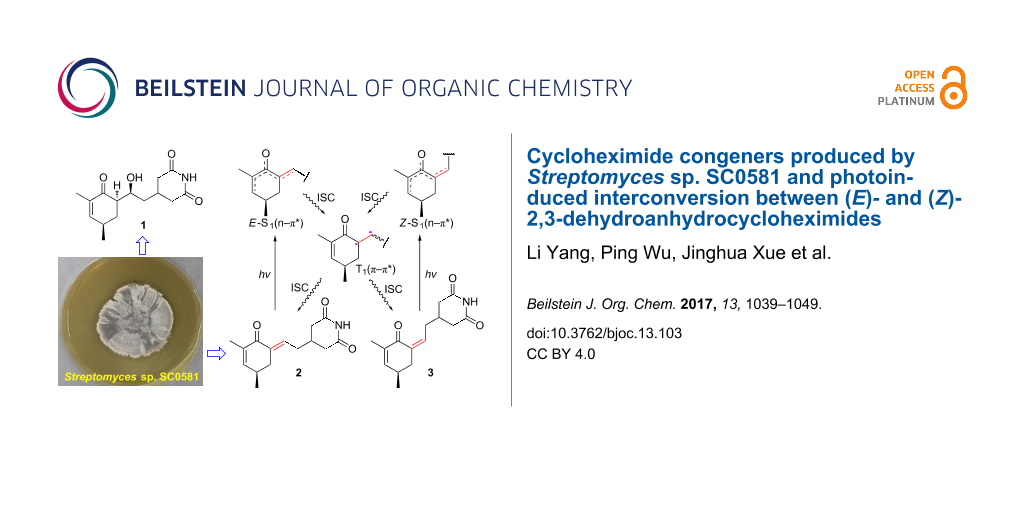
Abstract
Three new cycloheximide congeners, 2,3-dehydro-α-epi-isocycloheximide (1), (E)- and (Z)-2,3-dehydroanhydrocycloheximides (2 and 3), together with three known compounds, anhydroisoheximide (4), cycloheximide (5), and isocycloheximide (6), were obtained from the cultures of Streptomyces sp. SC0581. Their structures were elucidated by extensive spectroscopic analysis in combination with theoretical conformational analysis and ECD computations. The photoinduced interconversion between 2 and 3 was observed and verified and the possible reaction path and mechanism were proposed by theoretical computations. The antifungal and cytotoxic activities of 1–6 were evaluated and suggested that 2,3-dehydrogenation results in the loss of the activities and supported that the OH-α is important to the activities of cycloheximide congeners.

Graphical Abstract
Introduction
The glutarimide-containing antibiotics represent a fascinating class of natural products that exhibit a multitude of biological activities. The most famous representative of this family, cycloheximide (5), has been used for decades as an inhibitor of eukaryotic translation elongation [1-3]. Other members of this family show potent cell migration inhibition and antiviral activity [4-6], which continues to capture the attention from researchers in synthetic and biosynthetic chemistry, medicinal chemistry, and pharmacology. However, cycloheximide (5) has held back the clinical and agricultural applications due to its reproductive toxicity [7]. Identifying new analogues that offer similar activity without the toxic side-effects could provide a viable lead of therapeutic drugs or agricultural pesticides. During the course of our searching for bioactive microbial metabolites [8,9], a culture extract of Streptomyces sp. SC0581 was found to show antifungal activity against the phytopathogen Phytophthora infestans. Bioassay-guided fractionation of the extract led to the isolation of three new cycloheximide congeners (Figure 1), 2,3-dehydro-α-epi-isocycloheximide (1), (E)- and (Z)-2,3-dehydroanhydrocycloheximides (2 and 3), and one known but new naturally occurring cycloheximide congener, anhydroisoheximide (4) [6], together with cycloheximide (5) and isocycloheximide (6) [10]. Their structures were elucidated by spectroscopic analysis, theoretical conformational analysis, and ECD/TDDFT calculations. The photoinduced interconversion between 2 and 3 was observed and verified, and the possible reaction path and mechanism were proposed by theoretical computations. All the isolated compounds were evaluated for antifungal activity and cancer cell toxicity. Herein are reported the isolation, structural elucidation, and biological activities of these compounds and the interconversion between 2 and 3.
![[1860-5397-13-103-1]](/bjoc/content/figures/1860-5397-13-103-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
Structure elucidation
The molecular formula of compound 1 was established as C15H21NO4 based on the HRESI(+)-MS ion at m/z 302.1357 [M + Na]+ (calcd 302.1363). Its 1H and 13C NMR spectral data (Table 1), in combination with the HSQC spectrum, indicated the presence of a conjugated keto carbonyl (δC 202.6), two amide carbonyls (δC 175.5, 175.7), a trisubstituted olefin (δH 6.65; δC 134.2, 151.9), two methyls (δH 1.72, 1.16; δC 14.5, 20.3), four aliphatic methines with one being oxygenated (δH 4.26; δC 67.3), and four methylenes. Analysis of the 1H,1H-COSY and HMBC spectra, in particular the HMBC correlations from the olefinic methyl protons (δH 1.72, H3-7) to C1 (δC 202.6), C2 (δC 134.2), and C3 (δC 151.9), from the aliphatic methyl (δH 1.16, H3-8) to C3, C4 (δC 31.8), and C5 (δC 31.7), and from the oxymethine (δH 4.26, H-α) to C1 and C6 (δC 52.2), readily constructed a planar structure of 2,3-dehydrocycloheximide.
Table 1: 1H (500 MHz) and 13C (125 MHz) NMR data of 1–3 in CD3OD.
| 1 | 2 | 3 | ||||||
|---|---|---|---|---|---|---|---|---|
| position | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | ||
| 1 | 202.6, C | 190.5, C | 192.8, C | |||||
| 2 | 134.2, C | 136.2, C | 137.5, C | |||||
| 3 | 151.9, CH | 6.65 dq (3.7, 1.5) | 153.4, CH | 6.79 dq (3.6, 1.3) | 152.3, CH | 6.71 dq (3.2, 1.3) | ||
| 4 | 31.8, CH | 2.59 m | 32.3, CH | 2.59 m | 32.2, CH | 2.27 m | ||
| 5 | 31.7, CH2 |
eq 2.16 dtd (12.7, 4.3, 1.8)
ax 1.39 ddd (14.4, 12.7, 11.3) |
34.6, CH2 |
β 2.86 dd (14.3, 5.1)
α 2.30 m |
43.3, CH2 |
β 2.74 dd (13.4,5.1)
α 2.34 m |
||
| 6 | 52.2, CH | 2.54 dt (14.4, 4.3) | 136.1, C | 136.2, C | ||||
| 7 | 14.5, CH3 | 1.72 dd (2.4, 1.4) | 16.4, CH3 | 1.80 t (1.6) | 16.1, CH3 | 1.76 t (1.6) | ||
| 8 | 20.3, CH3 | 1.16 d (7.2) | 21.0, CH3 | 1.14 d (7.9) | 20.5, CH3 | 1.13 d (7.2) | ||
| α | 67.3, CH | 4.26 ddd (10.4, 4.3, 2.1) | 134.6, CH | 6.59 tt (7.6, 1.7) | 137.1, CH | 5.86 tt (7.7, 1.4) | ||
| β | 37.2, CH2 |
1.58 ddd (14.0, 10.4, 3.6)
1.33 ddd (14.0, 8.2, 2.1) |
33.4, CH2 | 2.31–2.42 m | 34.5, CH2 | 2.56 m | ||
| 2' | 175.7, C | 175.2, C | 175.4, C | |||||
| 3' | 38.0, CH2 |
2.69 m
2.39 m |
38.1, CH2 |
2.67 m
2.31–2.42 m |
38.2, CH2 |
2.60–2.66 m
2.31–2.42 m |
||
| 4' | 27.4, CH | 2.36 m | 31.6, CH | 2.31–2.42 m | 33.8, CH | 2.60–2.66 m | ||
| 5' | 36.5, CH2 |
2.79 m
2.37 m |
38.2, CH2 |
2.60–2.66 m
2.31–2.42 m |
38.3, CH2 |
2.60–2.66 m
2.31–2.42 m |
||
| 6' | 175.5, C | 175.2, C | 175.4, C | |||||
The relative configuration of the cyclohexenone ring in 1 was assigned by analysis of the 1H NMR proton coupling constants (Table 1) and NOESY correlations. The large proton coupling constants, JH6/H5ax = 14.4 Hz and JH5ax/H4 = 11.3 Hz, suggested that H6 and H4 were in pseudo-axial positions and on the same face of the cyclohexenone ring. This was further corroborated by the presence of strong NOE correlations of H3-8 with both H5ax and H5eq and of H5eq with both H6 and H4 in the NOESY spectrum (Figure 2). It was also in agreement with no NOE correlation being detected between H5ax and H6 or H4. However, straightforward analysis of the 1H NMR and NOESY spectra was unable to assign the relative configuration of Cα due to its location on the flexible side chain. In order to clarify the relative configuration of this chiral carbon, theoretical conformational analysis was carried out on two possible stereoisomers, (4R,6R,αS)-1 and (4R,6R,αR)-1. MMFF conformational search and subsequent geometry optimization using the DFT-D3 method at the B3LYP-D3/6-31G(d) level followed by a higher level of energy calculations at the B3LYP-D3/def2-TZVP level afforded 14 and 11 distinctive low-energy conformers (ΔG < 2.5 kcal/mol) for (4R,6R,αS)-1 and (4R,6R,αR)-1, respectively (Table S1, Supporting Information File 1). Among the low-energy conformers of (4R,6R,αS)-1, those having a gauche relationship between H-α/H-6 were found to be greatly dominant (accounting for about 96%) in the equilibrium mixture in MeOH, which could be categorized into two groups, (S)-1a and (S)-1b, based on the torsion angle of H6–C6–Cα–Hα (ΦH6/Hα) being around either −62° or +55° and each group constituted about 48% equilibrium populations. As can be seen in Figure 2, conformers in both (S)-1a and (S)-1b groups, represented by the second lowest energy minimum (S)-1a1 (ΔG = 0.02 kcal/mol) and the global energy minimum (S)-1b1, respectively, matched up well with the proton coupling constant between H6/Hα (J = 4.3 Hz) and the aforementioned NOESY correlations. Furthermore, conformers in group (S)-1a were also consistent with the diagnostic NOE interaction of Hα/H5eq observed in the NOESY spectrum. In turn, (4R,6R,αR)-1 afforded the dominant conformers (accounting for about 82% equilibrium populations), as represented by the global energy minimum (R)-1a1 (Figure 2), having an anti relationship (ΦH6/Hα ≈ 174°) between Hα/H6, which were inconsistent with the small JH6/Hα value (4.3 Hz) and the absence of an NOE correlation between Hα/H5ax in the experimental spectra. Accordingly, the relative configuration of 1 was assigned to be 4R*,6R*,αS*. In order to determine the absolute configuration, the low-energy conformers of both stereoisomers were subjected to TDDFT calculations of the electronic circular dichroism (ECD) spectra. As shown in Figure 3, the calculated ECD spectra of (4R,6R,αS)-1 and (4R,6R,αR)-1 were similar to one another and both in good agreement with the measured spectrum of 1, which indicated a R configuration of both C4 and C6 but gave no evidence for any of Cα. For the latter, the S configuration was assigned according to the relative configuration as deduced from above conformational analysis. Therefore, compound 1 was defined as 2,3-dehydro-α-epi-isocycloheximide.
![[1860-5397-13-103-2]](/bjoc/content/figures/1860-5397-13-103-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Representatives of the theoretical dominant conformers of (4R,6R,αS)-1 ((S)-1a1 and (S)-1b1) and (4R,6R,αR)-1 ((R)-1a1) in equilibrium populations (in MeOH) and key NOESY correlations (dashed arrows) of 1.
Figure 2: Representatives of the theoretical dominant conformers of (4R,6R,αS)-1 ((S)-1a1 and (S)-1b1) and (4R...
![[1860-5397-13-103-3]](/bjoc/content/figures/1860-5397-13-103-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Comparison of the experimental ECD spectrum of 1 with the M11/TZVP calculated spectra of (4R,6R,αS)-1 and (4R,6R,αR)-1 in MeOH (σ = 0.38 eV for both; shift = +15 and +10 nm, scaling factor = 0.50 and 1.0, respectively).
Figure 3: Comparison of the experimental ECD spectrum of 1 with the M11/TZVP calculated spectra of (4R,6R,αS)-...
Compounds 2 and 3 were obtained as a 7:3 equilibrium mixture. They could be separated by reverse phase HPLC using aqueous MeOH, but readily inter-converted and reached equilibrium in a couple of days at room temperature (in MeOH). The mixture gave a [M + Na]+ ion peak at m/z 284.1255 in the HRESI(+)-MS, appropriate for a molecular formula of C15H19NO3, having one H2O unit less than that of 1. The 1D NMR spectra of the mixture contained two sets of signals in the ratio of 7:3 (according to the 1H NMR proton integral values), corresponding to two isomeric molecules 2 and 3 (Table 1), respectively. Each set of proton and carbon signals resembled those of compound 1 except for replacement of the resonances for the hydroxymethine at Cα and the aliphatic methine at C6 in 1 by the signals for an additional trisubstituted olefin [δH 6.59; δC134.6 (CH), 136.1 (C) in 2 and δH 5.86; δC137.1 (CH), 136.2 (C) in 3], suggesting a gross structure of an anhydrous derivative of 1. This was supported by the HMBC correlations of the olefinic proton Hα with C1 (δC 190.5 in 2, 192.8 in 3), C4' (δC 31.6 in 2, 33.8 in 3), and C5 (2, δC 34.6 in 2, 43.3 in 3). The structural difference between 2 and 3 was identified in the NOESY spectrum. The presence of a strong NOE correlation between the minor Hα (δH 5.86) and H5β (δH 2.74) was observed while the same correlation between the major counterparts [δH 6.59 (Hα), 2.86 (H5β)] could not be found in the spectrum, revealing the geometrical configurations of the double bond between C6/Cα were E in 2 and Z in 3. In order to assign their absolute configurations, (R)-2 and (R)-3 were separately subjected to ECD/TDDFT calculations. As can be seen in Figure 4, the calculated ECD curve of (R)-2 agreed well with the experimental spectrum of the mixture, whereas, that of (R)-3 was largely inconsistent with the measured spectrum. Nevertheless, the summed ECD spectrum of (R)-2 and (R)-3 (7:3) showed the best fit with the experimental spectrum, indicating the R configuration of C4 in both compounds. Therefore, the structures of 2 and 3 were elucidated as (E)- and (Z)-2,3-dehydroanhydrocycloheximides, respectively. It is noted that a compound of unspecified absolute configuration was recently reported to possess the same planar structure as 3 [11].
![[1860-5397-13-103-4]](/bjoc/content/figures/1860-5397-13-103-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Comparison of the experimental ECD spectrum with the BH&HLYP/TZVP calculated spectra of the mixture of (R)-2/3 (7:3) and individual (R)-2 and (R)-3 in MeOH (σ = 0.38 eV, shift = +18 nm, scaling factor = 0.44, 0.14, and 0.13, respectively).
Figure 4: Comparison of the experimental ECD spectrum with the BH&HLYP/TZVP calculated spectra of the mixture...
Photoinduced interconversion between 2 and 3
When elucidating the structures of 2 and 3, we understood that the interconversion between 2 and 3 is likely a photochemical process as it is known that most Z/E isomerizations of carbon double bonds in conjugated olefins and α,β-enones are facilitated by this process [12-14]. However, as all experiments in the present study were carried out either in the dark (fermentation) or under indoor light (extraction and fractionation), with neither direct sunlight shining into nor any artificial high-energy light being applied in the laboratory, we doubted if the fast conversion was induced by our laboratory indoor light. Thus, compounds 2 and 3 in the mixture were separated by preparative HPLC and their MeOH solutions were exposed to the natural indoor light at room temperature and subjected to UPLC analysis every 12 h. As a result, the interconversion between 2 and 3 was found to occur before 12 h and reach equilibrium (estimated ratios between 2/3 were 52:48 in 2 and 53:47 in 3 according to peak area intensities) at 36 h (Figure 5), whereas no changes were detected for the solutions kept in the dark (Figure 5), confirming the interconversion is a photoinduced geometrical isomerization.
![[1860-5397-13-103-5]](/bjoc/content/figures/1860-5397-13-103-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: UPLC analysis of photoreaction products of 2 (around tR = 7.5 min) and 3 (around tR = 11.5 min).
Figure 5: UPLC analysis of photoreaction products of 2 (around tR = 7.5 min) and 3 (around tR = 11.5 min).
Compounds 2 and 3 possess a chiral 6-ethylidene-2-cyclohexenone chromophore. To the best of our knowledge, Z/E photoisomerizations of compounds with this kind of chromophore had been rarely reported. For better understanding the insights into this reaction, we conducted theoretical investigation using the truncated structures 2a and 3a (Figure 1). At first, the lowest-energy geometries of 2a and 3a in MeOH in the ground (S0), first singlet excited (S1), and first triplet excited (T1) states were optimized at the B3LYP/def2-SVP level. Starting from these energy minima, other equilibrium points rotating around the C6–Cα double bond in S0, S1, and T1 states were located by relaxed potential energy surface scans, except for geometries around torsion angles (C1–C6–Cα–Cβ, Φ) of 90° and −90° in the S1 surface, for which optimizations failed to converge. Due to the presence of the stereogenic center C4 in 2a and 3a, the twisted structures around Φ 90º and −90° are diastereomeric in chirality [15]. Thus, two twisted minima, P and P′ (Φ = 77º and −84°, respectively) in the T1 surface and two energy maxima (Φ = 90° and −90°, respectively) in S0 surface were located. The resultant potential energy surfaces of the three states and the geometries of key stationary points are shown in Figure 6. The key bond lengths and torsion angles Φ of these geometries are presented in Table 2. Compound 4 has a structure similar to 2 except for replacement of the C2–C3 double bond by a single bond. Its geometrical isomerization was not observed in the present study, but (E)-2-ethylidenecyclohexanone, which has a similar chromophore to that of 4, was reported to be able to give the Z-isomer upon photoirridiation [16]. For comparison purpose, the truncated structure (4a, Figure 1) of this compound was also calculated using the same methods. The lowest-energy geometries of (E)- and (Z)-4a in the S0, S1 ((E)-4a-S1, (Z)-4a-S1), and T1 (P-4a, P′-4a) surfaces and the two energy maxima ((E)-4a-T1, (Z)-4a-T1) in the T1 surface were optimized and their key geometrical parameters are also listed in Table 2. Analysis of geometrical parameters of key points in the excited and ground states (Table 2) showed that the S1 and T1 minima of 2a/3a correspond to stable 1(n–π*) and 3(π–π*) states of α,β-enones [14], respectively, and the T1 planar maxima, with increased the C1–O1 and C6–Cα bond lengths and a decreased C1–C6 bond length relative to those of T1 minima, show similarity to a T2 π–π* species [14]. Comparison of parameters of geometries in excited states between 2a/3a and 4a revealed that the electron delocalization are extended to C2 and C3 in the S1 minima 2b and 3b and due to this, their C1–C6 bond length is increased by about 0.04 Å while that of C6–Cα is decreased by about 0.03 Å with respect to those in (E)-4a-S1 and (Z)-4a-S1 (Table 2), implying that the presence of C2–C3 double bond in 2a/3a strengthens C6–Cα double bond and increases the rotational barrier of C6–Cα bond in the S1 (n–π*) state. In contrast, no significant differences between 2a/3a and 4a can be found in the T1 (π–π*) states, including the rotational barriers of C6–Cα bond (Table 2).
Table 2: Relative energies and selected geometrical parameters of key stationary points in S0, S1, and T1 potential energy surfaces of 2a/3a and 4a.
| ΔE | bond lengths (Å) | Φ | ||||||
|---|---|---|---|---|---|---|---|---|
| geometry | (kcal/mol) | C1–O1 | C1–C2 | C2–C3 | C1–C6 | C6–Cα | Cα–Cβ | (degrees) |
| 2a | 0.0 | 1.228 | 1.492 | 1.351 | 1.500 | 1.350 | 1.494 | −179.94 |
| 3a | 1.57 | 1.229 | 1.494 | 1.352 | 1.496 | 1.352 | 1.497 | 0.72 |
| 2b | 75.73 | 1.308 | 1.445 | 1.369 | 1.449 | 1.372 | 1.495 | 176.99 |
| 3b | 74.70 | 1.308 | 1.452 | 1.367 | 1.449 | 1.369 | 1.491 | −0.54 |
| P | 54.85 | 1.250 | 1.491 | 1.347 | 1.452 | 1.466 | 1.498 | 76.78 |
| P′ | 54.80 | 1.250 | 1.491 | 1.347 | 1.452 | 1.466 | 1.498 | −83.71 |
| 2c | 61.18 | 1.266 | 1.486 | 1.352 | 1.429 | 1.497 | 1.482 | 177.72 |
| 3c | 62.88 | 1.263 | 1.492 | 1.351 | 1.431 | 1.503 | 1.483 | −1.171 |
| (E)-4a | 0.0 | 1.222 | 1.532 | 1.536 | 1.505 | 1.351 | 1.494 | −179.95 |
| (Z)-4a | 1.40 | 1.222 | 1.533 | 1.540 | 1.501 | 1.351 | 1.497 | 1.04 |
| (E)-4a-S1 | 77.06 | 1.291 | 1.552 | 1.530 | 1.410 | 1.400 | 1.494 | 177.96 |
| (Z)-4a-S1 | 76.10 | 1.292 | 1.557 | 1.530 | 1.412 | 1.396 | 1.490 | 1.41 |
| P-4a | 53.19 | 1.241 | 1.535 | 1.535 | 1.456 | 1.469 | 1.495 | 79.08 |
| P′-4a | 53.19 | 1.241 | 1.535 | 1.535 | 1.456 | 1.469 | 1.495 | 83.24 |
| (E)-4a-T1 | 60.0 | 1.263 | 1.533 | 1.535 | 1.428 | 1.505 | 1.484 | −175.70 |
| (Z)-4a-T1 | 61.0 | 1.254 | 1.539 | 1.533 | 1.436 | 1.505 | 1.486 | 4.30 |
![[1860-5397-13-103-6]](/bjoc/content/figures/1860-5397-13-103-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Potential energy surfaces of 2a/3a in the S0, S1, and T1 states, geometries of key points in the surfaces, and proposed photoreaction path (dashed arrow: vibrational relaxation; wavy arrow: ISC; double headed arrow: energy gap between the two points).
Figure 6: Potential energy surfaces of 2a/3a in the S0, S1, and T1 states, geometries of key points in the su...
In respect to the photoreaction path, it is predicted that the Z/E isomerization of 2a/3a is impossible to take place along the S1 surface due to the presence of large rotational barrier (>25 kcal/mol) in accessing the twisted intermediates (Figure 6). Instead, radiationless relaxations via an intersystem crossing (ISC) from the S1 minima 2b and 3b to the T1 maxima 2c and 3c, respectively, are energetically and symmetrically reasonable [14,17] as well as are preferred according to El-Sayed’s rules [18]. Therefore, the isomerization of 2a/3a, after vertical excitation, is proposed to proceed in the route as shown in Figure 6, which involves radiationless decay from the S1 minima to the triplet manifold via an ISC and then from the T1 twisted minima to the ground state via a second ISC, leading to isomerization of the C6–Cα double bond. This route is generally similar to those proposed for isomerization of the C–C double bond in acyclic α,β-enones [14,17], but more complicated in details. Due to the presence of two T1 minima and two S0 maxima, there are two equivalent points for the second ISC, one around Φ = 90° the other around Φ = −90°, and two points being with slightly different energy gaps. Any of these two ISC points is accessible from either 2c or 3c and can decay to either 2a or 3a (Figure 6).
With the calculated energies, some observed results in experiments can also be explained. The slightly faster reaction of 3 and higher yield of 2 in equilibrium shown in Figure 5 are generally in accordance with the difference in energies between 2a and 3a (Table 2). They may also be related to the energy gaps between 2b/2c and 3b/3c (Figure 6), for which a more efficient ISC is expected for the smaller energy gap [14,17]. The weak reactivity of 4 in photoisomerization (not observed in the present study) can be attributable mainly to its higher vertical excitation energy (87.1 kcal/mol in 4a) relative to those of 2 (81.6 kcal/mol in 2a) and 3 (79.5 kcal/mol in 3a).
Antifungal activity and cytotoxicity
Compounds 1 and 4–6 and the mixture of 2 and 3 were evaluated for the in vitro antifungal activity against Colletotrichum gloeosporioides, Phytophthora infestans, and Saccharomyces cerevisiae using a previously described method [19]. The minimum inhibitory concentration (MIC) data were listed in Table 3. Compounds 5 and 6, as previously reported [20,21], displayed activity against all tested fungal strains and 5 showed a superior activity to 6, whereas compounds 1 and 4 and the mixture of 2 and 3 were found to be inactive (MIC > 100 μg/mL) against all tested strains, which supported the previous finding that the OH-α was important to the antifungal activity of cycloheximide derivatives (4 vs 6) [21,22], and suggested that 2,3-dehydrogenation might result in the loss of the activity (1 vs 5, 6). The cytotoxicity of these isolates against human lung epithelial carcinoma (A549), human cervical epithelial adenocarcinoma (HeLa), and human breast carcinoma (MCF-7) was also evaluated, using MTT assay [23] with doxorubicin as a positive control. As shown in Table 3, the isolates showed a similar activity profile with that of the antifungal activity, e.g., only 5 and 6 were active and others were inactive (>50 μM) toward all the tested cell lines. These findings suggested that these cycloheximide derivatives possibly exert the antifungal and cytotoxic activities via a similar mode of action.
Table 3: Antifungal activity and cytotoxicity of 1–6a.
| antifungal activity (MIC, μg/mL) | cytotoxicity (IC50, μM)b | ||||||
|---|---|---|---|---|---|---|---|
| compound | C. gloeosporioides | P. infestans | S. cerevisiae | A549 | Hela | MCF-7 | |
| 5 | 25.0 | 25.0 | 3.1 | 15.8 ± 3.4 | 15.6 ± 3.6 | 18.1 ± 0.2 | |
| 6 | 50.0 | 50.0 | 6.3 | 30.0 ± 3.8 | 21.9 ± 2.9 | 21.9 ± 2.9 | |
| doxorubicin | 0.12 ± 0.02 | 0.31 ± 0.02 | 1.00 ± 0.20 | ||||
aCompounds 1 and 4 and the mixture of 2 and 3 being inactive against all tested fungi (MIC > 100 μg/mL) and tumor cells (IC50 > 50 μM), are not listed. bValues represent means ± SD based on three individual experiments.
Conclusion
Three new (1–3) and three known (4–6) cycloheximide congeners were obtained from the cultures of Streptomyces sp. SC0581. The structure elucidation of the new compounds were achieved by spectroscopic analysis in combination with theoretical conformational analysis and ECD simulations, in which theoretical computations were shown to play a key role in solving challenges in assignments of relative and absolute configurations. Analysis of the antifungal and cancer cell toxic activity data of 1–6 suggested that change of the C2–C3 single bond to a double bond can lead to the loss of the activities and supported the OH-α is important to the activities of cycloheximide congeners. Furthermore, compounds 2 and 3, with a chiral 6-ethylidene-2-cyclohexenone chromophore, were found to undergo E/Z photoisomerization under the indoor light. Theoretical investigation showed the presence of the C2–C3 double bond in 2 and 3 strengthens the C6–Cα double bond and increases the rotational barrier of the C6–Cα bond in the S1 (n–π*) states due to the extended electron delocalization, whereas it scarcely affects the T1 (π–π*) states, compared to those in 4. It also revealed a photoisomerization route similar to that commonly found in acyclic α,β-enones [14,17], except for the presence of two equivalent points (around Φ 90° and −90°, respectively) for the ISC from T1 to S0 state, arising from the chiral nature of molecules.
Experimental
General experimental procedures
Optical rotations were recorded in MeOH on a Perkin-Elmer 343 spectropolarimeter. UV spectra and ECD spectra were obtained simultaneously on a Chirascan CD spectrometer (Applied Photophysics Ltd., England) using MeOH as solvent. 1H NMR, 13C NMR, and 2D NMR data were recorded on a Bruker Avance III 500 MHz spectrometer with TMS as internal standard. HRESIMS data were recorded on a Bruker maXis Q-TOF spectrometer. Preparative HPLC were carried out with a Shimadzu Shim-packed Pro-ODS column (20 mm × 25 cm) equipped with a Shimadzu LC-6AD pump and a Shimadzu RID-10A refractive index detector. UPLC analysis was performed on an Acquity H-Class UPLC system consisting of a quaternary solvent delivery system, an auto-sampler, and a DAD detector. For column chromatography, silica gel 60 (100–200 mesh, Qingdao Marine Chemical Ltd., Qingdao, People's Republic of China), YMC ODS (75 μm, YMC Co. Ltd., Kyoto, Japan) and Sephadex LH-20 (GE Healthcare, Uppsala, Sweden) were used. Analytical TLC were performed on HSGF254 silica gel plates (0.2 mm, Yantai Jiangyou silica gel Development Co. Ltd., Yantai, China); spots were visualized after spraying with 10% H2SO4 solution followed by heating.
Biological material
Streptomyces sp. SC0581, isolated from a soil sample collected from Dinghu Mountain Biosphere Reserve, Guangdong, People's Republic of China, was identified according to morphological characteristics and sequence analysis of the ITS region (GenBank accession no. KX687558). A reference strain maintained at −80 °C was deposited in the culture collection of South China Botanical Garden, Chinese Academy of Sciences, Guangzhou, People's Republic of China.
Fermentation, extraction and isolation
The strain was grown on the PDA medium at 28 °C for 10 days and 6 pieces of PDA culture plugs of the strain were inoculated into each of two 500 mL Erlenmeyer flasks containing 150 mL of seed medium (glucose 0.4%, malt extract 1.0%, yeast extract 0.4%, pH 5.5) and shaken on a rotatory (150 rpm) at 28 °C for 2 days. Then, the cultures were transferred into three 3 L flasks containing 1 L of seed medium and cultivated at the same culture conditions. Finally, 10 mL each of the culture broth was transferred into two hundred 500 mL flasks containing 60 mL of YMG medium and 60 g of wheat grains, and the fermentation was carried out stably in the dark at 28 °C for 40 days. The obtained solid fermentation cultures were extracted with 95% EtOH for three times and concentrated in vacuum. The resultant extract was successively partitioned between petroleum ether, EtOAc, and n-BuOH. The EtOAc and n-BuOH soluble fractions, showing the activity against P. infestans, were combined and subjected to silica gel column chromatography (CC) eluted with CHCl3/MeOH mixtures of increasing polarity (100:0 to 70:30) to give twenty fractions including two antifungal fractions, Fr. 1 (CHCl3/MeOH, 95:5) and Fr. 6 (CHCl3/MeOH, 90:10). Fr. 1 (6.0 g) was further separated by ODS CC using aqueous MeOH (10–90%) to obtain five subfractions (Frs. 1A–1E). Fr. 1B (40% MeOH) was purified by preparative HPLC using isocratic elution of aqueous CH3CN (20%) at a flow rate of 5 mL/min to afford 5 (9 mg, tR = 80.4 min), 6 (5 mg, tR = 60.0 min), and the mixture of 2 and 3 (4 mg, 2, tR = 105.5 min, 3, tR = 180.2 min). Fr. 1C (50% MeOH) was separated by preparative HPLC using 40% MeOH at 5 mL/min to yield 4 (15 mg, tR =110.8 min). Fr. 6 (4.4 g) was separated by ODS CC using aqueous MeOH (10–90%) followed by preparative HPLC using 40% MeOH at 5 mL/min to give 1 (10 mg, tR = 108.6 min).
2,3-Dehydro-α-epiisocycloheximide (1): Colorless oil; [α]D25 +1.8 (c 0.20, MeOH); UV (MeOH) λmax (log ε) 201 (3.40), 237 (3.14); CD (MeOH) λ(∆ε) 203 (−5.06), 237 (+1.62), 344 (−0.85); 1H and 13C NMR data, see Table 1; HRESIMS (m/z): [M + Na]+ calcd for C15H21NNaO4, 302.1363; found: 302.1357.
Mixture of (E)-2,3-dehydroanhydrocycloheximide (2) and (Z)-2,3-dehydroanhydrocycloheximide (3): Colorless oil; [α]D25 +25.5 (c 0.25, MeOH); UV (MeOH) λmax (log ε) 204 (4.04), 244 (3.58), 272 (3.49); CD (MeOH) λ(∆ε) 271 (+1.58); 1H and 13C NMR data, see Table 1; HRESIMS (m/z): [M + Na]+ calcd for C15H19NNaO3, 284.1257; found: 284.1255.
Evaluation of in vitro antifungal activity
Saccharomyces cerevisiae, Colletotrichum gloeosporioides, and Phytophthora infestans were used as the test fungal strains. The antifungal activity was evaluated using the microplate Alamar Blue assay (MABA) [19]. For the filamentous fungi, C. gloeosporioides and P. infestans, the strains were grown on PDA cultures for 7 days. The spore suspensions were harvested by flooding the colony with PDB medium, rubbing the surface with a sterile scraper, and filtering with four layers of gauze. The spore suspensions were subjected to quantification with a hemocytometer and adjusted with PDB medium to 1 × 105 CFU/mL for the microplate Alamar Blue assay. Test compounds were diluted with the DMSO to give two-fold gradient concentrations. 100 μL of spore suspension of each strain containing Alamar Blue (8%, v/v) and the compound solution (4%, v/v), was added into 96-well microtiter plate in triplicate. The final concentrations of tested compounds were 100, 50, 25, 12.5, 6.25 and 3.125 μg/mL. To negative control wells were added DMSO instead of the test compound, and blank control wells contained Alamar Blue but without spore suspension. The plate was incubated in the dark at 28 °C for 6–8 hours. When the color of negative control wells switched from blue to red, the final concentration of the well which was closest to the red one and remained blue was received as the minimal inhibitory concentration (MIC). With regard to the S. cerevisiae, the strain was incubated in PDB medium on a rotary shaker at 150 rpm in 28 °C for 12 hours. The suspensions of the strain were quantified with a hemocytometer and adjusted with PDB medium to 1 × 104 CFU/mL. The next steps of microplate Alamar Blue assay for S. cerevisiae were the same as mentioned above for the two filamentous fungi.
Evaluation of tumor cell toxicity
The evaluation was conducted as previously described [23].
Photoinduced interconversion between 2 and 3
The mixture of 2 and 3 (7:3) was separated by a SPOLAR C18 column (4.6 × 250 mm, 5 μm) using 35% MeOH at a flow rate of 1 mL/min, yielding compounds 2 (tR = 19.3 min) and 3 (tR = 28.5 min) in pure form. The whole separation process was performed in the dark. Then, two aliquots of MeOH solutions were prepared for each of 2 and 3. One aliquot of solution was placed under the natural indoor light (illumination ranging between 155–315 lux) at room temperature and the other was kept in the dark. The two aliquots of solutions were subjected to UPLC analysis every 12 hours on an Acquity UPLC BEH C18 column (2.1 × 100 mm, 1.7 μm) using 35% MeOH as mobile phase at a flow rate of 0.3 mL/min. The column temperature was controlled at 40 °C and detection wavelength was set at 280 nm.
Computational details
Molecular Merck force field (MMFF) calculations were done using the Spartan'14 program (Wavefunction Inc., Irvine, CA, USA). Density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations were performed with the Gaussian 09 program package [24]. For computations of ECD spectra, the conformers generated by a MMFF conformational search in an energy window of 10 kcal/mol were subjected to geometry optimization using the dispersion-corrected DFT-D3 method [25] at the B3LYP-D3/6-31G(d) level. Frequency calculations were carried out on those optimized conformers with relative energies (ΔE) less than 4.5 kcal/mol using the same level to verify that they were true minima and to estimate their relative thermal free energies (ΔG) at 298.15 K. The more accurate energies of these conformers in MeOH were obtained with the B3LYP-D3/def2-TZVP method. Solvent effects were taken into account by using polarizable continuum model (PCM). The TDDFT calculations were performed using the hybrid BHandHLYP [26], M11 [27], and/or PBE1PBE [28,29] functionals, and Ahlrichs’ basis set TZVP (triple zeta valence plus) polarization [30]. The number of excited states per each molecule was 24. The ECD spectra were generated by the program SpecDis [31] using a Gaussian band shape from dipole-length dipolar and rotational strengths. Equilibrium population of each conformer at 298.15 K was calculated from its relative free energies using Boltzmann statistics. The calculated spectra were generated from the low-energy conformers according to the Boltzmann weighting of each conformer in MeOH solution.
For theoretical investigations on the photoinduced interconversion between 2 and 3, the truncated structures 2a and 3a (Figure 1) were used. Geometries of 2a and 3a in MeOH solution in S0, S1, and T1 states were optimized by DFT (for S0 and T1) or TDDFT (for S1 only) calculations at the B3LYP/def2-SVP level. For diradical triplets, the spin-unrestricted formalism was used. Solvent effects were treated using PCM. To build 3D conformers of 2a and 3a, the global energy minima of 2 and 3, obtained in above ECD computations, were used and their glutarimide ring was replaced by a hydrogen atom. The built conformers were subjected to geometry optimizations to obtain the absolute energy minima of 2a and 3a in the S0, S1, and T1 states. Starting from these minima, other equilibrium points rotating around the C6–Cα double bond in surfaces of the three states were located by relaxed potential energy surface scans, except for those around Φ 90° and −90° in the S1 surface, for which optimizations failed to converge. Frequency calculations were also carried out on the significant points, including geometries 2a–2c, 3a–3c, P, and P′ and verified that they were either true minima or maxima (2c and 3c only). Structures (E)- and (Z)-4a were calculated with the same method and their stable energy minima in the S0, S1, and T1 states and T1 maxima were optimized (Table 2).
Supporting Information
| Supporting Information File 1: HRESIMS, 1D and 2D NMR spectra of compounds 1–3; energies, populations, and key torsion angles of low-energy conformers of compounds 1–3 by theoretical computations. | ||
| Format: PDF | Size: 936.0 KB | Download |
Acknowledgements
We thank Mr. Yunfei Yuan, South China Botanical Garden, Chinese Academy of Sciences, for NMR spectroscopic measurements, and Ms. Aijun Sun, South China Sea Institute of Oceanology, Chinese Academy of Sciences, for HRESIMS measurements. We gratefully acknowledge support from the Guangzhou Branch of the Supercomputing Center of Chinese Academy of Sciences. This study was supported by an NSFC grant (No. 81172942) and a research project from the Bureau of Science and Technology of Guangzhou Municipality (grant no. 201510010015).
References
-
Ennis, H. L.; Lubin, M. Science 1964, 146, 1474–1476. doi:10.1126/science.146.3650.1474
Return to citation in text: [1] -
Obrig, T. G.; Culp, W. J.; McKeehan, W. L.; Hardesty, B. J. Biol. Chem. 1971, 246, 174–181.
Return to citation in text: [1] -
Schneider-Poetsch, T.; Ju, J.; Eyler, D. E.; Dang, Y.; Bhat, S.; Merrick, W. C.; Green, R.; Shen, B.; Liu, J. O. Nat. Chem. Biol. 2010, 6, 209–217. doi:10.1038/nchembio.304
Return to citation in text: [1] -
Nakae, K.; Yoshimoto, Y.; Sawa, T.; Homma, Y.; Hamada, M.; Takeuch, T.; Imoto, M. J. Antibiot. 2000, 53, 1130–1136. doi:10.7164/antibiotics.53.1130
Return to citation in text: [1] -
Saito, N.; Suzuki, F.; Sasaki, K.; Ishida, N. Antimicrob. Agents Chemother. 1976, 10, 14–19. doi:10.1128/AAC.10.1.14
Return to citation in text: [1] -
Guo, H.-f.; Li, Y.-h.; Yi, H.; Zhang, T.; Wang, S.-q.; Tao, P.-z.; Li, Z.-r. J. Antibiot. 2009, 62, 639–642. doi:10.1038/ja.2009.87
Return to citation in text: [1] [2] -
Lawana, V.; Korrapati, M. C.; Mehendale, H. M. Cycloheximide. In Encyclopedia of Toxicology, 3rd ed.; Wexler, P., Ed.; Academic Press: Oxford, 2014; pp 1103–1105. doi:10.1016/B978-0-12-386454-3.00298-0
Return to citation in text: [1] -
Xue, J.; Wu, P.; Xu, L.; Wei, X. Org. Lett. 2014, 16, 1518–1521. doi:10.1021/ol500418f
Return to citation in text: [1] -
Wu, P.; Xue, J.; Yao, L.; Xu, L.; Li, H.; Wei, X. Org. Lett. 2015, 17, 4922–4925. doi:10.1021/acs.orglett.5b02536
Return to citation in text: [1] -
Jeffs, P. W.; McWilliams, D. J. Am. Chem. Soc. 1981, 103, 6185–6192. doi:10.1021/ja00410a034
Return to citation in text: [1] -
Yang, D.; Xu, Q. Y.; Deng, X. M.; Song, S. Y.; Hu, Z. Y.; Zheng, Z. H. Rec. Nat. Prod. 2015, 9, 336–341.
Return to citation in text: [1] -
Liu, R. S. H.; Hammond, G. S. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 11153–11158. doi:10.1073/pnas.210323197
Return to citation in text: [1] -
Tsuno, T.; Yoshida, M.; Iwata, T.; Sugiyama, K. Tetrahedron 2002, 58, 7681–7689. doi:10.1016/S0040-4020(02)00856-6
Return to citation in text: [1] -
Reguero, M.; Olivucci, M.; Bernardi, F.; Robb, M. A. J. Am. Chem. Soc. 1994, 116, 2103–2114. doi:10.1021/ja00084a056
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
García-Expósito, E.; González-Moreno, R.; Martín-Vilà, M.; Muray, E.; Rifé, J.; Bourdelande, J. L.; Branchadell, V.; Ortuño, R. M. J. Org. Chem. 2000, 65, 6958–6965. doi:10.1021/jo000549g
Return to citation in text: [1] -
Schneider, R. A.; Meinwald, J. J. Am. Chem. Soc. 1967, 89, 2023–2032. doi:10.1021/ja00985a011
Return to citation in text: [1] -
Marian, C. M. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 187–203. doi:10.1002/wcms.83
Return to citation in text: [1] [2] [3] [4] -
El-Sayed, M. A. J. Chem. Phys. 1963, 38, 2834–2838. doi:10.1063/1.1733610
Return to citation in text: [1] -
Cox, K. D.; Quello, K.; Deford, R. J.; Beckerman, J. L. Plant Dis. 2009, 93, 328–331. doi:10.1094/pdis-93-4-0328
Return to citation in text: [1] [2] -
Wang, J.; He, W.; Huang, X.; Tian, X.; Liao, S.; Yang, B.; Wang, F.; Zhou, X.; Liu, Y. J. Agric. Food Chem. 2016, 64, 2910–2916. doi:10.1021/acs.jafc.6b00527
Return to citation in text: [1] -
Huang, S.-X.; Yu, Z.; Robert, F.; Zhao, L.-X.; Jiang, Y.; Duan, Y.; Pelletier, J.; Shen, B. J. Antibiot. 2011, 64, 163–166. doi:10.1038/ja.2010.150
Return to citation in text: [1] [2] -
Ennis, H. L. Biochem. Pharmacol. 1968, 17, 1197–1206. doi:10.1016/0006-2952(68)90056-7
Return to citation in text: [1] -
Shi, J.-F.; Wu, P.; Jiang, Z.-H.; Wei, X.-Y. Eur. J. Med. Chem. 2014, 71, 219–228. doi:10.1016/j.ejmech.2013.11.012
Return to citation in text: [1] [2] -
Gaussian 09, revision C.01; Gaussian, Inc.: Wallingford CT, 2010.
Return to citation in text: [1] -
Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104. doi:10.1063/1.3382344
Return to citation in text: [1] -
Gaussian 92/DFT, Revision F.2 ed.; Gaussian, Inc.: Pittsburgh PA, 1993.
Return to citation in text: [1] -
Peverati, R.; Truhlar, D. G. J. Phys. Chem. Lett. 2011, 2, 2810–2817. doi:10.1021/jz201170d
Return to citation in text: [1] -
Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. Rev. Lett. 1996, 77, 3865–3868. doi:10.1103/PhysRevLett.77.3865
Return to citation in text: [1] -
Adamo, C.; Barone, V. J. Chem. Phys. 1999, 110, 6158–6169. doi:10.1063/1.478522
Return to citation in text: [1] -
Schäfer, A.; Huber, C.; Ahlrichs, R. J. Chem. Phys. 1994, 100, 5829–5835. doi:10.1063/1.467146
Return to citation in text: [1] -
Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. Chirality 2013, 25, 243–249. doi:10.1002/chir.22138
Return to citation in text: [1]
| 28. | Perdew, J. P.; Burke, K.; Ernzerhof, M. Phys. Rev. Lett. 1996, 77, 3865–3868. doi:10.1103/PhysRevLett.77.3865 |
| 29. | Adamo, C.; Barone, V. J. Chem. Phys. 1999, 110, 6158–6169. doi:10.1063/1.478522 |
| 30. | Schäfer, A.; Huber, C.; Ahlrichs, R. J. Chem. Phys. 1994, 100, 5829–5835. doi:10.1063/1.467146 |
| 31. | Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. Chirality 2013, 25, 243–249. doi:10.1002/chir.22138 |
| 1. | Ennis, H. L.; Lubin, M. Science 1964, 146, 1474–1476. doi:10.1126/science.146.3650.1474 |
| 2. | Obrig, T. G.; Culp, W. J.; McKeehan, W. L.; Hardesty, B. J. Biol. Chem. 1971, 246, 174–181. |
| 3. | Schneider-Poetsch, T.; Ju, J.; Eyler, D. E.; Dang, Y.; Bhat, S.; Merrick, W. C.; Green, R.; Shen, B.; Liu, J. O. Nat. Chem. Biol. 2010, 6, 209–217. doi:10.1038/nchembio.304 |
| 6. | Guo, H.-f.; Li, Y.-h.; Yi, H.; Zhang, T.; Wang, S.-q.; Tao, P.-z.; Li, Z.-r. J. Antibiot. 2009, 62, 639–642. doi:10.1038/ja.2009.87 |
| 14. | Reguero, M.; Olivucci, M.; Bernardi, F.; Robb, M. A. J. Am. Chem. Soc. 1994, 116, 2103–2114. doi:10.1021/ja00084a056 |
| 17. | Marian, C. M. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 187–203. doi:10.1002/wcms.83 |
| 8. | Xue, J.; Wu, P.; Xu, L.; Wei, X. Org. Lett. 2014, 16, 1518–1521. doi:10.1021/ol500418f |
| 9. | Wu, P.; Xue, J.; Yao, L.; Xu, L.; Li, H.; Wei, X. Org. Lett. 2015, 17, 4922–4925. doi:10.1021/acs.orglett.5b02536 |
| 14. | Reguero, M.; Olivucci, M.; Bernardi, F.; Robb, M. A. J. Am. Chem. Soc. 1994, 116, 2103–2114. doi:10.1021/ja00084a056 |
| 17. | Marian, C. M. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 187–203. doi:10.1002/wcms.83 |
| 7. | Lawana, V.; Korrapati, M. C.; Mehendale, H. M. Cycloheximide. In Encyclopedia of Toxicology, 3rd ed.; Wexler, P., Ed.; Academic Press: Oxford, 2014; pp 1103–1105. doi:10.1016/B978-0-12-386454-3.00298-0 |
| 14. | Reguero, M.; Olivucci, M.; Bernardi, F.; Robb, M. A. J. Am. Chem. Soc. 1994, 116, 2103–2114. doi:10.1021/ja00084a056 |
| 17. | Marian, C. M. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 187–203. doi:10.1002/wcms.83 |
| 4. | Nakae, K.; Yoshimoto, Y.; Sawa, T.; Homma, Y.; Hamada, M.; Takeuch, T.; Imoto, M. J. Antibiot. 2000, 53, 1130–1136. doi:10.7164/antibiotics.53.1130 |
| 5. | Saito, N.; Suzuki, F.; Sasaki, K.; Ishida, N. Antimicrob. Agents Chemother. 1976, 10, 14–19. doi:10.1128/AAC.10.1.14 |
| 6. | Guo, H.-f.; Li, Y.-h.; Yi, H.; Zhang, T.; Wang, S.-q.; Tao, P.-z.; Li, Z.-r. J. Antibiot. 2009, 62, 639–642. doi:10.1038/ja.2009.87 |
| 15. | García-Expósito, E.; González-Moreno, R.; Martín-Vilà, M.; Muray, E.; Rifé, J.; Bourdelande, J. L.; Branchadell, V.; Ortuño, R. M. J. Org. Chem. 2000, 65, 6958–6965. doi:10.1021/jo000549g |
| 14. | Reguero, M.; Olivucci, M.; Bernardi, F.; Robb, M. A. J. Am. Chem. Soc. 1994, 116, 2103–2114. doi:10.1021/ja00084a056 |
| 12. | Liu, R. S. H.; Hammond, G. S. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 11153–11158. doi:10.1073/pnas.210323197 |
| 13. | Tsuno, T.; Yoshida, M.; Iwata, T.; Sugiyama, K. Tetrahedron 2002, 58, 7681–7689. doi:10.1016/S0040-4020(02)00856-6 |
| 14. | Reguero, M.; Olivucci, M.; Bernardi, F.; Robb, M. A. J. Am. Chem. Soc. 1994, 116, 2103–2114. doi:10.1021/ja00084a056 |
| 14. | Reguero, M.; Olivucci, M.; Bernardi, F.; Robb, M. A. J. Am. Chem. Soc. 1994, 116, 2103–2114. doi:10.1021/ja00084a056 |
| 11. | Yang, D.; Xu, Q. Y.; Deng, X. M.; Song, S. Y.; Hu, Z. Y.; Zheng, Z. H. Rec. Nat. Prod. 2015, 9, 336–341. |
| 10. | Jeffs, P. W.; McWilliams, D. J. Am. Chem. Soc. 1981, 103, 6185–6192. doi:10.1021/ja00410a034 |
| 16. | Schneider, R. A.; Meinwald, J. J. Am. Chem. Soc. 1967, 89, 2023–2032. doi:10.1021/ja00985a011 |
| 21. | Huang, S.-X.; Yu, Z.; Robert, F.; Zhao, L.-X.; Jiang, Y.; Duan, Y.; Pelletier, J.; Shen, B. J. Antibiot. 2011, 64, 163–166. doi:10.1038/ja.2010.150 |
| 22. | Ennis, H. L. Biochem. Pharmacol. 1968, 17, 1197–1206. doi:10.1016/0006-2952(68)90056-7 |
| 19. | Cox, K. D.; Quello, K.; Deford, R. J.; Beckerman, J. L. Plant Dis. 2009, 93, 328–331. doi:10.1094/pdis-93-4-0328 |
| 20. | Wang, J.; He, W.; Huang, X.; Tian, X.; Liao, S.; Yang, B.; Wang, F.; Zhou, X.; Liu, Y. J. Agric. Food Chem. 2016, 64, 2910–2916. doi:10.1021/acs.jafc.6b00527 |
| 21. | Huang, S.-X.; Yu, Z.; Robert, F.; Zhao, L.-X.; Jiang, Y.; Duan, Y.; Pelletier, J.; Shen, B. J. Antibiot. 2011, 64, 163–166. doi:10.1038/ja.2010.150 |
| 27. | Peverati, R.; Truhlar, D. G. J. Phys. Chem. Lett. 2011, 2, 2810–2817. doi:10.1021/jz201170d |
| 25. | Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104. doi:10.1063/1.3382344 |
| 19. | Cox, K. D.; Quello, K.; Deford, R. J.; Beckerman, J. L. Plant Dis. 2009, 93, 328–331. doi:10.1094/pdis-93-4-0328 |
| 23. | Shi, J.-F.; Wu, P.; Jiang, Z.-H.; Wei, X.-Y. Eur. J. Med. Chem. 2014, 71, 219–228. doi:10.1016/j.ejmech.2013.11.012 |
| 23. | Shi, J.-F.; Wu, P.; Jiang, Z.-H.; Wei, X.-Y. Eur. J. Med. Chem. 2014, 71, 219–228. doi:10.1016/j.ejmech.2013.11.012 |
| 14. | Reguero, M.; Olivucci, M.; Bernardi, F.; Robb, M. A. J. Am. Chem. Soc. 1994, 116, 2103–2114. doi:10.1021/ja00084a056 |
| 17. | Marian, C. M. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 187–203. doi:10.1002/wcms.83 |
© 2017 Yang et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)