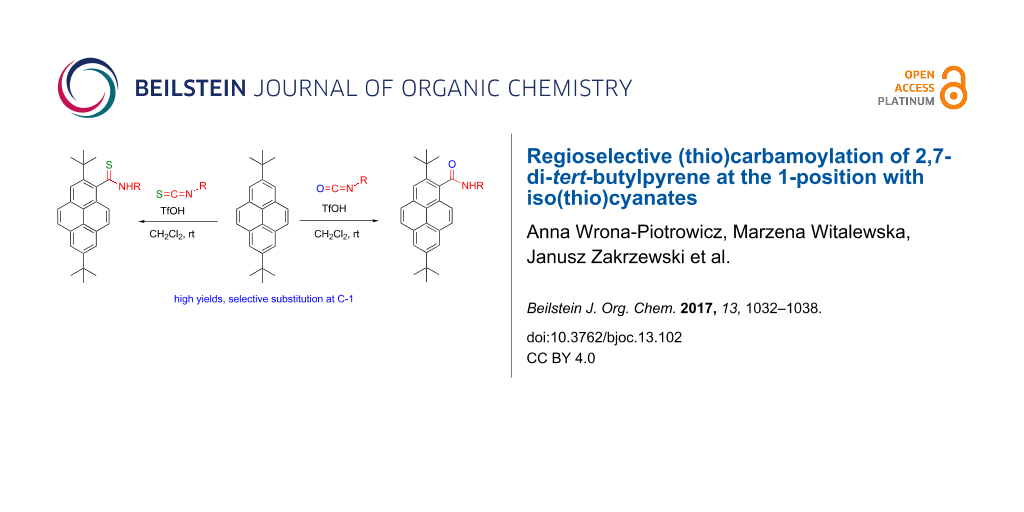
Abstract
It has been found that 2,7-di-tert-butylpyrene reacts with aliphatic iso(thio)cyanates in the presence of trifluoromethanesulfonic acid to exclusively afford the corresponding 1-substituted (thio)amides in high yields. For aromatic iso(thio)cyanates the reaction is less regioselective, although substitution at the 1-position prevails. For ethoxycarbonyl isothiocyanate, apart from the 1-substituted thioamide, 1,8-disubstituted thioamide and 2,7-di-tert-butylpyrene-1-carbonitrile are formed (especially at longer reaction times).

Graphical Abstract
Introduction
Direct functionalization of pyrene 1 has attracted a great deal of attention in recent years because it is the most straightforward route to novel organic molecular materials for optoelectronic devices (OLEDs, field-effect transistors, fluorescent sensors, dye lasers, etc.) [1-7]. Since 1 is an electron-rich arene, aromatic electrophilic substitution seems to be the simplest method for this purpose, and a plethora of substituted pyrenes have been synthesized in this way [1]. As is shown in Figure 1, the most reactive in such reactions are positions 1, 3, 6 and 8 of 1. However, Friedel–Crafts alkylation of 1 with an excess of sterically hindered tert-butyl chloride leads to 2,7-di-tert-butylpyrene (2) [8]. This compound, owing to the presence of two bulky and electron-donating tert-butyl groups, displays different reactivity towards electrophiles. It has been reported that nitration and bromination of 2 take place at the 1-position (however, the bromine atom in 1-bromopyrene can migrate into the 4-position in the presence of AlCl3) [8,9], whereas Friedel–Crafts acylation and Vilsmeier formylation take place at the 4-position [10].
![[1860-5397-13-102-1]](/bjoc/content/figures/1860-5397-13-102-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Sites of electrophilic attack in 1 and 2.
Figure 1: Sites of electrophilic attack in 1 and 2.
We recently reported an efficient method for the synthesis of pyrene-1-carbothioamides via reaction of pyrene with isothiocyanates in the presence of trifluoromethanesulfonic (triflic) acid (TfOH) [11,12]. Since 2 was used as a starting material in the syntheses of various pyrenyl fluorophores exhibiting unique photophysical properties [4,10,13-20], we thought it would be of interest to study its reactivity in the above reaction (and to extend its scope for isocyanates). An additional reason for such a study was the fact that thioamides (and amides) are versatile starting materials in the syntheses of various products (especially heterocycles) [21,22]. Furthermore, aromatic amides undergo a variety of directed C–H bond functionalizations [23]. Herein we report that the reaction of 2 with aliphatic isocyanates and isothiocyanates proceeds regioselectively at the 1-position (Scheme 1).
![[1860-5397-13-102-i1]](/bjoc/content/inline/1860-5397-13-102-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Triflic acid promoted reaction of 2 with iso(thio)cyanates.
Scheme 1: Triflic acid promoted reaction of 2 with iso(thio)cyanates.
Results and Discussion
The reaction was carried out at room temperature in dichloromethane using 1.7 equiv of isocyanate or isothiocyanate and 3.3 equiv of TfOH. For the isocyanates, the reaction was completed in less than 5 min, whereas the reaction with isothiocyanates required ≈30 min for completion. For the aliphatic isocyanates or isothiocyanates it afforded exclusively C1-substituted (thio)amides (as deduced from the splitting pattern of the pyrenyl protons in their 1H NMR spectra, containing one pair of 1-proton doublets displaying a meta-coupling constant (≈2Hz, H6 and H8), two pairs of 1-proton doublets displaying ortho-coupling constants (≈7 Hz, H4, H5 and H9, H10) and one 1-proton singlet (H3).
Unexpectedly, the reaction of 2 with aromatic iso(thio)cyanates was less selective and led to inseparable mixtures of 1-subtituted (main components) and 4-substituted products 3e,m,n. However, for the reaction with p-methoxyphenyl isothiocyanate we were able to obtain, by crystallization, the major product, C(1)-substituted thioamide 3n, contaminated with only ≈5% of its C4-substituted counterpart. We tentatively explain this poorer regioselectivity by possible π–π interaction of pyrene with the arene ring of the protonated aromatic iso(thio)cyanate during formation of the Wheland complex, which may direct the electrophilic attack partly to the 4-position of the pyrene moiety.
To the best of our knowledge, the reaction reported here is the first example of a C1-selective substitution of 2 leading to the formation of a C–C bond.
The presence of two tert-butyl groups significantly increases the reactivity of 2 in comparison with 1. A control experiment in which an equimolar mixture of 1 and 2 was treated in dichloromethane at rt with one equiv of tert-butyl isothiocyanate and 2 equiv of TfOH for 30 min resulted in practically exclusive formation of thioamide 3d (the amount of the product of thiocarbamoylation of 1 was estimated as <2%).
The reaction of 2 with ethoxycarbonyl isothiocyanate (2 equiv) in the presence of TfOH ran in a more complex way (Scheme 2, Table 1).
![[1860-5397-13-102-i2]](/bjoc/content/inline/1860-5397-13-102-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Triflic acid promoted reaction of 2 with ethoxycarbonyl isothiocyanate.
Scheme 2: Triflic acid promoted reaction of 2 with ethoxycarbonyl isothiocyanate.
Table 1: Reaction of 2 with ethoxycarbonyl isothiocyanate (2 equiv) and TfOH (4 equiv) in CH2Cl2 at room temperature.
| Entry | Reaction time | 3o (%) | 4 (%) | 5 (%) |
|---|---|---|---|---|
| 1 | 10 min | 78 | 9 | 7 |
| 2 | 2 h | 46 | 17 | 13 |
| 3 | 4 h | 40 | 21 | 18 |
| 4 | 6 h | 39 | 33 | 20 |
| 5 | 8 h | 36 | 30 | 21 |
| 6 | 16 h | 29 | 26 | 32 |
| 7 | 3 days | 15 | 17 | 38 |
| 8 | 7 days | 10 | 14 | 54 |
| 9 | 7daysa | 9 | 3 | 43 |
| 10 | 12 days | 7 | 9 | 19 |
a3 Equivalents of isothiocyanate were used.
After 10 min, 1,8-bis-carbothioamide 4, containing ≈20% of other regioisomers (9%) and nitrile 5 (7%) were isolated by column chromatography besides the expected 3o (78%). The amounts of 4 and 5 significantly increased along with the reaction time (Table 1). The highest yield of 4 (33%) was found after 6 h, whereas a 7-day reaction afforded 5 in a moderate (54%) isolated yield.
Attempts to separate the regioisomeric bis-thioamides failed, but repeated chromatography and recrystallization from dichloromethane/hexane allowed isolation of the practically pure main regioisomer, 1,8-dithioamide 4. Its structure was confirmed by single-crystal X-ray diffraction, which revealed a cisoidal conformation of this molecule in the crystal (Figure 2, for details, see Supporting Information File 1 and Supporting Information File 2). However, the 1H NMR spectrum of 4 at room temperature was more complex than expected and contained broadened signals, suggesting the presence of other rotamers in CDCl3 solution.
![[1860-5397-13-102-2]](/bjoc/content/figures/1860-5397-13-102-2.png?scale=2.0&max-width=1024&background=FFFFFF)
The mechanism of the formation of nitrile 5 in the reaction under study here is so far unclear. The control experiment showed that under reaction conditions the secondary thioamide 3o undergoes transformation into 5, but to our knowledge this reaction, which must involve cleavage of the C–N bond, has no precedent in the literature (in contrast to the well-known formation of nitriles from primary thioamides, involving formal elimination of H2S [24]).
Encouraged by the above results, we decided to check whether TfOH-promoted Friedel–Crafts acylation of 2 will also occur at the 1-position. For this purpose, we examined the reaction of 2 with acetic acid and trifluoroacetic anhydride, (TFAA)/TfOH, according to our protocol used for the acylation of pyrene with alkynoic acids [25]. However, in this case we observed exclusive formation of C-4 substituted ketone 6 (Scheme 3).
![[1860-5397-13-102-i3]](/bjoc/content/inline/1860-5397-13-102-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
In the 1H NMR spectrum of 6 we can see two pairs of doublets split with Jmeta (1.8 Hz), assignable to H(1),H(3) and H(6),H(8) and a pair of doublets split with Jortho (9 Hz) assignable to H(9),H(10 as well as a singlet (H5).
In our opinion, the observed difference in the regioselectivity of the (thio)carbamoylation and acylation of 2 may be due to different bulkiness of the reacting electrophile: the electrophilic center of the protonated iso(thio)cyanate is relatively unhindered and able to attack the electronically activated but sterically hindered pyrene 1-position, whereas the bulkier protonated acetyl trifluoroacetate (the postulated electrophile in the examined Friedel–Crafts acylation) attacks sterically the less hindered 4-position.
Conclusion
We found that triflic acid-promoted (thio)carbamoylation of 2 with aliphatic iso(thio)cyanates occurs selectively at the 1- position, whereas Friedel–Crafts acylation promoted by this acid leads to 4-substitution. The reaction is less regioselective for aromatic iso(thio)cyanates. We believe that the described reaction opens up new synthetic routes to novel fluorophores having a sterically hindered pyrene framework.
Experimental
General
All reagents were purchased from Sigma-Aldrich and used without further purification. Solvents were purified before use by reported methods. Column chromatography was carried out on silica gel 60 (0.040–0.063 mm, 230–400 mesh, Fluka). 1H and 13C NMR spectra were recorded in CDCl3 at 600 MHz for 1H and 150 MHz for 13C at room temperature. Chemical shifts are reported in ppm and are referenced relative to solvent signals.
2,7-Di-tert-butylpyrene-1-carboxamides. General procedure
Analogous as described in [12]. Isocyanate (1.2 mmol) and TfOH (348 μL, 4 mmol) were added to a solution of 2,7-di-tert-butylpyrene (2, 314 mg, 1 mmol) in CH2Cl2 (10 mL) at room temperature. After stirring for 5 min, the reaction mixture was poured into ice-water (50 mL) and extracted several times with CH2Cl2. The combined extracts were dried over anhydrous Na2SO4 and evaporated. The products were isolated by flash chromatography (eluent: CH2Cl2).
2,7-Di-tert-butyl-N-ethylpyrene-1-carboxamide (3a). White solid (293 mg, 76%). Mp 258–259 °C; 1H NMR (600 MHz, CDCl3) δ 8.26 (s, 1H), 8.20 (d, J = 1.8 Hz, 1H), 8.18 (d, J = 1.8 Hz, 1H), 8.03 (m, 3H), 7.98 (d, J = 9.0 Hz, 1H), 5.87 (t, J = 5.4 Hz, 1H), 3.74 (m, 1H), 3.64 (m, 1H), 1.67 (s, 9H), 1.58 (s, 9H), 1.36 (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 171.8, 149.2, 144.0, 131.3, 131.1, 130.9, 130.3, 128.7, 128.4, 128.2, 127.2, 124.4, 123.6, 122.7, 122.7, 122.4, 122.4, 37.1, 35.2, 35.1, 32.3, 31.9, 14.4; IR (KBr, cm−1): 3414, 3259, 2958, 1629, 1534, 1300, 1224, 880; anal. calcd. for C27H31NO: C, 84.11; H, 8.10; N, 3.63; O, 4.15; found: C, 84.07; H, 8.12; N, 3.71.
(For characterization data of other synthesized amides, see Supporting Information File 1).
2,7-Di-tert-butylpyrene-1-carbothioamides. General procedure
Analogous as described in [12]. Isothiocyanate (1.2 mmol) and TfOH (348 μL, 4 mmol) were added to a solution of 2,7-di-tert-butylpyrene (314 mg, 1 mmol, ) in CH2Cl2 (10 mL) at room temperature. After stirring for 30 min, the reaction mixture was poured into ice-water (50 mL) and extracted several times with CH2Cl2. The combined extracts were dried over anhydrous Na2SO4 and evaporated. For the reaction with ethoxycarbonyl isothiocyanate, chromatography afforded two fractions. The less polar fraction contained monothioamide 3o and nitrile 5, whereas the more polar fraction contained dithioamides 4. The separation of 3o and 5 required the second chromatography with CH2Cl2/hexanes (1:1) as the eluent. The yields of 3o, 4 and 5 are gathered in Table 1.
2,7-Di-tert-butyl-N-ethylpyrene-1-carbothioamide (3f). White solid (378 mg, 94%). Mp 222–223 °C; 1H NMR (600 MHz, CDCl3) δ 8.28 (s, 1H), 8.19 (d, J = 1.2 Hz, 1H), 8.17 (d, J = 1.2 Hz, 1H), 8.08 (d, J = 9.6 Hz, 1H), 8.02 (d, J = 9.0 Hz, 1H), 8.01 (d, J = 9.0 Hz, 1H), 7.95 (d, J = 9.0 Hz, 1H), 7.59 (s, 1H), 4.09 (m, 1H), 3.96 (m, 1H), 1.72 (s, 9H), 1.58 (s, 9H), 1.43 (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 203.4, 149.2, 142.5, 137.1, 131.0, 130.9, 130.3, 128.4, 128.0, 127.7, 127.2, 124.3, 124.2, 122.9, 122.7, 122.3, 41.0, 37.8, 35.2, 32.6, 31.9, 12.8; IR (KBr, cm−1): 3424, 3164, 2961, 1603, 1540, 1391, 1334, 1223, 881; anal. calcd. for C27H31NS: C, 80.75; H, 7.78; N, 3.49; S, 7.98; found: C, 80.70; H, 7.80; N, 3.51, S, 7.99.
Ethyl (2,7-di-tert-butylpyrene-1-carbonothioyl)carbamate (3o). Yellow solid. Mp 97–98 °C; 1H NMR (600 MHz, CDCl3) δ 9.82 (s, 1H), 8.33 (s, 1H), 8.19 (d, J = 13.2 Hz, 2H), 8.05 (s, 2H), 7.99 (m, 2H), 3.92 (q, J = 7.2 Hz, 2H), 1.74 (m, 9H), 1.59 (m, 9H), 0.97 (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 211.0, 149.0, 148.9, 141.9, 134.9, 131.0, 130.8, 130.2, 128.4, 127.8, 127.4, 125.9, 124.4, 123.4, 122.7, 122.6, 122.5, 122.2, 63.0, 37.6, 35.2, 32.5, 31.9, 13.8; IR (KBr, cm−1): 3447, 3383, 2961, 2906, 2868, 1769, 1479, 1227, 1137, 1043, 881; anal. calcd. for C28H31NO2S: C, 75.47; H, 7.01; N, 3.14; O, 7.18; S, 7.20; found: C, 75.34; H, 7.18; N, 3.21, S, 7.14.
Diethyl (2,7-di-tert-butyl.pyrene-1,8-dicarbonothioyl)carbamate (4). Yellow solid. Mp 240–241 °C; 1H NMR (600 MHz, CDCl3) δ 9.75 (s, 1H), 9.72 (s, 1H), 8.28 (m, 2H), 8.02 (s, 2H), 7.95 (s, 2H), 3.90 (m, 4H), 1.69 (m, 18H), 0.97 (m, 3H), 0.88 (m, 3H). The 13C NMR spectrum was not measured due to limited solubility of this compound. IR (KBr, cm−1): 3432, 3385, 3162, 2964, 2910, 1773, 1492, 1228, 1168, 1149, 1110, 1038; anal. calcd. for C32H34N2O4S2: C, 66.64; H, 6.29; N, 4.86; O, 11.10; S, 11.12; found: C, 66.59; H, 6.33; N, 4.81, S, 11.03.
(For characterization data of other synthesized thioamides, see Supporting Information File 1).
2,7-Di-tert-butylpyrene-1-carbonitrile (5). Yellow solid. Mp 176–177 °C; 1H NMR (600 MHz, CDCl3) δ 8.50 (d, J = 9.0 Hz, 1H), 8.30 (d, J = 1.2 Hz, 1H), 8.28 (d, J = 1.2 Hz, 1H), 8.18 (d, J = 9.6 Hz, 1H), 8.17 (s, 1H), 8.14 (d, J = 9.0 Hz, 1H), 7.98 (d, J = 9.0 Hz, 1H), 1.81 (s, 9H), 1.62 (s, 9H); 13C NMR (150 MHz, CDCl3) δ 150.9, 149.9, 134.7, 133.4, 130.55, 130.53, 130.50, 130.0, 126.9, 124.0, 124.0, 123.9, 122.1, 121.9, 121.5, 119.8, 103.9, 36.2, 35.3, 31.8, 30.8; IR (KBr, cm−1): 2958, 2905, 2872, 2198, 1600, 1465, 1364, 1228, 879, 733; anal. calcd. for C25H25N: C, 88.45; H, 7.42; N, 4.13; found: C, 88.39; H, 7.51; N, 4.06.
4-Acetyl-2,7-di-tert-butylpyrene (6). 2,7-Di-tert-butylpyrene (2, 628 mg, 2 mmol) and triflic acid (174 μL, 2 mmol) were added at 0 °C to a solution of acetic acid (126 μL, 2.2 mmol) and trifluoroacetic anhydride (278 μL, 2 mmol) in CH2Cl2 (20 mL). The reaction mixture was warmed to room temperature and stirred for 2 h. After this time the reaction mixture was poured into ice-water (50 mL) and extracted several times with CH2Cl2. The combined extracts were dried over anhydrous Na2SO4 and evaporated. Flash chromatography (eluent: CH2Cl2) afforded the product. Yellow solid. (310 mg, 87%). Mp 121–122 °C; 1H NMR (600 MHz, CDCl3) δ 9.23 (d, J = 1.8 Hz, 1H), 8.59 (s, 1H), 8.29 (d, J = 1.2 Hz, 1H), 8.28 (d, J = 1.8 Hz, 1H), 8.24 (d, J = 1.2 Hz, 1H), 8.05 (d, J = 9.0 Hz, 1H), 8.01 (d, J = 9.0 Hz, 1H), 2.94 (s, 3H), 1.60 (s, 18H); 13C NMR (150 MHz, CDCl3) δ 201.9, 149.51, 149.07, 134.8, 132.3, 130.80, 130.78, 128.9, 128.3, 127.1, 126.7, 124.5, 123.9, 123.6, 123.5, 122.9, 121.9, 35.5, 35.2, 31.93, 31.88, 29.9; IR (KBr, cm–1): 2958, 2901, 2870, 1666, 1601, 1391, 1225, 1200, 892; anal. calcd. for C26H28O: C, 87.60; H, 7.98; O, 4.48; found: C, 87.57; H, 8.05.
Supporting Information
| Supporting Information File 1: Characterization data, copies of 1H, 13C NMR and IR spectra for synthesized compounds, and crystallographic structure and refinement data of 4. | ||
| Format: PDF | Size: 2.1 MB | Download |
| Supporting Information File 2: Crystallographic data for 4. | ||
| Format: CIF | Size: 5.4 KB | Download |
References
-
Feng, X.; Hu, J.-Y.; Redshaw, C.; Yamato, T. Chem. – Eur. J. 2016, 22, 11898–11916. doi:10.1002/chem.201600465
Return to citation in text: [1] [2] -
Figueira-Duarte, T. M.; Müllen, K. Chem. Rev. 2011, 111, 7260–7314. doi:10.1021/cr100428a
Return to citation in text: [1] -
Casas-Solvas, J. M.; Howgego, J. D.; Davis, A. P. Org. Biomol. Chem. 2014, 12, 212–232. doi:10.1039/C3OB41993B
Return to citation in text: [1] -
Mateo-Alonso, A. Chem. Soc. Rev. 2014, 43, 6311–6324. doi:10.1039/C4CS00119B
Return to citation in text: [1] [2] -
Niko, Y.; Cho, Y.; Kawauchi, S.; Konishi, G.-i. RSC Adv. 2014, 4, 36480–36484. doi:10.1039/C4RA06282E
Return to citation in text: [1] -
Piotrowicz, M.; Zakrzewski, J.; Métivier, R.; Brosseau, A.; Makal, A.; Woźniak, K. J. Org. Chem. 2015, 80, 2573–2581. doi:10.1021/jo502619k
Return to citation in text: [1] -
Chercka, D.; Yoo, S.-J.; Baumgarten, M.; Kim, J.-J.; Müllen, K. J. Mater. Chem. C 2014, 2, 9083–9086. doi:10.1039/C4TC01801J
Return to citation in text: [1] -
Rodenburg, L.; Brandsma, R.; Tintel, C.; van Thuijl, J.; Lugtenburg, J.; Cornelisse, J. Recl. Trav. Chim. Pays-Bas 1986, 105, 156–161. doi:10.1002/recl.19861050504
Return to citation in text: [1] [2] -
Yamato, T.; Fujimoto, M.; Miyazawa, A.; Matsuo, K. J. Chem. Soc., Perkin Trans. 1 1997, 1201–1208. doi:10.1039/a606200h
Return to citation in text: [1] -
Bock, H.; Subervie, D.; Mathey, P.; Pradhan, A.; Sarkar, P.; Dechambenoit, P.; Hillard, E. A.; Durola, F. Org. Lett. 2014, 16, 1546–1549. doi:10.1021/ol500154k
Return to citation in text: [1] [2] -
Wrona-Piotrowicz, A.; Zakrzewski, J.; Métivier, R.; Brosseau, A.; Makal, A.; Woźniak, K. RSC Adv. 2014, 4, 56003–56012. doi:10.1039/C4RA07045C
Return to citation in text: [1] -
Wrona-Piotrowicz, A.; Zakrzewski, J.; Gajda, A.; Gajda, T.; Makal, A.; Brosseau, A.; Métivier, R. Beilstein J. Org. Chem. 2015, 11, 2451–2458. doi:10.3762/bjoc.11.266
Return to citation in text: [1] [2] [3] -
Ozaki, K.; Kawasumi, K.; Shibata, M.; Ito, H.; Itami, K. Nat. Commun. 2015, 6, No. 6251. doi:10.1038/ncomms7251.
Return to citation in text: [1] -
Duan, Z.; Hoshino, D.; Yang, Z.; Yano, H.; Ueki, H.; Liu, Y.; Ohuchi, H.; Takayanagi, Y.; Zhao, G.; Nishioka, Y. Mol. Cryst. Liq. Cryst. 2011, 538, 199–207. doi:10.1080/15421406.2011.564018
Return to citation in text: [1] -
Gonell, S.; Poyatos, M.; Peris, E. Chem. – Eur. J. 2014, 20, 9716–9724. doi:10.1002/chem.201304952
Return to citation in text: [1] -
García, R.; More, S.; Melle-Franco, M.; Mateo-Alonso, A. Org. Lett. 2014, 16, 6096–6099. doi:10.1021/ol5029332
Return to citation in text: [1] -
Paudel, A.; Hu, J.-Y.; Yamato, T. J. Chem. Res., Synop. 2008, 457–460. doi:10.3184/030823408X338710
Return to citation in text: [1] -
Jang, K.; Ranasinghe, A. D.; Heske, C.; Lee, D.-C. Langmuir 2010, 26, 13630–13636. doi:10.1021/la101921c
Return to citation in text: [1] -
Mochida, K.; Kawasumi, K.; Segawa, Y.; Itami, K. J. Am. Chem. Soc. 2011, 133, 10716–10719. doi:10.1021/ja202975w
Return to citation in text: [1] -
Jiang, L.; Papageorgiou, A. C.; Oh, S. C.; Sağlam, Ö.; Reichert, J.; Duncan, D. A.; Zhang, Y.-Q.; Klappenberger, F.; Guo, Y.; Allegretti, F.; More, S.; Bhosale, R.; Mateo-Alonso, A.; Barth, J. V. ACS Nano 2016, 10, 1033–1041. doi:10.1021/acsnano.5b06340
Return to citation in text: [1] -
Jagodziński, T. S. Chem. Rev. 2003, 103, 197–228. doi:10.1021/cr0200015
Return to citation in text: [1] -
Volkov, A.; Tinnis, F.; Slagbrand, T.; Trillo, P.; Adolfsson, H. Chem. Soc. Rev. 2017, 45, 6685–6697. doi:10.1039/C6CS00244G
Return to citation in text: [1] -
Zhu, R.-Y.; Farmer, M. E.; Chen, Y.-Q.; Yu, J.-Q. Angew. Chem., Int. Ed. 2016, 55, 10578–10599. doi:10.1002/anie.201600791
Return to citation in text: [1] -
Yamaguchi, K.; Yajima, K.; Mizuno, N. Chem. Commun. 2012, 48, 11247–11249. doi:10.1039/c2cc36635e
Return to citation in text: [1] -
Flamholc, R.; Plażuk, D.; Zakrzewski, J.; Métivier, R.; Nakatani, K.; Makal, A.; Woźniak, K. RSC Adv. 2014, 4, 31594–31601. doi:10.1039/C4RA03961K
Return to citation in text: [1]
| 1. | Feng, X.; Hu, J.-Y.; Redshaw, C.; Yamato, T. Chem. – Eur. J. 2016, 22, 11898–11916. doi:10.1002/chem.201600465 |
| 2. | Figueira-Duarte, T. M.; Müllen, K. Chem. Rev. 2011, 111, 7260–7314. doi:10.1021/cr100428a |
| 3. | Casas-Solvas, J. M.; Howgego, J. D.; Davis, A. P. Org. Biomol. Chem. 2014, 12, 212–232. doi:10.1039/C3OB41993B |
| 4. | Mateo-Alonso, A. Chem. Soc. Rev. 2014, 43, 6311–6324. doi:10.1039/C4CS00119B |
| 5. | Niko, Y.; Cho, Y.; Kawauchi, S.; Konishi, G.-i. RSC Adv. 2014, 4, 36480–36484. doi:10.1039/C4RA06282E |
| 6. | Piotrowicz, M.; Zakrzewski, J.; Métivier, R.; Brosseau, A.; Makal, A.; Woźniak, K. J. Org. Chem. 2015, 80, 2573–2581. doi:10.1021/jo502619k |
| 7. | Chercka, D.; Yoo, S.-J.; Baumgarten, M.; Kim, J.-J.; Müllen, K. J. Mater. Chem. C 2014, 2, 9083–9086. doi:10.1039/C4TC01801J |
| 10. | Bock, H.; Subervie, D.; Mathey, P.; Pradhan, A.; Sarkar, P.; Dechambenoit, P.; Hillard, E. A.; Durola, F. Org. Lett. 2014, 16, 1546–1549. doi:10.1021/ol500154k |
| 8. | Rodenburg, L.; Brandsma, R.; Tintel, C.; van Thuijl, J.; Lugtenburg, J.; Cornelisse, J. Recl. Trav. Chim. Pays-Bas 1986, 105, 156–161. doi:10.1002/recl.19861050504 |
| 9. | Yamato, T.; Fujimoto, M.; Miyazawa, A.; Matsuo, K. J. Chem. Soc., Perkin Trans. 1 1997, 1201–1208. doi:10.1039/a606200h |
| 8. | Rodenburg, L.; Brandsma, R.; Tintel, C.; van Thuijl, J.; Lugtenburg, J.; Cornelisse, J. Recl. Trav. Chim. Pays-Bas 1986, 105, 156–161. doi:10.1002/recl.19861050504 |
| 12. | Wrona-Piotrowicz, A.; Zakrzewski, J.; Gajda, A.; Gajda, T.; Makal, A.; Brosseau, A.; Métivier, R. Beilstein J. Org. Chem. 2015, 11, 2451–2458. doi:10.3762/bjoc.11.266 |
| 1. | Feng, X.; Hu, J.-Y.; Redshaw, C.; Yamato, T. Chem. – Eur. J. 2016, 22, 11898–11916. doi:10.1002/chem.201600465 |
| 23. | Zhu, R.-Y.; Farmer, M. E.; Chen, Y.-Q.; Yu, J.-Q. Angew. Chem., Int. Ed. 2016, 55, 10578–10599. doi:10.1002/anie.201600791 |
| 25. | Flamholc, R.; Plażuk, D.; Zakrzewski, J.; Métivier, R.; Nakatani, K.; Makal, A.; Woźniak, K. RSC Adv. 2014, 4, 31594–31601. doi:10.1039/C4RA03961K |
| 21. | Jagodziński, T. S. Chem. Rev. 2003, 103, 197–228. doi:10.1021/cr0200015 |
| 22. | Volkov, A.; Tinnis, F.; Slagbrand, T.; Trillo, P.; Adolfsson, H. Chem. Soc. Rev. 2017, 45, 6685–6697. doi:10.1039/C6CS00244G |
| 12. | Wrona-Piotrowicz, A.; Zakrzewski, J.; Gajda, A.; Gajda, T.; Makal, A.; Brosseau, A.; Métivier, R. Beilstein J. Org. Chem. 2015, 11, 2451–2458. doi:10.3762/bjoc.11.266 |
| 4. | Mateo-Alonso, A. Chem. Soc. Rev. 2014, 43, 6311–6324. doi:10.1039/C4CS00119B |
| 10. | Bock, H.; Subervie, D.; Mathey, P.; Pradhan, A.; Sarkar, P.; Dechambenoit, P.; Hillard, E. A.; Durola, F. Org. Lett. 2014, 16, 1546–1549. doi:10.1021/ol500154k |
| 13. | Ozaki, K.; Kawasumi, K.; Shibata, M.; Ito, H.; Itami, K. Nat. Commun. 2015, 6, No. 6251. doi:10.1038/ncomms7251. |
| 14. | Duan, Z.; Hoshino, D.; Yang, Z.; Yano, H.; Ueki, H.; Liu, Y.; Ohuchi, H.; Takayanagi, Y.; Zhao, G.; Nishioka, Y. Mol. Cryst. Liq. Cryst. 2011, 538, 199–207. doi:10.1080/15421406.2011.564018 |
| 15. | Gonell, S.; Poyatos, M.; Peris, E. Chem. – Eur. J. 2014, 20, 9716–9724. doi:10.1002/chem.201304952 |
| 16. | García, R.; More, S.; Melle-Franco, M.; Mateo-Alonso, A. Org. Lett. 2014, 16, 6096–6099. doi:10.1021/ol5029332 |
| 17. | Paudel, A.; Hu, J.-Y.; Yamato, T. J. Chem. Res., Synop. 2008, 457–460. doi:10.3184/030823408X338710 |
| 18. | Jang, K.; Ranasinghe, A. D.; Heske, C.; Lee, D.-C. Langmuir 2010, 26, 13630–13636. doi:10.1021/la101921c |
| 19. | Mochida, K.; Kawasumi, K.; Segawa, Y.; Itami, K. J. Am. Chem. Soc. 2011, 133, 10716–10719. doi:10.1021/ja202975w |
| 20. | Jiang, L.; Papageorgiou, A. C.; Oh, S. C.; Sağlam, Ö.; Reichert, J.; Duncan, D. A.; Zhang, Y.-Q.; Klappenberger, F.; Guo, Y.; Allegretti, F.; More, S.; Bhosale, R.; Mateo-Alonso, A.; Barth, J. V. ACS Nano 2016, 10, 1033–1041. doi:10.1021/acsnano.5b06340 |
| 11. | Wrona-Piotrowicz, A.; Zakrzewski, J.; Métivier, R.; Brosseau, A.; Makal, A.; Woźniak, K. RSC Adv. 2014, 4, 56003–56012. doi:10.1039/C4RA07045C |
| 12. | Wrona-Piotrowicz, A.; Zakrzewski, J.; Gajda, A.; Gajda, T.; Makal, A.; Brosseau, A.; Métivier, R. Beilstein J. Org. Chem. 2015, 11, 2451–2458. doi:10.3762/bjoc.11.266 |
| 24. | Yamaguchi, K.; Yajima, K.; Mizuno, N. Chem. Commun. 2012, 48, 11247–11249. doi:10.1039/c2cc36635e |
© 2017 Wrona-Piotrowicz et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)