of In and Zr. Interestingly, in the case of gallium at 0 °C and Sn(IV) at 25 °C a diastereomer of 5a also formed in 20 and 29% yields, respectively. Generally, with nearly all metals, the yield of 5a increased dramatically when the temperature was raised to 25 °C, but dropped at higher temperatures. The decreased yield at 67 °C may emerge from the irreversible formation of the aldol condensation product 6ac, which is obtained in 73% yield in presence of AlCl3, on expense of 5a.
The X-ray structure of racemic 5a (from ethanol) could not be solved due to the presence of a solid racemate in the orthorhombic space group P212121 (no. 19). Three axial hydroxy groups, whose probability of allocation at C2 and C6 is 0.5, respectively, appear to be attached to the pyran ring. This can easily be explained by the superposition of two enantiomers which are statistically and isoconformationally incorporated in the crystal lattice. Separation of two enantiomers was achieved by using chiral column chromatography (Chiralpak AD, Daicel) followed by recrystallization from H2O/MeOH (1:4) providing the appropriate crystal for X-ray analysis. Accordingly, the enantiopure crystal of 5a was unambiguously assigned to a tetrahydro-2H-pyran-2,4-diol structure with all phenyl and methyl groups occupying equatorial positions while the hydroxy groups are placed in axial positions with an allocation probability of 1.0 each (Figure 1). Within the crystal lattice the molecules arrange in a chain along the a-axis, so that each molecule is twisted by 180° against each other. Additionally, there are alternating inter- and intramolecular hydrogen bonds between the hydroxy groups.
![[1860-5397-12-80-1]](/bjoc/content/figures/1860-5397-12-80-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Crystal structure of enantiopure 5a [40].
Figure 1: Crystal structure of enantiopure 5a [40].
Mechanistic aspects
To shed light on the mechanism, the metal to enolate ratio was varied (Table 2), while keeping the optimum temperature for each metal as determined in the previous experiments. The reaction was already successful with two equivalents of enolate per metal fragment. However, higher yields were obtained at higher loadings. For example, zirconium worked best with three enolate units and tin with four. Surprisingly, an excess of an enolate had different effects on the reactions depending on the metals. While the yield was decreased with zirconium, it increased both with aluminum and indium. In the latter case, >99% yield was obtained.
Table 2: Dependence on the stoichiometric amount of propiophenone (1a) enolate with regard to the metal (reaction time: 2 h, reaction temperature: 25 °C, 5a/6a in %).
| Entry | Metal chloride | Amount of propiophenone (1a) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 2 equiv | 3 equiv | 4 equiv | 6 equiv | ||||||
| 5a | 6a | 5a | 6a | 5a | 6a | 5a | 6a | ||
| 1 | ZrCl4 | 33 | 48 | 76 | 23 | 68 | 22 | – | – |
| 2 | SnCl4 | 20 | 45 | 14 | 35 | 36 | 22 | – | – |
| 3 | AlCl3 | – | – | 66 | 8 | – | – | 66 | 34 |
| 4 | InCl3 | – | – | 85 | 13 | – | – | >99 | – |
In the same manner the influence of the amount of aldehyde was examined. As can be seen from the data in Table 3 the ratio of 5a to 6a decreases dramatically by increasing the amount of aldehyde two-fold, owing to the formation of higher amounts of the monoaldol product in presence of excess aldehyde (Scheme 2).
Table 3: Stoichiometry dependence on the amount of aldehyde (reaction time: 2 h, reaction temperature: 25 °C, 5a/6a in %).
| Entry | Metal | In presence of benzaldehyde (3a) in | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 equiv | 1.5 equiv | 2 equiv | 4 equiv | ||||||
| 5a | 6a | 5a | 6a | 5a | 6a | 5a | 6a | ||
| 1 | Zr | 65 | 7.0 | 40 | 16 | 19 | 31 | 5 | 60 |
| 2 | Al | 66 | 8 | 42 | 39 | 30 | 40 | – | – |
| 3 | In | 78 | 13 | 53 | 30 | 34 | 42 | – | – |
In order to check whether this outcome is the result of thermodynamic control, a second aldehyde was added to the reaction mixture after 2 h. The larger the amount of aldehyde in the reaction, the higher is the yield of monoaldol product 6a, which supports a thermodynamically controlled equilibrium as further confirmed by the following observations: (1) with gallium(III) two diastereomeric tetrahydro-2H-pyran-2,4-diols are formed at 0 °C and only one (i.e., 5a) at elevated temperature; (2) at higher temperature, the product yield of 5a is gradually reduced on account of new 6ac (only in the case of AlCl3), which is expected to be formed irreversibly from the metal-bound monoaldolate; (3) when excess benzaldehyde (3a) was added to the reaction with AlCl3, the yield of domino product 5a gradually decreased from, for example, 42% to 30% with 1.5 and 2.0 equivalents of benzaldehyde, respectively.
Likewise, the amount of metal chloride influences the yield of 5a. The higher the amount of metal chloride the less likely is the molecular preorganization, which is necessary for the reaction. The reactions carried out with 1.0, 0.75 and 0.5 equivalents of zirconium provided 5a in 76, 42 and 14% yield, respectively. Reducing the amount to 0.25 equivalents of zirconium furnished only 3% yield.
Even the concentration influences the yield of 5a. The optimum concentration is 375 mM, in which the reaction afforded 76% yield of 5a. In case of 750 mM, the yield decreases drastically to 40%. However, lower concentrations such as 250 mM and 187 mM do not have such an obvious influence and furnish the expected product in 61 and 59% yields, respectively.
A time dependency study clearly showed that the best yields were achieved after 2 hours and longer reaction times did not lead to improved yields (Table 4).
Table 4: Time dependency of domino aldol reactions in the presence of various metal chlorides (reaction temperature: 25 °C, 5a/6a in %).
| Entry | Time | AlCl3 | GaCl3 | InCl3 | ZrCl4 | SnCl4 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 5a | 6a | 5a | 6a | 5a | 6a | 5a | 6a | 5a | 6a | ||
| 1 | 10 min | 34 | 14 | 15 | 70 | 56 | 9 | 23 | 26 | 15 (20)a | 40 |
| 2 | 30 min | 39 | 11 | 22 | 61 | 60 | 10 | 21 | 20 | 21 (19)a | 18 |
| 3 | 60 min | 48 | 10 | 42 | 40 | 68 | 10 | 43 | 16 | 34 (29)a | 17 |
| 4 | 120 min | 66 | 8 | 70 | 19 | 87 | - | 68 | 22 | 36 (29)a | 22 |
| 5 | 1 day | – | – | − | – | 73 | 3 | 67 | 6 | – | – |
| 6 | 5 days | – | – | – | – | 75 | 5 | 64 | 5 | – | – |
aSecond diastereomer.
Variation of enolate and aromatic aldehyde
Subsequently, various aldehydes were tested in the domino aldol reaction with propiophenone enolate in combination with different metals (Table 5). All aromatic aldehydes, even those containing strongly coordinating substituents such as the dimethylamino group, are accepted in this transformation. With anthracene-9-carbaldehyde (3f), however, the yields drastically decreased, most likely due to steric hindrance. Although benzaldehyde (3a) was not used, product 5a appeared in the reaction of anthracene-9-carbaldehyde (3f) with Al and Zr metals, a finding that requires an explanation (vide infra). The NMR investigations suggest the same relative configuration of 5b−j as in 5a since coupling constants, the shift of the 2-CH3 group and the coupling constant J(4-H, 5-H) agreed.
The facile formation of domino products from aromatic aldehydes proposed to use this reaction also with aromatic dialdehydes (3j−l). As anticipated the reaction proceeded smoothly with terephthalaldehyde (3j) giving rise to product 5j (see Scheme 2 and Table 5), while isophthalaldehyde (3k) provided a mixture of isomers, which were not separable. The steric congestion in o-phthalaldehyde (3l) precluded the formation of the domino-aldol product. However, it is known that o-phthalaldehyde (3l) provides the corresponding aldol product in the presence of base [41].
Table 5: Reactions of various aldehydes with propiophenone metal enolate (reaction time: 2 h, reaction temperature: 25 °C).
| Entry | Product | Aldehyde | Yield of 5 [%] | |||
|---|---|---|---|---|---|---|
| AlCl3 | InCl3 | SnCl4 | ZrCl4 | |||
| 1 | 5b |
![[Graphic 1]](/bjoc/content/inline/1860-5397-12-80-i9.svg?max-width=637&scale=1.0)
3b |
14 | 47 | 34 | 40 |
| 2 | 5c |
![[Graphic 2]](/bjoc/content/inline/1860-5397-12-80-i10.svg?max-width=637&scale=1.0)
3c |
44 | 94 | 36 | 60a |
| 3 | 5d |
![[Graphic 3]](/bjoc/content/inline/1860-5397-12-80-i11.svg?max-width=637&scale=1.0)
3d |
17 | 72 | – | 45 |
| 4 | 5e |
![[Graphic 4]](/bjoc/content/inline/1860-5397-12-80-i12.svg?max-width=637&scale=1.0)
3e |
– | 50 | 28a | 45 |
| 5 | 5f |
![[Graphic 5]](/bjoc/content/inline/1860-5397-12-80-i13.svg?max-width=637&scale=1.0)
3f |
13b | 55 | – | 29b |
| 6 | 5g |
![[Graphic 6]](/bjoc/content/inline/1860-5397-12-80-i14.svg?max-width=637&scale=1.0)
3g |
– | 62 | – | 66 |
| 7 | 5h |
![[Graphic 7]](/bjoc/content/inline/1860-5397-12-80-i15.svg?max-width=637&scale=1.0)
3h |
– | 50 | – | 51 |
| 8 | 5i |
![[Graphic 8]](/bjoc/content/inline/1860-5397-12-80-i16.svg?max-width=637&scale=1.0)
3i |
–c | 53 | –c | –c |
| 9 | 5jd |
![[Graphic 9]](/bjoc/content/inline/1860-5397-12-80-i17.svg?max-width=637&scale=1.0)
3j |
– | 35 | – | 30 |
| 10 | 5k |
![[Graphic 10]](/bjoc/content/inline/1860-5397-12-80-i18.svg?max-width=637&scale=1.0)
3k |
– | –e | – | –e |
| 11 | 5l |
![[Graphic 11]](/bjoc/content/inline/1860-5397-12-80-i19.svg?max-width=637&scale=1.0)
3l |
– | –f | – | –f |
aRef. [37]. bAdditionally 10% (Al) and 20% (Zr) of 5a are formed. cThe reaction was not carried out. d1,4-Bis-(2,4-dimethyl-3,5-diphenyl-3,5-dihydroxytetrahydropyranyl)-benzene. eInseparable mixture. fNo reaction.
The variability in the ketone moiety proved to be rather restricted (Scheme 3). While in the case of propiophenone (1a) and butyrophenone (1f) moderate to good yields (≥50%) were obtained, the reaction with acetophenone (1e) did not furnish any domino aldol product at all. The only acyclic aliphatic ketone that led to the formation of the domino aldol product was pinacolone (1d), which gave the tetrahydro-2H-pyran-2,4-diol 7d in 20% yield (Scheme 3, Table 6). The reactions of the cyclic ketones were only successful in the case of cyclohexanone (1h), while transformations with five- (1g) and seven- (1i) membered rings failed most likely due to strain.
![[1860-5397-12-80-i3]](/bjoc/content/inline/1860-5397-12-80-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of various ketones (1b−i) with benzaldehyde (3a) in the presence of InCl3 and ZrCl4.
Scheme 3: Reaction of various ketones (1b−i) with benzaldehyde (3a) in the presence of InCl3 and ZrCl4.
Table 6: Variation of the ketone in the domino aldol reaction with benzaldehyde (3a) in the presence of InCl3 and ZrCl4 (reaction time: 2 h, reaction temperature: 25 °C).
| Entry | Product | Ketone | Yield of 7 [%] | Product | Ketone | Yield of 7 [%] | ||
|---|---|---|---|---|---|---|---|---|
| InCl3 | ZrCl4 | InCl3 | ZrCl4 | |||||
| 1 | 7b |
![[Graphic 12]](/bjoc/content/inline/1860-5397-12-80-i20.svg?max-width=637&scale=1.0)
1b |
–a | 40 | 7fb |
![[Graphic 13]](/bjoc/content/inline/1860-5397-12-80-i21.svg?max-width=637&scale=1.0)
1f |
70 | 50 |
| 2 | 7c |
![[Graphic 14]](/bjoc/content/inline/1860-5397-12-80-i22.svg?max-width=637&scale=1.0)
1c |
–a | 45 | 7g |
![[Graphic 15]](/bjoc/content/inline/1860-5397-12-80-i23.svg?max-width=637&scale=1.0)
1g |
– | – |
| 3 | 7d |
![[Graphic 16]](/bjoc/content/inline/1860-5397-12-80-i24.svg?max-width=637&scale=1.0)
1d |
20 | – | 7h |
![[Graphic 17]](/bjoc/content/inline/1860-5397-12-80-i25.svg?max-width=637&scale=1.0)
1h |
5 | 5 |
| 4 | 7e |
![[Graphic 18]](/bjoc/content/inline/1860-5397-12-80-i26.svg?max-width=637&scale=1.0)
1e |
– | – | 7i |
![[Graphic 19]](/bjoc/content/inline/1860-5397-12-80-i27.svg?max-width=637&scale=1.0)
1i |
– | – |
aThe reaction was not carried out. bRef. [42].
The structure of product 7h, which precipitated from the crude mixture in crystalline form, was solved by single crystal X-ray analysis. The crystals are in the space group P21/c. It forms "fibers" with alternating incorporation of the two enantiomers of 7h and they are held together by hydrogen bonds (intramolecular: 1.90 Å, intermolecular: 1.93 Å) (Figure 2).
![[1860-5397-12-80-2]](/bjoc/content/figures/1860-5397-12-80-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: (a) Crystal structure of 7h and (b) its arrangement in the crystal [43].
Figure 2: (a) Crystal structure of 7h and (b) its arrangement in the crystal [43].
Various aldehydes were also probed in the domino aldol reaction with the indium enolate of butyrophenone (1f, Scheme 4). Reactions involving 2-furfural, cinnamaldehyde, butyraldehyde, isobutyraldehyde and 2-phenylpropanal did not provide the corresponding tetrahydro-2H-pyrans, while the reaction with aldehydes possessing p-NMe2 (3b), p-F (3c) and p-MeO (3d) substituted phenyl units worked in reasonable yields affording 8b–d.
![[1860-5397-12-80-i4]](/bjoc/content/inline/1860-5397-12-80-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reaction of n-butyrophenone (1f) with various aldehydes (3b−d) in presence of InCl3 (reaction time: 2 h, reaction temperature: 25 °C).
Scheme 4: Reaction of n-butyrophenone (1f) with various aldehydes (3b−d) in presence of InCl3 (reaction time:...
The electronic influence of substituents at the aldehyde and/or ketone moiety was more systematically analyzed using series of benzaldehydes (3a, 3c and 3d) and propiophenones (1a–c) both substituted by H, OMe and F at the para-phenyl position. Yields increased with time as expected. The introduction of two F or MeO substituents leads to a decrease of the yield compared with 5a (Scheme 5, Table 7). The best yields were obtained within a series in the case of ketone and aldehyde possessing a donor–acceptor situation.
![[1860-5397-12-80-i5]](/bjoc/content/inline/1860-5397-12-80-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Domino aldol reactions of different aldehydes and ketones possessing p-H, p-F and p-MeO substituents at the phenyl units with ZrCl4. Products see Table 7.
Scheme 5: Domino aldol reactions of different aldehydes and ketones possessing p-H, p-F and p-MeO substituent...
Table 7: Time dependent domino aldol reactions of different aldehydes and ketones having H, F and MeO units at para-position of phenyl units using ZrCl4. Yields are in % and numbers in brackets are for mono aldol products (reaction time: 2 h, reaction temperature: 25 °C).
![[Graphic 20]](/bjoc/content/inline/1860-5397-12-80-i28.svg?max-width=637&scale=1.0)
|
![[Graphic 21]](/bjoc/content/inline/1860-5397-12-80-i29.svg?max-width=637&scale=1.0)
|
![[Graphic 22]](/bjoc/content/inline/1860-5397-12-80-i30.svg?max-width=637&scale=1.0)
|
||||||||
| 1a | 1b | 1c | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
![[Graphic 23]](/bjoc/content/inline/1860-5397-12-80-i31.svg?max-width=637&scale=1.0)
3a–c |
H (3a) | F (3c) | MeO (3d) | H (3a) | F (3c) | MeO (3d) | H (3a) | F (3c) | MeO (3d) | |
| Entry | Time [min] | 5a | 5c | 5d | 7a | 9a | 9b | 7b | 9c | 9d |
| 1 | 5 |
5
(+10) |
9
(+9) |
7
(+12) |
5
(+15) |
5
(+18) |
8
(+14) |
25
(+7) |
26
(+11) |
15
(+19) |
| 2 | 10 |
8
(+16) |
23
(+10) |
10
(+15) |
6
(+20) |
8
(+19) |
10
(+15) |
28
(+8) |
26
(+11) |
17
(+15) |
| 3 | 20 |
19
(+16) |
27
(+8) |
15
(+17) |
8
(+16) |
12
(+15) |
14
(+17) |
29
(+7) |
28
(+20) |
20
(+9) |
| 4 | 30 |
27
(+18) |
35
(+10) |
16
(+16) |
12
(+16) |
15
(+19) |
20
(+16) |
32
(+12) |
32
(+12) |
25
(+11) |
| 5 | 45 |
44
(+19) |
51
(+9) |
20
(+13) |
19
(+19) |
20
(+17) |
30
(+17) |
37
(+6) |
36
(+17) |
29
(+10) |
| 6 | 60 |
60
(+20) |
52
(+10) |
35
(+12) |
24
(+18) |
21
(+18) |
37
(+15) |
40
(+6) |
44
(+11) |
30
(+9) |
| 7 | 120 |
76
(+8) |
62
(+7) |
45
(+5) |
40
(+20) |
29
(+17) |
54
(+14) |
45
(+4) |
58
(+14) |
32
(+14) |
Domino aldol reaction by using a CYTOS™ microreactor
Using the CYTOS™ microreactor, a continuous reactor, the following results were obtained, which are well in agreement with the outcome of the batch experiments. Since a slow flow rate of 1 mL min−1 caused precipitation of the compounds and consequently blocking the reactor, flow rates from 2 mL min−1 onwards were used to run the reaction (Table 8). The breakdown of the yields with increasing flow rates is easily explained with the short reaction time.
Table 8: Reaction of benzaldehyde (3a) with indium propiophenone enolate in the CYTOSTM Labsystem (5a/6a in %).
| Entry | Flow rate [mL min−1] | 22 °C | 34 °C | 47 °C | Average rxn. time [min] | |||
|---|---|---|---|---|---|---|---|---|
| 5a | 6a | 5a | 6a | 5a | 6a | |||
| 1 | 1 | − | − | − | − | − | − | 60 |
| 2 | 2 | 62 | 12 | 59 | 10 | 50 | 14 | 30 |
| 3 | 3 | 63 | 14 | 58 | 14 | 48 | 12 | 20 |
| 4 | 5 | 21 | 12 | 20 | 9 | 12 | 6 | 12 |
| 5 | 9 | 20 | 8 | 22 | 6 | 25 | 5 | 6 |
Discussion
The experimental results indicated that two diastereomeric tetrahydro-2H-pyran-2,4-diols were formed at 0 °C in the presence of gallium and at 25 °C with Sn(IV), but only one (i.e., 5a) at elevated temperature. Such finding is indicative of thermodynamic control in the reaction. In the first step, presumably a metal di-, tri- or tetraenolate is formed based on the ratio of enolate to metal chloride. Because in principle a metal dienolate is sufficient as the nucleophilic component, additional enolate ligands may simply act as "innocent bystander ligands" in the reaction cascade. Since we have been able to obtain 5a with GaCl3 in a diastereomerically pure form at 25 °C – although at low temperature a sizeable amount of 20% of a second diastereomer was formed – it is reasonable to assume a reversible formation of the metal-bound tetrahydro-2H-pyran-2,4-diol. Under thermodynamic control all large substituents R (methyl, phenyl) are placed in the equatorial position which leads for all metal ions excluding Ga to only 1 out of 16 possible diastereoisomers.
To shed more light on the mechanism, DFT calculations were carried out using the Gaussian 09 program [44]. Gas-phase optimization of geometries was performed by using the B3LYP [45-47] method with Pople’s split-valence 6-31G(d) basis set on C, H, O atoms and double-ζ quality basis set (LANL2DZ) [48-50] containing Hay and Wadt’s effective core potential (ECP) on hexa-coordinate indium [51] as implemented in Gaussian 09 owing to the predicted good results in our earlier work [30]. The remaining coordination sites of indium were occupied by two THF molecules. The minima and transition states of the calculated structures were verified by analyzing the harmonic vibrational frequencies, using analytical second derivatives. To predict the energies plausibly, as recommended for organometallic compounds, single point calculations with M06 functional [52] were performed using the same basis sets (Scheme 6).
![[1860-5397-12-80-i6]](/bjoc/content/inline/1860-5397-12-80-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: DFT calculations on the formation of A3, hydrolysis of which provides 5a, at M06/6-31G(d)/LANL2DZ//B3LYP/6-31G(d)/LANL2DZ level (ΔGrel with unscaled zpe are in kcal mol−1).
Scheme 6: DFT calculations on the formation of A3, hydrolysis of which provides 5a, at M06/6-31G(d)/LANL2DZ//...
Complexation of the metal enolate E with benzaldehyde (PhCHO) is followed by the exergonic first aldol addition showing a small activation barrier of 1.82 kcal mol−1 via a half−chair like transition state (TS-C-A1), which is in accord with the anti-selective aldol addition of titanium enolates [53,54]. TS-C-A1 leads to the formation of anti-aldolate A1, possessing ΔGrel of −6.60 kcal mol−1. In the next step, A1 is attacked by a second enolate at higher temperature via the bicyclic transition state TS-A1-A2 (ΔGrel = 4.18 kcal mol−1) with a chair–chair conformation. In the last step, intramolecular cyclization with a relative TS energy of 1.22 kcal mol−1 (TS-A2-A3) takes place, which furnishes the metal-bound hemiacetal in a boat conformation (A3) with a relative free energy of −7.49 kcal mol−1. Hydrolysis of A3 provides tetrahydro-2H-pyran-2,4-diol 5a.
Computationally predicted A2 has a lower free energy than the hemiacetal A3, which is responsible for the formation of product 5a. This finding suggests that the hydrolysis occurs on the stage of A2 furnishing A2OH. Afterwards, intramolecular ring closure of A2OH with a relative activation barrier of 23.8 kcal mol−1 leads to 5a in an exergonic process (Scheme 7). At higher temperature, the condensation product 6ac emerges from the dehydration of 6a, taking place via an irreversible reaction, which is accountable for the decrease of the yield at higher temperatures.
![[1860-5397-12-80-i7]](/bjoc/content/inline/1860-5397-12-80-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: The follow-up reactions of A2OH and 6a at M06/6-31G(d)//B3LYP/6-31G(d) level (ΔGrel with unscaled zpe are in kcal mol−1).
Scheme 7: The follow-up reactions of A2OH and 6a at M06/6-31G(d)//B3LYP/6-31G(d) level (ΔGrel with unscaled z...
The mechanism depicted in Scheme 6 is in agreement with the following set of requirements with regard to the metal center: it should (a) exhibit Lewis acidity, (b) be sufficiently electropositive, and (c) display a sufficiently large ion radius so that the reaction cascade can take place in the periphery of the metal. The latter requirement is apparently prohibitive for a boron(III)-mediated reaction because the ion radius of the B3+ ion is very small (25 pm). In the case of tin(II), a high yield of 6a (up to 90%) was observed but formation of 5a was not detected which indicates a hindrance for the second aldol addition. Presumably due to the low charge density of the Sn2+ ions the second carbonyl function is not sufficiently activated for the last ketone–ketone–aldol step. For lanthanum and cerium and maybe even for tin(II) the size may cause problems since these ions are too big. The distance between the reactants is probably too large for a bond formation (Table 9).
Table 9: Effect of the properties of metals on the yields of 5a (reaction time: 2 h).
| Metal | χa | Ionic radiusb [pm] | Charge density Z2/r [e2Å−1] | Yield 5a [%] |
|---|---|---|---|---|
| SnCl2 | 1.72 | 102 (CN 2) | 3.92 | – |
| ZrCl4 | 1.22 | 80 (CN 5) | 20.00 | 76 |
| SnCl4 | 1.72 | 76 (CN 5) | 21.05 | 34 |
| TiCl4 | 1.32 | 56 (CN 5) | 28.57 | 56 |
| BCl3 | 2.01 | 25 (CN 4) | 36.00 | – |
| AlCl3 | 1.47 | 53 (CN 4) | 16.98 | 52 |
| GaCl3 | 1.82 | 61 (CN 4) | 14.80 | 93 |
| InCl3 | 1.49 | 76 (CN 4) | 11.84 | 85 |
| LaCl3 | 1.08 | 117 (CN 6) | 7.69 | – |
| CeCl3 | 1.08 | 115 (CN 6) | 7.82 | – |
| Sc(OTf)3 | 1.20 | 89 (CN 6) | 10.11 | – |
a χ: Electronegativity (According to Allred and Rochow). b CN = Coordination number [55].
In the case of 9-anthracenylaldehyde (3f) employing Zr or Al, formation of 5a, which was not observed in the presence of indium, is detected. Most likely the size of the anthracenyl moiety decreases the rate of the second aldol reaction or the hemiacetal formation, so that deprotonation and subsequently a retro-aldol reaction takes place (Scheme 8). The formation of a domino aldol product with two anthracenyl residues was not observed most probably due to the steric demand of the anthracenyl unit.
![[1860-5397-12-80-i8]](/bjoc/content/inline/1860-5397-12-80-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Proposed mechanism for the formation of benzaldehyde in the reaction of 9-anthracenylaldehyde (3f) with Zr and Al.
Scheme 8: Proposed mechanism for the formation of benzaldehyde in the reaction of 9-anthracenylaldehyde (3f) ...
As illustrated in Scheme 8, the conversion of the aldehyde to the ketone moiety was also witnessed with benzaldehyde (3a) and 4-methoxybenzaldehyde (3d) in the reaction with 4'-fluoropropiophenone (1b) enolate and when reacting 4-methoxybenzaldehyde (3d) with propiophenone (1a) enolate. Here, there is also no interaction between the metal center and the aryl ring of the aldehyde possible. So, a competition between the second aldol or hemiacetal formation and the deprotonation should be considered.
Conclusion
The present results demonstrate a domino aldol reaction working with several substrates and metals that is far superior to the other method with TiCp2 [20,22], which could only be realized with one single substrate resulting in a formal trimerization. The variations in the metal fragment are promising with regard to the development of an enantioselective version of the above reaction and further variations in the substrates. DFT calculations unveil the mechanism for the stereoselective formation of 5a.
Supporting Information
Experimental section, copies of 1H and 13C NMR spectra of compounds, Cartesian coordinates and CIF files of 5a and 7h.
| Supporting Information File 1: Experimental section, copies of 1H and 13C NMR spectra of compounds and Cartesian coordinates. | ||
| Format: PDF | Size: 3.1 MB | Download |
| Supporting Information File 2: CIF file of compound 5a. | ||
| Format: CIF | Size: 16.5 KB | Download |
| Supporting Information File 3: CIF file of compound 7h. | ||
| Format: CIF | Size: 12.4 KB | Download |
Acknowledgements
Support from the DFG (Graduiertenkolleg, SFB), Degussa AG and the Fonds der Chemischen Industrie is gratefully acknowledged. We thank Dr. Thomas Koy and Wolfgang Henn for valuable preparative work and helpful discussions in the early stage of this manuscript. We are indebted to the High-Performance-Computing (HPC) Linux Cluster HorUS of University of Siegen for computational support.
References
-
Wurtz, C. A. Bull. Soc. Chim. Fr. 1872, 17, 436–442.
Return to citation in text: [1] -
Nielsen, A. T.; Houlihan, W. J. The Aldol Condensation. In Organic Reactions; Cope, A. C., Ed.; Wiley: New York, 1968; Vol. 16, pp 1 ff.
Return to citation in text: [1] -
Mekelburger, H. B.; Wilcox, C. S. Formation of Enolates. In Comprehensive Organic Synthesis. Selectivity, Strategy, and Efficiency in Modern Organic Chemistry; Trost, B. M.; Fleming, I., Eds.; Pergamon Press: Oxford, 1991; Vol. 2, pp 99–131.
Return to citation in text: [1] -
Heathcock, C. H. Modern Enolate Chemistry: Regio- and Stereoselective Formation of Enolates and the Consequence of Enolate Configuration on Subsequent Reactions. In Modern Synthetic Methods; Scheffold, R., Ed.; VHCA: Basel, 1992; Vol. 6, pp 1 ff.
Return to citation in text: [1] -
Mahrwald, R. Aldol Reactions; Springer Science+Business Media B.V.: Heidelberg, 2009. doi:10.1007/978-1-4020-8701-1
Return to citation in text: [1] -
Evans, D. A.; Fitch, D. M.; Smith, T. E.; Cee, V. J. J. Am. Chem. Soc. 2000, 112, 10033–10046. doi:10.1021/ja002356g
Return to citation in text: [1] -
Crimmins, M. T.; Katz, J. D.; Washburn, D. G.; Allwein, S. P.; McAtee, L. F. J. Am. Chem. Soc. 2002, 124, 5661–5663. doi:10.1021/ja0262683
Return to citation in text: [1] -
Tanaka, H.; Sawayama, A. M.; Wandless, T. J. J. Am. Chem. Soc. 2003, 125, 6864–6865. doi:10.1021/ja035429f
Return to citation in text: [1] -
Song, H. Y.; Joo, J. M.; Kang, J. W.; Kim, D.-S.; Jung, C.-K.; Kwak, H. S.; Park, J. H.; Lee, E.; Hong, C. Y.; Jeong, S.; Jeon, K.; Park, J. H. J. Org. Chem. 2003, 68, 8080–8087. doi:10.1021/jo034930n
Return to citation in text: [1] -
Kalesse, M.; Cordes, M.; Symkenberg, G.; Lu, H.-H. Nat. Prod. Rep. 2014, 31, 563–594. doi:10.1039/c3np70102f
Return to citation in text: [1] -
Shiina, I. Chem. Rec. 2014, 14, 144–183. doi:10.1002/tcr.201300022
Return to citation in text: [1] -
Mukaiyama, T.; Banno, K.; Narasaka, K. J. Am. Chem. Soc. 1974, 96, 7503–7509. doi:10.1021/ja00831a019
Return to citation in text: [1] -
Mukaiyama, T. Org. React. 1982, 28, 203–331.
Return to citation in text: [1] -
Braun, M. Formation of C–C- Bonds by Addition of Enolates to Carbonyl Groups. In Stereoselective Synthesis: C-C Bond Formation by Addition to C=O, C=N and Reactions Involving Olefinic Double Bonds; Hoffmann, W.; Mulzer, J.; Schaumann, E.; Hoffmann, J., Eds.; Houben-Weyl, Methods of Organic Chemistry, Vol. E 21b; Thieme Verlag: Stuttgart, 1995; pp 1603–1666.
Return to citation in text: [1] -
Machajewski, T. D.; Wong, C.-H. Angew. Chem., Int. Ed. 2000, 39, 1352–1375. doi:10.1002/(SICI)1521-3773(20000417)39:8<1352::AID-ANIE1352>3.0.CO;2-J
Return to citation in text: [1] -
Andrushko, V.; Andrushko, N., Eds. Stereoselective Synthesis of Drugs and Natural Products; John Wiley & Sons: Hoboken, 2013; Vol. 1, pp 215–249.
Return to citation in text: [1] -
Wang, X.; Meng, Q.; Nation, A. J.; Leighton, J. L. J. Am. Chem. Soc. 2002, 124, 10672–10673. doi:10.1021/ja027655f
See for aldol reactions as a first step in a domino process.
Return to citation in text: [1] -
Bazin, S.; Feray, L.; Siri, D.; Naubron, J.-V.; Bertrand, M. P. Chem. Commun. 2002, 2506–2507. doi:10.1039/B206695E
See for aldol reactions as a second step in a domino process.
Return to citation in text: [1] -
Davies, H. M. L.; Lian, Y. Acc. Chem. Res. 2012, 45, 923–935. doi:10.1021/ar300013t
See for aldol reactions as a later step in a domino process.
Return to citation in text: [1] -
Ghorai, M. K.; Halder, S.; Das, S. J. Org. Chem. 2015, 80, 9700–9712. doi:10.1021/acs.joc.5b01768
Return to citation in text: [1] [2] -
Barba, F.; de la Fuente, J. L. J. Org. Chem. 1996, 61, 8662–8663. doi:10.1021/jo952158l
Return to citation in text: [1] -
Yun, S.-S.; Suh, I.-H.; Choi, S.-S.; Lee, S. Chem. Lett. 1998, 27, 985–986. doi:10.1246/cl.1998.985
Return to citation in text: [1] [2] -
Ghosh, A. K.; Shevlin, M. The Development of Titanium Enolate-based Aldol Reactions. In Modern Aldol Reactions; Mahrwald, R., Ed.; Wiley-VCH: Weinheim, 2008; pp 63–125.
Return to citation in text: [1] -
Kanno, K.-I.; Takahashi, T. Zr(IV) and Hf(IV) Lewis Acids. In Acid Catalysis in Modern Organic Synthesis; Yamamoto, H.; Ishihara, K., Eds.; Wiley-VCH: Weinheim, 2008; Vol. 2, pp 825–858.
Return to citation in text: [1] -
Mukaiyama, T.; Matsuo, J.-I. Boron and silicon enolates in crossed aldol reaction. In Modern Aldol Reactions; Mahrwald, R., Ed.; Wiley-VCH: Weinheim, 2008; pp 127–160.
Return to citation in text: [1] -
Renzetti, A.; Marrone, A.; Gérard, S.; Sapi, J.; Nakazawa, H.; Re, N.; Fontana, A. Phys. Chem. Chem. Phys. 2015, 17, 8964–8972. doi:10.1039/C4CP05412A
Return to citation in text: [1] -
Dias, L. C.; Aguilar, A. M. Chem. Soc. Rev. 2008, 37, 451–469. doi:10.1039/B701081H
Return to citation in text: [1] -
Donohoe, T. J. Contemp. Org. Synth. 1996, 3, 1–18. doi:10.1039/CO9960300001
Return to citation in text: [1] -
Evans, D. A.; Janey, J. M.; Magomedov, N.; Tedrow, J. S. Angew. Chem., Int. Ed. 2001, 40, 1884–1888. doi:10.1002/1521-3773(20010518)40:10<1884::AID-ANIE1884>3.0.CO;2-9
Return to citation in text: [1] -
Cinar, M. E.; Schmittel, M. J. Org. Chem. 2015, 80, 8175–8182. doi:10.1021/acs.joc.5b01256
and references therein.
Return to citation in text: [1] [2] [3] -
Dénès, F.; Pérez-Luna, A.; Chemla, F. Chem. Rev. 2010, 110, 2366–2447. doi:10.1021/cr800420x
and references therein.
Return to citation in text: [1] [2] -
Schmittel, M.; Burghart, A.; Malisch, W.; Reising, J.; Söllner, R. J. Org. Chem. 1998, 63, 396–400. doi:10.1021/jo971650x
Return to citation in text: [1] -
Schmittel, M.; Burghart, A.; Werner, H.; Laubender, M.; Söllner, R. J. Org. Chem. 1999, 64, 3077–3085. doi:10.1021/jo981793z
Return to citation in text: [1] -
Schmittel, M.; Ghorai, M. K.; Haeuseler, A.; Henn, W.; Koy, T.; Söllner, R. Eur. J. Org. Chem. 1999, 2007–2010. doi:10.1002/(SICI)1099-0690(199909)1999:9<2007::AID-EJOC2007>3.0.CO;2-2
Return to citation in text: [1] [2] -
Schmittel, M.; Ghorai, M. K. Synlett 2001, 1992–1994. doi:10.1055/s-2001-18766
Return to citation in text: [1] -
Schmittel, M.; Haeuseler, A.; Nilges, T.; Pfitzner, A. Chem. Commun. 2003, 34–35. doi:10.1039/B209536J
Return to citation in text: [1] -
Haeuseler, A.; Henn, W.; Schmittel, M. Synthesis 2003, 2576–2589. doi:10.1055/s-2003-42450
Return to citation in text: [1] [2] [3] -
Kobayashi, S.; Hachiya, I. J. Org. Chem. 1994, 59, 3590–3596. doi:10.1021/jo00092a017
Return to citation in text: [1] -
Cuperly, D.; Petrignet, J.; Crévisy, C.; Grée, R. Chem. – Eur. J. 2006, 12, 3261–3274. doi:10.1002/chem.200501555
Return to citation in text: [1] -
CCDC 1470044 contains the supplementary crystallographic data for this paper. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif
Return to citation in text: [1] -
Giorgi, G.; Arroyo, F. J.; López-Alvarado, P.; Menéndez, J. C. Tetrahedron 2011, 67, 5582–5589. doi:10.1016/j.tet.2011.05.115
Return to citation in text: [1] -
Schmittel, M.; Söllner, R.; Drauz, K.; Günther, K. Tetrahydropyran-2,4-diole, Verfahren zu deren Herstellung und Verwendung. Deutsches Patent- und Markenamt DE 19911 198 A1, May 11, 2000.
Return to citation in text: [1] -
CCDC 1470045 contains the supplementary crystallographic data for this paper. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif
Return to citation in text: [1] -
Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford CT, 2010.
Return to citation in text: [1] -
Becke, A. D. J. Chem. Phys. 1993, 98, 1372–1377. doi:10.1063/1.464304
Return to citation in text: [1] -
Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913
Return to citation in text: [1] -
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/PhysRevB.37.785
Return to citation in text: [1] -
Hay, P. J.; Wadt, W. R. J. Chem. Phys. 1985, 82, 270–283. doi:10.1063/1.448799
Return to citation in text: [1] -
Wadt, W. R.; Hay, P. J. J. Chem. Phys. 1985, 82, 284–298. doi:10.1063/1.448800
Return to citation in text: [1] -
Hay, P. J.; Wadt, W. R. J. Chem. Phys. 1985, 82, 299–310. doi:10.1063/1.448975
Return to citation in text: [1] -
Wells, R. L.; Kher, S. S.; Baldwin, R. A.; White, P. S. Polyhedron 1994, 13, 2731–2735. doi:10.1016/S0277-5387(00)83430-0
See for: Indium trichloride crystallizes from THF as InCl3(THF)3.
Return to citation in text: [1] -
Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x
Return to citation in text: [1] -
Annunziata, R.; Cinquini, M.; Cozzi, F.; Borgia, A. L. J. Org. Chem. 1992, 57, 6339–6342. doi:10.1021/jo00049a053
Return to citation in text: [1] -
Kanemasa, S.; Mori, T.; Tatsukawa, A. Tetrahedron Lett. 1993, 51, 8293–8296. doi:10.1016/S0040-4039(00)61414-4
Return to citation in text: [1] -
Huheey, J. E.; Keiter, E. A.; Keiter, R. L. Inorganic Chemistry: Principles of Structure and Reactivity, 4th ed.; Harper Collins: New York, 1993.
Return to citation in text: [1]
| 53. | Annunziata, R.; Cinquini, M.; Cozzi, F.; Borgia, A. L. J. Org. Chem. 1992, 57, 6339–6342. doi:10.1021/jo00049a053 |
| 54. | Kanemasa, S.; Mori, T.; Tatsukawa, A. Tetrahedron Lett. 1993, 51, 8293–8296. doi:10.1016/S0040-4039(00)61414-4 |
| 55. | Huheey, J. E.; Keiter, E. A.; Keiter, R. L. Inorganic Chemistry: Principles of Structure and Reactivity, 4th ed.; Harper Collins: New York, 1993. |
| 20. | Ghorai, M. K.; Halder, S.; Das, S. J. Org. Chem. 2015, 80, 9700–9712. doi:10.1021/acs.joc.5b01768 |
| 22. | Yun, S.-S.; Suh, I.-H.; Choi, S.-S.; Lee, S. Chem. Lett. 1998, 27, 985–986. doi:10.1246/cl.1998.985 |
| 1. | Wurtz, C. A. Bull. Soc. Chim. Fr. 1872, 17, 436–442. |
| 2. | Nielsen, A. T.; Houlihan, W. J. The Aldol Condensation. In Organic Reactions; Cope, A. C., Ed.; Wiley: New York, 1968; Vol. 16, pp 1 ff. |
| 3. | Mekelburger, H. B.; Wilcox, C. S. Formation of Enolates. In Comprehensive Organic Synthesis. Selectivity, Strategy, and Efficiency in Modern Organic Chemistry; Trost, B. M.; Fleming, I., Eds.; Pergamon Press: Oxford, 1991; Vol. 2, pp 99–131. |
| 4. | Heathcock, C. H. Modern Enolate Chemistry: Regio- and Stereoselective Formation of Enolates and the Consequence of Enolate Configuration on Subsequent Reactions. In Modern Synthetic Methods; Scheffold, R., Ed.; VHCA: Basel, 1992; Vol. 6, pp 1 ff. |
| 5. | Mahrwald, R. Aldol Reactions; Springer Science+Business Media B.V.: Heidelberg, 2009. doi:10.1007/978-1-4020-8701-1 |
| 20. | Ghorai, M. K.; Halder, S.; Das, S. J. Org. Chem. 2015, 80, 9700–9712. doi:10.1021/acs.joc.5b01768 |
| 21. | Barba, F.; de la Fuente, J. L. J. Org. Chem. 1996, 61, 8662–8663. doi:10.1021/jo952158l |
| 22. | Yun, S.-S.; Suh, I.-H.; Choi, S.-S.; Lee, S. Chem. Lett. 1998, 27, 985–986. doi:10.1246/cl.1998.985 |
| 34. | Schmittel, M.; Ghorai, M. K.; Haeuseler, A.; Henn, W.; Koy, T.; Söllner, R. Eur. J. Org. Chem. 1999, 2007–2010. doi:10.1002/(SICI)1099-0690(199909)1999:9<2007::AID-EJOC2007>3.0.CO;2-2 |
| 17. |
Wang, X.; Meng, Q.; Nation, A. J.; Leighton, J. L. J. Am. Chem. Soc. 2002, 124, 10672–10673. doi:10.1021/ja027655f
See for aldol reactions as a first step in a domino process. |
| 18. |
Bazin, S.; Feray, L.; Siri, D.; Naubron, J.-V.; Bertrand, M. P. Chem. Commun. 2002, 2506–2507. doi:10.1039/B206695E
See for aldol reactions as a second step in a domino process. |
| 19. |
Davies, H. M. L.; Lian, Y. Acc. Chem. Res. 2012, 45, 923–935. doi:10.1021/ar300013t
See for aldol reactions as a later step in a domino process. |
| 37. | Haeuseler, A.; Henn, W.; Schmittel, M. Synthesis 2003, 2576–2589. doi:10.1055/s-2003-42450 |
| 12. | Mukaiyama, T.; Banno, K.; Narasaka, K. J. Am. Chem. Soc. 1974, 96, 7503–7509. doi:10.1021/ja00831a019 |
| 13. | Mukaiyama, T. Org. React. 1982, 28, 203–331. |
| 14. | Braun, M. Formation of C–C- Bonds by Addition of Enolates to Carbonyl Groups. In Stereoselective Synthesis: C-C Bond Formation by Addition to C=O, C=N and Reactions Involving Olefinic Double Bonds; Hoffmann, W.; Mulzer, J.; Schaumann, E.; Hoffmann, J., Eds.; Houben-Weyl, Methods of Organic Chemistry, Vol. E 21b; Thieme Verlag: Stuttgart, 1995; pp 1603–1666. |
| 15. | Machajewski, T. D.; Wong, C.-H. Angew. Chem., Int. Ed. 2000, 39, 1352–1375. doi:10.1002/(SICI)1521-3773(20000417)39:8<1352::AID-ANIE1352>3.0.CO;2-J |
| 16. | Andrushko, V.; Andrushko, N., Eds. Stereoselective Synthesis of Drugs and Natural Products; John Wiley & Sons: Hoboken, 2013; Vol. 1, pp 215–249. |
| 30. |
Cinar, M. E.; Schmittel, M. J. Org. Chem. 2015, 80, 8175–8182. doi:10.1021/acs.joc.5b01256
and references therein. |
| 31. |
Dénès, F.; Pérez-Luna, A.; Chemla, F. Chem. Rev. 2010, 110, 2366–2447. doi:10.1021/cr800420x
and references therein. |
| 32. | Schmittel, M.; Burghart, A.; Malisch, W.; Reising, J.; Söllner, R. J. Org. Chem. 1998, 63, 396–400. doi:10.1021/jo971650x |
| 33. | Schmittel, M.; Burghart, A.; Werner, H.; Laubender, M.; Söllner, R. J. Org. Chem. 1999, 64, 3077–3085. doi:10.1021/jo981793z |
| 34. | Schmittel, M.; Ghorai, M. K.; Haeuseler, A.; Henn, W.; Koy, T.; Söllner, R. Eur. J. Org. Chem. 1999, 2007–2010. doi:10.1002/(SICI)1099-0690(199909)1999:9<2007::AID-EJOC2007>3.0.CO;2-2 |
| 35. | Schmittel, M.; Ghorai, M. K. Synlett 2001, 1992–1994. doi:10.1055/s-2001-18766 |
| 36. | Schmittel, M.; Haeuseler, A.; Nilges, T.; Pfitzner, A. Chem. Commun. 2003, 34–35. doi:10.1039/B209536J |
| 37. | Haeuseler, A.; Henn, W.; Schmittel, M. Synthesis 2003, 2576–2589. doi:10.1055/s-2003-42450 |
| 6. | Evans, D. A.; Fitch, D. M.; Smith, T. E.; Cee, V. J. J. Am. Chem. Soc. 2000, 112, 10033–10046. doi:10.1021/ja002356g |
| 7. | Crimmins, M. T.; Katz, J. D.; Washburn, D. G.; Allwein, S. P.; McAtee, L. F. J. Am. Chem. Soc. 2002, 124, 5661–5663. doi:10.1021/ja0262683 |
| 8. | Tanaka, H.; Sawayama, A. M.; Wandless, T. J. J. Am. Chem. Soc. 2003, 125, 6864–6865. doi:10.1021/ja035429f |
| 9. | Song, H. Y.; Joo, J. M.; Kang, J. W.; Kim, D.-S.; Jung, C.-K.; Kwak, H. S.; Park, J. H.; Lee, E.; Hong, C. Y.; Jeong, S.; Jeon, K.; Park, J. H. J. Org. Chem. 2003, 68, 8080–8087. doi:10.1021/jo034930n |
| 10. | Kalesse, M.; Cordes, M.; Symkenberg, G.; Lu, H.-H. Nat. Prod. Rep. 2014, 31, 563–594. doi:10.1039/c3np70102f |
| 11. | Shiina, I. Chem. Rec. 2014, 14, 144–183. doi:10.1002/tcr.201300022 |
| 38. | Kobayashi, S.; Hachiya, I. J. Org. Chem. 1994, 59, 3590–3596. doi:10.1021/jo00092a017 |
| 39. | Cuperly, D.; Petrignet, J.; Crévisy, C.; Grée, R. Chem. – Eur. J. 2006, 12, 3261–3274. doi:10.1002/chem.200501555 |
| 26. | Renzetti, A.; Marrone, A.; Gérard, S.; Sapi, J.; Nakazawa, H.; Re, N.; Fontana, A. Phys. Chem. Chem. Phys. 2015, 17, 8964–8972. doi:10.1039/C4CP05412A |
| 28. | Donohoe, T. J. Contemp. Org. Synth. 1996, 3, 1–18. doi:10.1039/CO9960300001 |
| 29. | Evans, D. A.; Janey, J. M.; Magomedov, N.; Tedrow, J. S. Angew. Chem., Int. Ed. 2001, 40, 1884–1888. doi:10.1002/1521-3773(20010518)40:10<1884::AID-ANIE1884>3.0.CO;2-9 |
| 30. |
Cinar, M. E.; Schmittel, M. J. Org. Chem. 2015, 80, 8175–8182. doi:10.1021/acs.joc.5b01256
and references therein. |
| 25. | Mukaiyama, T.; Matsuo, J.-I. Boron and silicon enolates in crossed aldol reaction. In Modern Aldol Reactions; Mahrwald, R., Ed.; Wiley-VCH: Weinheim, 2008; pp 127–160. |
| 31. |
Dénès, F.; Pérez-Luna, A.; Chemla, F. Chem. Rev. 2010, 110, 2366–2447. doi:10.1021/cr800420x
and references therein. |
| 24. | Kanno, K.-I.; Takahashi, T. Zr(IV) and Hf(IV) Lewis Acids. In Acid Catalysis in Modern Organic Synthesis; Yamamoto, H.; Ishihara, K., Eds.; Wiley-VCH: Weinheim, 2008; Vol. 2, pp 825–858. |
| 23. | Ghosh, A. K.; Shevlin, M. The Development of Titanium Enolate-based Aldol Reactions. In Modern Aldol Reactions; Mahrwald, R., Ed.; Wiley-VCH: Weinheim, 2008; pp 63–125. |
| 27. | Dias, L. C.; Aguilar, A. M. Chem. Soc. Rev. 2008, 37, 451–469. doi:10.1039/B701081H |
| 37. | Haeuseler, A.; Henn, W.; Schmittel, M. Synthesis 2003, 2576–2589. doi:10.1055/s-2003-42450 |
| 40. | CCDC 1470044 contains the supplementary crystallographic data for this paper. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif |
| 41. | Giorgi, G.; Arroyo, F. J.; López-Alvarado, P.; Menéndez, J. C. Tetrahedron 2011, 67, 5582–5589. doi:10.1016/j.tet.2011.05.115 |
| 30. |
Cinar, M. E.; Schmittel, M. J. Org. Chem. 2015, 80, 8175–8182. doi:10.1021/acs.joc.5b01256
and references therein. |
| 52. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 48. | Hay, P. J.; Wadt, W. R. J. Chem. Phys. 1985, 82, 270–283. doi:10.1063/1.448799 |
| 49. | Wadt, W. R.; Hay, P. J. J. Chem. Phys. 1985, 82, 284–298. doi:10.1063/1.448800 |
| 50. | Hay, P. J.; Wadt, W. R. J. Chem. Phys. 1985, 82, 299–310. doi:10.1063/1.448975 |
| 51. |
Wells, R. L.; Kher, S. S.; Baldwin, R. A.; White, P. S. Polyhedron 1994, 13, 2731–2735. doi:10.1016/S0277-5387(00)83430-0
See for: Indium trichloride crystallizes from THF as InCl3(THF)3. |
| 45. | Becke, A. D. J. Chem. Phys. 1993, 98, 1372–1377. doi:10.1063/1.464304 |
| 46. | Becke, A. D. J. Chem. Phys. 1993, 98, 5648–5652. doi:10.1063/1.464913 |
| 47. | Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B 1988, 37, 785–789. doi:10.1103/PhysRevB.37.785 |
| 42. | Schmittel, M.; Söllner, R.; Drauz, K.; Günther, K. Tetrahydropyran-2,4-diole, Verfahren zu deren Herstellung und Verwendung. Deutsches Patent- und Markenamt DE 19911 198 A1, May 11, 2000. |
| 43. | CCDC 1470045 contains the supplementary crystallographic data for this paper. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif |
© 2016 Cinar et al; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)