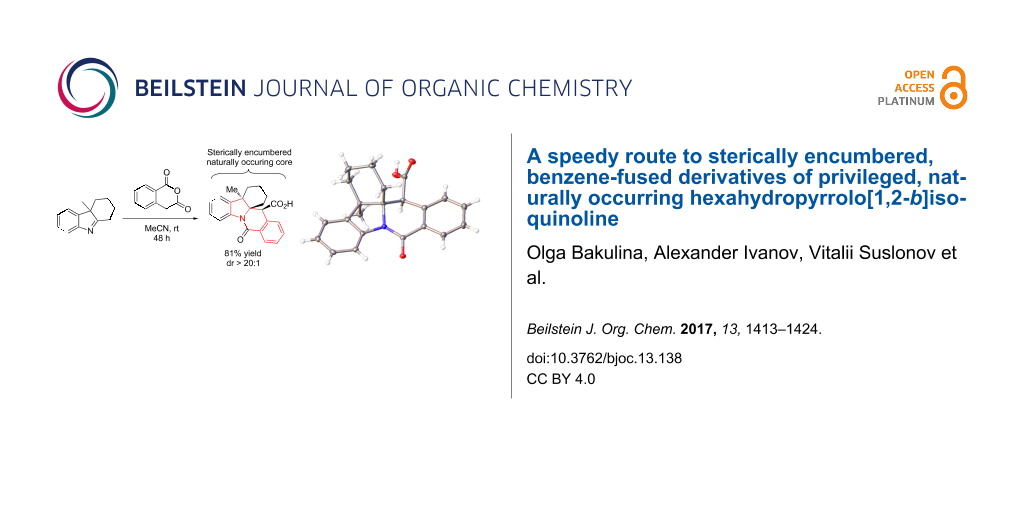
Abstract
A series of 15 benzene-fused hexahydropyrrolo[1,2-b]isoquinolonic acids with substantial degree of steric encumbrance has been prepared via a novel variant of the Castagnoli–Cushman reaction of homophthalic anhydride (HPA) and various indolenines. The employment of a special kind of a cyclic imine component reaction allowed, for the first time, isolating a Mannich-type adduct between HPA and an imine component which has been postulated but never obtained in similar reactions.

Graphical Abstract
Introduction
The reaction of imines (prepared in a separate step or generated in situ) with α-C–H dicarboxylic acid anhydrides (known as the Castagnoli–Cushman reaction or CCR [1]) offers a direct entry into lactam frameworks 1 of various sizes (traditionally, δ- and γ- [2,3] and, more recently, ε-lactams [4,5]) containing a carboxylic acid functionality. Employment of homophthalic anhydride (HPA) in this reaction delivers medicinally important, most often trans-configured [6,7] tetrahydroisoquinolonic acids 2 (Figure 1) which have found utility as probe compounds or therapeutic agents in diverse areas such as neuroprotection [8], diabetes [9], and cancer [10].
![[1860-5397-13-138-1]](/bjoc/content/figures/1860-5397-13-138-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The Castagnoli–Cushman reaction (CCR).
Figure 1: The Castagnoli–Cushman reaction (CCR).
The use of cyclic imines (or surrogates thereof such as isoquinoline [11]) in the CCR is quite scarce in the literature [12-14]. As an example, 1-pyrroline (in the form of its trimer 3) has been reported by Smith and co-workers [15-17] to condense efficiently with HPA analogs to deliver hexahydropyrrolo[1,2-b]isoquinolones 4. The hexahydropyrrolo[1,2-b]isoquinoline core in general is ubiquitous to many natural products exemplified by tylophorine (5) [18], lycorine (6) [19] and its entire family of alkaloids, including zephyranthine (7) [20] and galantine (8) [21]. Considering the plethora of biological activities displayed by the lycorine and tylophorine alkaloids (such as pro-apoptotic [22], antiviral [23], hypoxia-inducible factor-1 inhibitory [24]), the scaffold can be confidently regarded as privileged [25]. Recently, we [26] and others [27] reported the use of indolenines as non-classical inputs for the Joullié–Ugi reaction and for subsequent preparation [28] of sterically encumbered, constrained peptidomimetic frameworks. Diversely substituted indolenines 9 are easy to prepare via the Fischer indole synthesis [26] and their use in the CCR can be expected to result in hexahydropyrrolo[1,2-b]isoquinolone derivatives fused with benzene 10 that have pronounced three-dimensional features and potentially contain several quaternary carbon centers (Figure 2). The first aspect has been recently recognized [29] as a central principle in drug design ensuring effective interaction of small molecules with protein targets and lower off-target effects. The presence of quaternary carbons is characteristic of the natural products domain and is also gaining prominence in medicinal chemistry [30]. Herein, we disclose the results obtained and observations made in the course of our attempt to involve 9 in reactions with HPA. Notably, due to its non-planar tetracyclic character, the hexahydropyrrolo[1,2-b]isoquinolone fused with benzene scaffold (present in 10) clearly appears related to (though topologically distinct from) the natural and synthetic camptothecin-like topoisomerase inhibitors [31].
![[1860-5397-13-138-2]](/bjoc/content/figures/1860-5397-13-138-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Assembly of hexahydropyrrolo[1,2-b]isoquinoline core via the CCR and its occurrence in natural and synthetic bioactive compounds.
Figure 2: Assembly of hexahydropyrrolo[1,2-b]isoquinoline core via the CCR and its occurrence in natural and ...
Results and Discussion
The study commenced with the synthesis of set of 2-H as well as 2-substituted indolenines 9a–t using a previously published procedure (Figure 3) [26]. Acetonitrile was previously found [32] to be an effective solvent promoting the CCR of HPA with acyclic amines at room temperature. As it also facilitated the isolation of the tetrahydroisoquinolonic acid product, it was chosen to study the CCR of 9a–t. The latter tend to precipitate in a pure form (often as a single diastereomer) from the reaction mixture and can be conveniently isolated by filtration. This prior observation also held true for the reaction between HPA and most of indolenines 9.
![[1860-5397-13-138-3]](/bjoc/content/figures/1860-5397-13-138-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Indolenine substrates 9a–t investigated in this work. aPrepared for the first time (the rest are known compounds, see Experimental section). bUnstable to isolation; was taken in the CCR step as a solution in chloroform.
Figure 3: Indolenine substrates 9a–t investigated in this work. aPrepared for the first time (the rest are kn...
As shown in Table 1, all of the 2-unsubstituted indolenines 9a–g and many 2-substituted one 9h–o furnished the expected respective tetracyclic tetrahydroisoquinolonic acids 10 on treatment with HPA (1.0 equiv) in acetonitrile (2 mL/mmol). Notably, when the CCR with HPA was repeated for 9h in other solvents (toluene, chloroform or DMF) at room temperature, this resulted in a similar product yield and diastereomeric ratio. However, the isolation of 10h from the respective reaction mixtures was distinctly cumbersome, which only confirmed acetonitrile to be the solvent of choice for these reactions.
Table 1: Indolo[1,2-b]isoquinolonic acids 10 obtained via the CCR of indolenines 9.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-138-i7.svg?max-width=637&scale=1.0)
|
|||||
| Entry | 9 | Product 10 | Time | Isolated yield, %a | anti/syn |
|---|---|---|---|---|---|
| 1 | 9a |
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-138-i8.svg?max-width=637&scale=1.0)
10a |
2 h | 56 | >20:1b |
| 2 | 9b |
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-138-i9.svg?max-width=637&scale=1.0)
10b |
2 h | 73 | >20:1b |
| 3 | 9c |
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-138-i10.svg?max-width=637&scale=1.0)
10c |
24 h | 66 | 3.3:1c |
| 4 | 9d |
![[Graphic 5]](/bjoc/content/inline/1860-5397-13-138-i11.svg?max-width=637&scale=1.0)
10d |
3 h | 84 | >20:1 |
| 5 | 9e |
![[Graphic 6]](/bjoc/content/inline/1860-5397-13-138-i12.svg?max-width=637&scale=1.0)
10e |
48 h | 66 | 0.8:1d |
| 6 | 9f |
![[Graphic 7]](/bjoc/content/inline/1860-5397-13-138-i13.svg?max-width=637&scale=1.0)
10f |
72 h | 79 | 2:1d |
| 7 | 9g |
![[Graphic 8]](/bjoc/content/inline/1860-5397-13-138-i14.svg?max-width=637&scale=1.0)
10g‘ |
16 h | 87 | 1.4:1d |
| 8 | 9h |
![[Graphic 9]](/bjoc/content/inline/1860-5397-13-138-i15.svg?max-width=637&scale=1.0)
10h |
16 h | 86 | 4.3:1c |
| 9 | 9i |
![[Graphic 10]](/bjoc/content/inline/1860-5397-13-138-i16.svg?max-width=637&scale=1.0)
10i |
48 h | 72 | 3:1d |
| 10 | 9j |
![[Graphic 11]](/bjoc/content/inline/1860-5397-13-138-i17.svg?max-width=637&scale=1.0)
10j |
48 h | 73 | >20:1 |
| 11 | 9k |
![[Graphic 12]](/bjoc/content/inline/1860-5397-13-138-i18.svg?max-width=637&scale=1.0)
10k |
48 h | 57 | 6.5:1c |
| 12 | 9l |
![[Graphic 13]](/bjoc/content/inline/1860-5397-13-138-i19.svg?max-width=637&scale=1.0)
10l |
48 h | 81 | >20:1 |
| 13 | 9m |
![[Graphic 14]](/bjoc/content/inline/1860-5397-13-138-i20.svg?max-width=637&scale=1.0)
10m |
72 h | 79 | 6:1c |
| 14 | 9n |
![[Graphic 15]](/bjoc/content/inline/1860-5397-13-138-i21.svg?max-width=637&scale=1.0)
10n |
25 d | 93 | >20:1 |
| 15 | 9o |
![[Graphic 16]](/bjoc/content/inline/1860-5397-13-138-i22.svg?max-width=637&scale=1.0)
10o |
50 d | 52 | 11:1 |
| 16 | 9p | – | 7 d | 0 | – |
| 17 | 9q | – | 7 d | 0 | – |
| 18 | 9r | – | 7 d | 0 | – |
| 19 | 9s | – | 7 d | 0 | – |
| 20 | 9t | – | 7 d | 0 | – |
aIsolated yield of the product precipitate from the reaction mixture. bAdditional quantity of the anti- and/or syn-diastereomer(s) isolated from the filtrate as respective methyl esters 10' (see Experimental section). cPure anti-diastereomer obtained by crystallization. dIsolated and characterized as respective methyl esters 10'.
A number of observations emerged from examination of the results in Table 1. In all cases (except entry 7 where carboxylic acid 10g did not precipitate from the reaction mixture and was isolated as respective methyl ester 10g'), the major diastereomer was shown to possess the (RS,RS)-configuration (vide infra) and is referred to as ‘anti‘ throughout this article (considering orientation of carboxylic group relative to C11a–C12 bond of the five-membered ring); the minor, (RS,SR)-configured diastereomer is referred to as ‘syn’ (Figure 4). A good to excellent yield of pure anti-diastereomer was obtained with 9a,b, 9d, 9j, 9l, 9n,o (Table 1, entries 1, 2, 4, 10, 12, 14 and 15) by simple filtration. We have also shown that in some of these cases (Table 1, entries 1 and 2) an additional quantity of anti- and/or syn-configured CCR product could be recovered from the filtrate in the form of respective methyl esters after O-methylation and chromatographic separation (see Experimental section): syn-10a' (7%), anti-10a' (12%), syn-10b' (13%). In those cases when the carboxylic acid precipitate contained a significant proportion of the syn-configured CCR product (Table 1, entries 3, 8, 11 and 13, anti/syn ratio ranging from 3.3:1 to 6.5:1), the latter was removed by crystallization and the respective pure anti-diastereomers (anti-10c, 10h, 10k and 10m) were obtained and characterized. In certain instances (Table 1, entries 5–7, and 9), isolation of pure diastereomeric CCR adducts was achieved by total esterification of the carboxylic acid product mixture and chromatographic separation of the respective methyl esters (the 0.8:1 anti/syn-10e mixture stereoconverged to anti-10e' on esterification, vide infra).
![[1860-5397-13-138-4]](/bjoc/content/figures/1860-5397-13-138-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Anti- and syn-diastereomers of 10 and 10'.
Figure 4: Anti- and syn-diastereomers of 10 and 10'.
The trans-diastereoselectivity of the CCR is well documented in the literature [33] and is, therefore, unsurprising. However, the stereocontrol achieved in this reaction over 3 stereocenters present in 10l (obtained in 81% yield as a single diastereomer) is certainly quite noteworthy and was confirmed by X-ray analysis (Figure 5).
![[1860-5397-13-138-5]](/bjoc/content/figures/1860-5397-13-138-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Single-crystal X-ray structure of compound 10l.
Figure 5: Single-crystal X-ray structure of compound 10l.
The tolerance of the reaction to the substitution pattern in the aromatic portion of the indolenines appears rather broad, both in terms of the electronic character of substituents and steric effects – although substituents in position 7 of the indolenine significantly affect the conformational behavior of the respective CCR adducts 10d,e (vide infra). Notably, free phenolic hydroxy function is well tolerated (9k → 10k) which is in line with literature reports [33]. However, the steric situation around the five-membered ring of indolenines had a profound effect on the reaction times and even the ability of certain indolenines to act as competent partners in the CCR. 2-Substituted indolenines 9h–o required significantly longer times (from 16 h to 50 days) to be converted to the respective CCR products, compared to their 2-unsubstituted counterparts. Indolenines 9p–t failed to undergo the reaction with HPA either at rt or at reflux temperature in acetonitrile or toluene. Attempts to trigger the reaction with these substrates by excluding solvent altogether [34] or applying microwave irradiation (up to 2 h at 200 °C in MeCN) were also unsuccessful. Any appreciable conversion led to predominant formation of HPA dimer 11 and product of its decarboxylation 12 (Scheme 1). The formation of these two products (observed by 1H NMR) was recently reported by Knapp et al. [35] as a result from the treatment of HPA with a strong base (which was absent in our case). The failure to activate sterically hindered indolenines toward the CCR using forcing conditions justifies conducting the reaction at room temperature, which led to clean conversions and good product yields despite the excruciatingly long reaction times (Table 1, entries 14 and 15).
![[1860-5397-13-138-i1]](/bjoc/content/inline/1860-5397-13-138-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Formation of unwanted products 11 and 12 in lieu of the CCR with 9p–t.
Scheme 1: Formation of unwanted products 11 and 12 in lieu of the CCR with 9p–t.
In order to ensure a correct stereochemical assignment of all major (anti) and minor (syn) products obtained in the reactions discussed herein, we obtained single-crystal X-ray structures of sixteen CCR adducts synthesized in this work and correlated this structural information with the NMR behavior of these compounds. The range of the coupling constant J(H11-H11a) values appears a straightforward criterion for relative stereochemistry assignment in compounds 10 derived from 2H-indolenines. As summarized in Figure 6 (see also Table S1 in Supporting Information File 1), anti-diastereomers display this coupling constant around 13 Hz, while it is in the 3-4 Hz range for syn-isomers.
![[1860-5397-13-138-6]](/bjoc/content/figures/1860-5397-13-138-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Typical J(H11-H11a)-values and corresponding dihedral angles for syn- and anti-diastereomers of compounds 10 derived from 2H-indolenines.
Figure 6: Typical J(H11-H11a)-values and corresponding dihedral angles for syn- and anti-diastereomers of com...
According to the X-ray analysis, the dihedral angle values in the C11H–C11aH fragment lie within the 165–170° and 55–58° range for anti- and syn-diastereomers, respectively (Supporting Information File 1, Table S1). Thus, for most of the 2H-indolenine-derived compounds (except for 10d,e), there appears to be a good correlation between the relative stereochemistry of 10, J(H11-H11a) values observed in the 1H NMR spectra of their solutions and said dihedral angle in crystals. Surprisingly, compounds 10d,e (derived from more sterically congested 7-substituted indolenines 9d,e) display the J(H11-H11a) values of 3.0 Hz for both diastereomers, which is inconsistent with the regular values of corresponding dihedral angles (166.3° and 169.7° for anti-10d and anti-10e, respectively; 57.5° for syn-10e) measurable in the X-ray structures of these compounds. We reasoned that such a phenomenon could be rationalized by a different conformer population in the solution compared to solid state. This hypothesis was preliminary confirmed by the results of variable-temperature NMR experiments summarized in Supporting Information File 1 (Figures S1–S6). Another potentially useful criterion for stereochemistry assignment of compounds 10 derived from 2-methylindolenines 10h–k, 10m,n is the range of 13C chemical shifts observed for the angular methyl group in their syn- and anti-diastereomers (Supporting Information File 1, Figure 7, Table S2). These findings were also verified by the available X-ray information on these compounds. Finally, the relative stereochemistry of compound 10o (for which neither X-ray structures nor reliable NMR criteria were available) was assigned on the basis of the through-space interactions observable in its NOESY spectrum and the value of a 3JCH coupling constant (Supporting Information File 1, Figure 8, Figures S7–9).
![[1860-5397-13-138-7]](/bjoc/content/figures/1860-5397-13-138-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: The difference in the 13C NMR chemical shifts of the angular methyl group between syn- and anti-diastereomers of 11a-Me in compounds 10h–k, 10m,n.
Figure 7: The difference in the 13C NMR chemical shifts of the angular methyl group between syn- and anti-dia...
![[1860-5397-13-138-8]](/bjoc/content/figures/1860-5397-13-138-8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: Criteria for stereochemistry assignment of anti-10o.
Figure 8: Criteria for stereochemistry assignment of anti-10o.
Besides the anomalous NMR behavior discussed above, compound 10e displayed a number of unusual tendencies which may shed some light on the diastereomeric integrity of CCR adducts in general as well as on the mechanism of their formation. Compound 10e initially formed as a 0.8:1 mixture of anti/syn diastereomers (Table 1). However, it was promptly noted that this mixture converges to thermodynamically more stable anti-10e (the isomerization occurs on heating to 80 °C or, more slowly, even at room temperature). Esterification of anti/syn-10e in the presence of potassium carbonate led to the formation of anti-10e' as a sole product, presumably, via a base-promoted enolization and subsequent isomerization of the syn-10e (10e', Scheme 2).
![[1860-5397-13-138-i2]](/bjoc/content/inline/1860-5397-13-138-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Syn/anti isomerization of compound 10e.
Scheme 2: Syn/anti isomerization of compound 10e.
Enolization is thought to be a key event in the formation of the CCR products which can occur via two alternative mechanisms: (a) N-acylation of the imine component followed by intramolecular Mannich reaction or (b) Mannich-type addition of the HPA enolate to a protonated imine component followed by intramolecular aminolysis of the cyclic anhydride moiety in Mannich adduct 13 (Scheme 3) [1].
![[1860-5397-13-138-i3]](/bjoc/content/inline/1860-5397-13-138-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Alternative mechanistic pathways for the CCR.
Scheme 3: Alternative mechanistic pathways for the CCR.
Investigation of the CCR leading to the formation of 10e undertaken in this work led to a serendipitous important observation that exposure of 9e to an equimolar amount of HPA in acetonitrile at low temperature (5 °C) and higher dilution (two-fold compared to that used throughout this study) over 2 days led to a predominant formation of the respective Mannich adduct 13e which crystallized out as a single diastereomer from the reaction mixture along with syn/anti-10e mixture and was separated from the latter mechanically under a microscope. Adduct 13e (which has been postulated in the literature as a principal intermediate in the CCR [1,28] but never isolated) was characterized by means of 1H and 13C NMR spectroscopy as well as X-ray crystallography. The isolation, for the first time, of the Mannich-type adduct 13 between HPA and an imine clearly attests to the viability of mechanistic pathway (b) shown in Scheme 3.
When left at room temperature for 12 h as a solution in CDCl3, single diastereomer 13e fully converted itself into a ca. 1:1 anti/syn-10e (Scheme 4).
![[1860-5397-13-138-i4]](/bjoc/content/inline/1860-5397-13-138-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Formation and fate of Mannich adduct 13e.
Scheme 4: Formation and fate of Mannich adduct 13e.
Conversion of diastereomerically pure 13e into a mixture of diastereomers can be rationalized by a faster formation of kinetic product syn-10e preceded by enolization, in competition with direct albeit slow conversion 13e → anti-10e (Scheme 5).
![[1860-5397-13-138-i5]](/bjoc/content/inline/1860-5397-13-138-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Mechanistic rationale for the 13e→ syn/anti-10e conversion.
Scheme 5: Mechanistic rationale for the 13e→ syn/anti-10e conversion.
We mentioned that tetracyclic compounds 10 carry resemblance to natural as well as synthetic camptothecin-like topoisomerase inhibitors (vide supra). Compounds 10 are endowed with HPA-derived carboxylic acid functionality which may facilitate or prevent compounds’ binding to DNA or DNA-topoisomerase complex. Depending on the medicinal chemistry context, compounds 10 can be decarboxylated to deliver sterically encumbered tetracyclic lactams 14 lacking the carboxylic acid group as we showed for exemplary compound 10h. The 4.3:1 anti/syn mixture of diastereomers of 10h rapidly and cleanly lost the carboxylic function at 200 °C and gave 83% yield of racemic compound 14h (Scheme 6).
![[1860-5397-13-138-i6]](/bjoc/content/inline/1860-5397-13-138-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Decarboxylation of anti/syn-10h.
Scheme 6: Decarboxylation of anti/syn-10h.
Conclusion
We have described a novel variant of the Castagnoli–Cushman reaction employing indolenines as cyclic imine components in the reaction with homophthalic anhydride (HPA). The compounds obtained contain a benzene-fused, privileged, naturally occurring hexahydropyrrolo[1,2-b]isoquinoline core and are distinctly encumbered from steric perspective. The novel compounds have been characterized by NMR spectroscopy, high-resolution mass spectrometry and X-ray crystallography and certain regularities in the NMR behavior have been established, leading a set of rules for stereochemical assignment based on the NMR data. A Mannich-type adduct between HPA and an imine (previously only postulated as a crucial intermediate en route to CCR products) has been isolated for the first time and fully characterized. Certain insights into the role of enolization equilibria in the formation and diastereomeric integrity of the CCR adducts has been obtained. The synthetic methodology described herein significantly expands the scope and tests the limits of the sterically permitted Castagnoli–Cushman reaction.
Experimental
General information. NMR spectroscopic data were recorded with a 400 MHz (400.13 MHz for 1H and 100.61 MHz for 13C) and a 500 MHz (500.03 MHz for 1H and 125.7 MHz for 13C) spectrometers in DMSO-d6 or in CDCl3 and were referenced to residual solvent proton signals (δH = 7.26 and 2.50 ppm, respectively) and solvent carbon signals (δC = 77.2 and 39.5 ppm, respectively). Coupling constants, J are reported in Hz. Melting points were determined with an automated heat block instrument and are uncorrected. Mass spectra were recorded with a HRMS-ESI-qTOF spectrometer (electrospray ionization mode). X-ray single crystal analyses were performed with monochromated Mo Kα or Cu Kα radiation, respectively. Column chromatography was performed on silica gel 60 (230–400 mesh). For TLC analysis UV254 silica gel coated plates were used. MeCN was distilled from P2O5 and stored over molecular sieves 4 Å. Homophthalic anhydride was acquired from a commercial source, stored at 5 °C and used as received. All indolenines 9 were stored in sealed vials at 5 °C in the dark.
CCDC 1503093 (13e), 1503094 (anti-10c), 1503095 (anti-10d), 1503096 (anti-10g'), 1503097 (anti-10e), 1503098 (anti-10a'), 1503099 (anti-10f), 1503100 (syn-10a'), 1503101 (syn-10f), 1503102 (anti-10e'), 1503103 (anti-10n), 1503104 (anti-10i), 1503105 (syn-10e), 1503106 (anti-10l), 1470399 (anti-10b), 1470389 (anti-10h), 1461790 (anti-10j) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk.
Indolenines 9. Indolenines 9a,b [26], 9d–g [26], 9h [36], 9i [37], 9j [38], 9k [39], 9p [40] are known compounds and were prepared from the arylhydrazine hydrochlorides and respective aldehydes or ketones according to the literature protocols.
General procedure 1. Synthesis of indolenines 9c,l,n,o,q–t. To a screw-cap vial containing suspension of corresponding arylhydrazine hydrochloride in glacial AcOH (15 mL) the carbonyl compound (1.1 equiv) was added in one portion. The reaction mixture was stirred at 55–60 °C for 4 h (or at reflux for 2–16 h) and concentrated in vacuo at 40 °C. The residue was diluted with DCM (50 mL) and filtered through Celite. The resulting solution was passed through a short pad of silica, washed with sat. NaHCO3 and water. The organic layer was dried over Na2SO4 and concentrated under reduced pressure to give pure indolenines 9c,l,s. Indolenines 9n,o,q,r,t were subjected to extra column chromatography.
General procedure 2. Synthesis of compounds 10. A mixture of homophthalic anhydride (1 equiv) and the corresponding indolenine 9a–t (1 equiv) was placed in a sealed screw-cap vial, dissolved in dry acetonitrile (2 mL per 1 mmol) and stirred at room temperature for the time indicated in Table 1. The reaction mixture was cooled to −14 °C, the resulting precipitate was filtered and washed with a minimum amount of cold acetonitrile to give pure compound 10.
General procedure 3. Synthesis of methyl esters 10'. The Castagnoli–Cushman product 10 was dissolved in dry acetone (10 mL per 1 mmol). Methyl iodide (2.5 equiv) and K2CO3 (2.5 equiv) were added to the solution and the resulting suspension was stirred for 24 h at room temperature. The volatiles were removed in vacuo. The residue was dissolved in DCM, washed with water, brine, dried over Na2SO4 and concentrated to give crude methyl ester 10', which was purified by column chromatography on silica gel.
Supporting Information
| Supporting Information File 1: Detailed experimental procedures, analytical data and copies of 1H and 13C NMR spectra for all new compounds; crystallographic data for compounds 10 and 13e; results of correlational and variable temperature NMR experiments. | ||
| Format: PDF | Size: 6.9 MB | Download |
Acknowledgements
This research was supported by the Russian Scientific Fund (project grant 14-50-00069). NMR, mass spectrometry and X-ray studies were performed at the Research Centre for Magnetic Resonance, the Centre for Chemical Analysis and Materials Research of Saint Petersburg State University Research Park and the Centre for X-ray Diffraction Methods. The authors are grateful to Dr. Sergey Miltsov for providing 2,3,3-trimethylindolenines employed in this study.
References
-
Krasavin, M.; Dar’in, D. Tetrahedron Lett. 2016, 57, 1635. doi:10.1016/j.tetlet.2016.03.021
Return to citation in text: [1] [2] [3] -
Castagnoli, N., Jr. J. Org. Chem. 1969, 34, 3187. doi:10.1021/jo01262a081
Return to citation in text: [1] -
Cushman, M.; Castagnoli, N., Jr. J. Org. Chem. 1973, 38, 440. doi:10.1021/jo00943a007
Return to citation in text: [1] -
Adamovskyi, M. I.; Ryabukhin, S. V.; Sibgatulin, D. A.; Rusanov, E.; Grygorenko, O. O. Org. Lett. 2017, 19, 130. doi:10.1021/acs.orglett.6b03426
Return to citation in text: [1] -
Bakulina, O.; Dar’in, D.; Krasavin, M. Synlett 2017, 28, 1165. doi:10.1055/s-0036-1588714
Return to citation in text: [1] -
Wang, L.; Liu, J.; Tian, H.; Qian, C.; Sun, J. Adv. Synth. Catal. 2005, 347, 689. doi:10.1002/adsc.200404211
Return to citation in text: [1] -
Mohammadi, M. H.; Mohammadi, A. A. Synth. Commun. 2011, 41, 523. doi:10.1080/00397911003611786
Return to citation in text: [1] -
Karimi, A. R.; Momeni, H. R.; Pashazadeh, R. Tetrahedron Lett. 2012, 53, 3440. doi:10.1016/j.tetlet.2012.04.089
Return to citation in text: [1] -
Humphries, P. S.; Benbow, J. W.; Bonin, P. D.; Boyer, D.; Doran, S. D.; Frisbie, R. K.; Piotrowski, D. W.; Balan, G.; Bechle, B. M.; Conn, E. L.; Dirico, K. J.; Oliver, R. M.; Soeller, W. C.; Southers, J. A.; Yang, X. Bioorg. Med. Chem. Lett. 2009, 19, 2400. doi:10.1016/j.bmcl.2009.03.082
Return to citation in text: [1] -
Rothweiler, U.; Czarna, A.; Krajewski, M.; Ciombor, J.; Kalinski, C.; Khazak, V.; Ross, G.; Skobeleva, N.; Weber, L.; Holak, T. A. ChemMedChem 2008, 3, 1118. doi:10.1002/cmdc.200800025
Return to citation in text: [1] -
Dar’in, D.; Bakulina, O.; Chizhova, M.; Krasavin, M. Org. Lett. 2015, 17, 3930. doi:10.1021/acs.orglett.5b02014
Return to citation in text: [1] -
Liu, H.; Li, G.; Wang, J.; Liu, J. Corydaline derivatives useful for reducing lipid levels. PCT Int. Appl. WO2010075469, Dec 22, 2009.
Chem. Abstr. 2010, 153, 145700.
Return to citation in text: [1] -
Georgieva, A.; Stanoeva, E.; Spassov, S.; Macicek, J.; Angelova, O.; Haimova, M.; De Kimpe, N. Tetrahedron 1991, 47, 3375. doi:10.1016/S0040-4020(01)86402-4
Return to citation in text: [1] -
Stanoeva, E.; Georgieva, A.; Avramova, S.; Burdzhiev, N.; Lázár, L. J. Heterocycl. Chem. 2015, 52, 130. doi:10.1002/jhet.1965
Return to citation in text: [1] -
Smith, F. T.; DeRuiter, J.; Carter, D. A. J. Heterocycl. Chem. 1989, 26, 1815. doi:10.1002/jhet.5570260653
Return to citation in text: [1] -
Smith, F. T.; Atigadda, R. V. J. Heterocycl. Chem. 1991, 28, 1813. doi:10.1002/jhet.5570280728
Return to citation in text: [1] -
Venkatram, A.; Colley, T.; DeRuiter, J.; Smith, F. J. Heterocycl. Chem. 2005, 42, 297. doi:10.1002/jhet.5570420220
Return to citation in text: [1] -
Mulchandani, N. B.; Venkatachalam, S. R. Phytochemistry 1976, 15, 1561. doi:10.1016/S0031-9422(00)88937-2
Return to citation in text: [1] -
Padwa, A.; Brodney, M. A.; Lynch, S. M. J. Org. Chem. 2001, 66, 1716. doi:10.1021/jo0014109
Return to citation in text: [1] -
Sun, Z.; Zhou, M.; Li, X.; Meng, X.; Peng, F.; Zhang, H.; Shao, Z. Chem. – Eur. J. 2014, 20, 6112. doi:10.1002/chem.201400178
Return to citation in text: [1] -
Schramm, A.; Saxena, P.; Chlebek, J.; Cahlikova, L.; Baburin, I.; Hering, S.; Hamburger, M. Planta Med. 2014, 80, 740. doi:10.1055/s-0034-1368590
Return to citation in text: [1] -
Wang, C.; Wang, Q.; Li, X.; Jin, X.; Xu, P.; Xu, N.; Xu, A.; Xu, Y.; Zheng, S.; Zheng, J.; Liu, C.; Huang, P. Biochem. Biophys. Res. Commun. 2017, 483, 197. doi:10.1016/j.bbrc.2016.12.168
Return to citation in text: [1] -
Guo, Y.; Wang, Y.; Cao, L.; Wang, P.; Qing, J.; Zheng, Q.; Shang, L.; Yin, Z.; Sun, Y. Antimicrob. Agents Chemother. 2016, 60, 913. doi:10.1128/AAC.02274-15
Return to citation in text: [1] -
Chen, C.-Y.; Zhu, G.-Y.; Wang, J.-R.; Jiang, Z.-H. RSC Adv. 2016, 6, 79958. doi:10.1039/c6ra16455b
Return to citation in text: [1] -
Evans, B. E.; Rittle, K. E.; Bock, M. G.; DiPardo, R. M.; Freidinger, R. M.; Whitter, W. L.; Lundell, G. F.; Veber, D. F.; Anderson, P. S.; Chang, R. S. L.; Lotti, V. J.; Cerino, D. J.; Chen, T. B.; Kling, P. J.; Kunkel, K. A.; Springer, J. P.; Hirshfield, J. J. Med. Chem. 1988, 31, 2235. doi:10.1021/jm00120a002
Return to citation in text: [1] -
Golubev, P.; Bakulina, O.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2016, 3969. doi:10.1002/ejoc.201600656
Return to citation in text: [1] [2] [3] [4] [5] -
Estévez, V.; Kloeters, L.; Kwietniewska, N.; Vicente-García, E.; Ruijter, E.; Orru, R. V. A. Synlett 2017, 28, 376. doi:10.1055/s-0036-1588084
Return to citation in text: [1] -
Golubev, P.; Krasavin, M. Eur. J. Org. Chem. 2017, 1740. doi:10.1002/ejoc.201700152
Return to citation in text: [1] [2] -
Lovering, F.; Bikker, J.; Humblet, C. J. Med. Chem. 2009, 52, 6752. doi:10.1021/jm901241e
Return to citation in text: [1] -
Ling, T.; Rivas, F. Tetrahedron 2016, 72, 6729. doi:10.1016/j.tet.2016.09.002
Return to citation in text: [1] -
Luo, W.-K.; Shi, X.; Zhou, W.; Yang, L. Org. Lett. 2016, 18, 2036. doi:10.1021/acs.orglett.6b00646
Return to citation in text: [1] -
Sarnpitak, P.; Krasavin, M. Tetrahedron Lett. 2014, 55, 2299. doi:10.1016/j.tetlet.2014.02.099
Return to citation in text: [1] -
Georgieva, A.; Stanoeva, E.; Karafilova, K.; Spassov, S.; Angelova, O.; Haimova, M.; De Kimpe, N.; Boelens, M. Tetrahedron 1994, 50, 9399. doi:10.1016/S0040-4020(01)85515-0
Return to citation in text: [1] [2] -
Lepikhina, A.; Bakulina, O.; Dar'in, D.; Krasavin, M. RSC Adv. 2016, 6, 83808. doi:10.1039/C6RA19196G
Return to citation in text: [1] -
Hong, J.; Wang, Z.; Levin, A.; Emge, T. J.; Floyd, D. M.; Knapp, S. Tetrahedron Lett. 2015, 56, 3001. doi:10.1016/j.tetlet.2014.10.027
Return to citation in text: [1] -
Ji, C.; Ma, L.; Yin,, M.; Yang, W.; Pan, K. Chem. – Asian J. 2016, 11, 2316. doi:10.1002/asia.201600818
Return to citation in text: [1] -
Szalóki, G.; Sanguinet, L. J. Org. Chem. 2015, 80, 3949. doi:10.1021/acs.joc.5b00282
Return to citation in text: [1] -
Balmond, E. I.; Tautgeg, B. K.; Faulkner, A. L.; Or, V. W.; Hodur, B. M.; Shaw, J. T.; Louie, A. Y. J. Org. Chem. 2016, 81, 8744. doi:10.1021/acs.joc.6b01193
Return to citation in text: [1] -
Schultz-Senft, M.; Gates, P. J.; Soennichsen, F. J.; Stanbitz, A. Dyes Pigm. 2017, 136, 292. doi:10.1016/j.dyepig.2016.08.039
Return to citation in text: [1] -
Prostoba, Y.; Coelho, P. J.; Pina, J.; Seixas de Melo, J. J. Photochem. Photobiol., A: Chem. 2010, 216, 59. doi:10.1016/j.jphotochem.2010.09.006
Return to citation in text: [1]
| 33. | Georgieva, A.; Stanoeva, E.; Karafilova, K.; Spassov, S.; Angelova, O.; Haimova, M.; De Kimpe, N.; Boelens, M. Tetrahedron 1994, 50, 9399. doi:10.1016/S0040-4020(01)85515-0 |
| 33. | Georgieva, A.; Stanoeva, E.; Karafilova, K.; Spassov, S.; Angelova, O.; Haimova, M.; De Kimpe, N.; Boelens, M. Tetrahedron 1994, 50, 9399. doi:10.1016/S0040-4020(01)85515-0 |
| 34. | Lepikhina, A.; Bakulina, O.; Dar'in, D.; Krasavin, M. RSC Adv. 2016, 6, 83808. doi:10.1039/C6RA19196G |
| 1. | Krasavin, M.; Dar’in, D. Tetrahedron Lett. 2016, 57, 1635. doi:10.1016/j.tetlet.2016.03.021 |
| 8. | Karimi, A. R.; Momeni, H. R.; Pashazadeh, R. Tetrahedron Lett. 2012, 53, 3440. doi:10.1016/j.tetlet.2012.04.089 |
| 22. | Wang, C.; Wang, Q.; Li, X.; Jin, X.; Xu, P.; Xu, N.; Xu, A.; Xu, Y.; Zheng, S.; Zheng, J.; Liu, C.; Huang, P. Biochem. Biophys. Res. Commun. 2017, 483, 197. doi:10.1016/j.bbrc.2016.12.168 |
| 37. | Szalóki, G.; Sanguinet, L. J. Org. Chem. 2015, 80, 3949. doi:10.1021/acs.joc.5b00282 |
| 6. | Wang, L.; Liu, J.; Tian, H.; Qian, C.; Sun, J. Adv. Synth. Catal. 2005, 347, 689. doi:10.1002/adsc.200404211 |
| 7. | Mohammadi, M. H.; Mohammadi, A. A. Synth. Commun. 2011, 41, 523. doi:10.1080/00397911003611786 |
| 23. | Guo, Y.; Wang, Y.; Cao, L.; Wang, P.; Qing, J.; Zheng, Q.; Shang, L.; Yin, Z.; Sun, Y. Antimicrob. Agents Chemother. 2016, 60, 913. doi:10.1128/AAC.02274-15 |
| 38. | Balmond, E. I.; Tautgeg, B. K.; Faulkner, A. L.; Or, V. W.; Hodur, B. M.; Shaw, J. T.; Louie, A. Y. J. Org. Chem. 2016, 81, 8744. doi:10.1021/acs.joc.6b01193 |
| 4. | Adamovskyi, M. I.; Ryabukhin, S. V.; Sibgatulin, D. A.; Rusanov, E.; Grygorenko, O. O. Org. Lett. 2017, 19, 130. doi:10.1021/acs.orglett.6b03426 |
| 5. | Bakulina, O.; Dar’in, D.; Krasavin, M. Synlett 2017, 28, 1165. doi:10.1055/s-0036-1588714 |
| 20. | Sun, Z.; Zhou, M.; Li, X.; Meng, X.; Peng, F.; Zhang, H.; Shao, Z. Chem. – Eur. J. 2014, 20, 6112. doi:10.1002/chem.201400178 |
| 26. | Golubev, P.; Bakulina, O.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2016, 3969. doi:10.1002/ejoc.201600656 |
| 2. | Castagnoli, N., Jr. J. Org. Chem. 1969, 34, 3187. doi:10.1021/jo01262a081 |
| 3. | Cushman, M.; Castagnoli, N., Jr. J. Org. Chem. 1973, 38, 440. doi:10.1021/jo00943a007 |
| 21. | Schramm, A.; Saxena, P.; Chlebek, J.; Cahlikova, L.; Baburin, I.; Hering, S.; Hamburger, M. Planta Med. 2014, 80, 740. doi:10.1055/s-0034-1368590 |
| 36. | Ji, C.; Ma, L.; Yin,, M.; Yang, W.; Pan, K. Chem. – Asian J. 2016, 11, 2316. doi:10.1002/asia.201600818 |
| 12. |
Liu, H.; Li, G.; Wang, J.; Liu, J. Corydaline derivatives useful for reducing lipid levels. PCT Int. Appl. WO2010075469, Dec 22, 2009.
Chem. Abstr. 2010, 153, 145700. |
| 13. | Georgieva, A.; Stanoeva, E.; Spassov, S.; Macicek, J.; Angelova, O.; Haimova, M.; De Kimpe, N. Tetrahedron 1991, 47, 3375. doi:10.1016/S0040-4020(01)86402-4 |
| 14. | Stanoeva, E.; Georgieva, A.; Avramova, S.; Burdzhiev, N.; Lázár, L. J. Heterocycl. Chem. 2015, 52, 130. doi:10.1002/jhet.1965 |
| 18. | Mulchandani, N. B.; Venkatachalam, S. R. Phytochemistry 1976, 15, 1561. doi:10.1016/S0031-9422(00)88937-2 |
| 1. | Krasavin, M.; Dar’in, D. Tetrahedron Lett. 2016, 57, 1635. doi:10.1016/j.tetlet.2016.03.021 |
| 28. | Golubev, P.; Krasavin, M. Eur. J. Org. Chem. 2017, 1740. doi:10.1002/ejoc.201700152 |
| 11. | Dar’in, D.; Bakulina, O.; Chizhova, M.; Krasavin, M. Org. Lett. 2015, 17, 3930. doi:10.1021/acs.orglett.5b02014 |
| 19. | Padwa, A.; Brodney, M. A.; Lynch, S. M. J. Org. Chem. 2001, 66, 1716. doi:10.1021/jo0014109 |
| 26. | Golubev, P.; Bakulina, O.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2016, 3969. doi:10.1002/ejoc.201600656 |
| 10. | Rothweiler, U.; Czarna, A.; Krajewski, M.; Ciombor, J.; Kalinski, C.; Khazak, V.; Ross, G.; Skobeleva, N.; Weber, L.; Holak, T. A. ChemMedChem 2008, 3, 1118. doi:10.1002/cmdc.200800025 |
| 35. | Hong, J.; Wang, Z.; Levin, A.; Emge, T. J.; Floyd, D. M.; Knapp, S. Tetrahedron Lett. 2015, 56, 3001. doi:10.1016/j.tetlet.2014.10.027 |
| 9. | Humphries, P. S.; Benbow, J. W.; Bonin, P. D.; Boyer, D.; Doran, S. D.; Frisbie, R. K.; Piotrowski, D. W.; Balan, G.; Bechle, B. M.; Conn, E. L.; Dirico, K. J.; Oliver, R. M.; Soeller, W. C.; Southers, J. A.; Yang, X. Bioorg. Med. Chem. Lett. 2009, 19, 2400. doi:10.1016/j.bmcl.2009.03.082 |
| 15. | Smith, F. T.; DeRuiter, J.; Carter, D. A. J. Heterocycl. Chem. 1989, 26, 1815. doi:10.1002/jhet.5570260653 |
| 16. | Smith, F. T.; Atigadda, R. V. J. Heterocycl. Chem. 1991, 28, 1813. doi:10.1002/jhet.5570280728 |
| 17. | Venkatram, A.; Colley, T.; DeRuiter, J.; Smith, F. J. Heterocycl. Chem. 2005, 42, 297. doi:10.1002/jhet.5570420220 |
| 1. | Krasavin, M.; Dar’in, D. Tetrahedron Lett. 2016, 57, 1635. doi:10.1016/j.tetlet.2016.03.021 |
| 26. | Golubev, P.; Bakulina, O.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2016, 3969. doi:10.1002/ejoc.201600656 |
| 24. | Chen, C.-Y.; Zhu, G.-Y.; Wang, J.-R.; Jiang, Z.-H. RSC Adv. 2016, 6, 79958. doi:10.1039/c6ra16455b |
| 39. | Schultz-Senft, M.; Gates, P. J.; Soennichsen, F. J.; Stanbitz, A. Dyes Pigm. 2017, 136, 292. doi:10.1016/j.dyepig.2016.08.039 |
| 25. | Evans, B. E.; Rittle, K. E.; Bock, M. G.; DiPardo, R. M.; Freidinger, R. M.; Whitter, W. L.; Lundell, G. F.; Veber, D. F.; Anderson, P. S.; Chang, R. S. L.; Lotti, V. J.; Cerino, D. J.; Chen, T. B.; Kling, P. J.; Kunkel, K. A.; Springer, J. P.; Hirshfield, J. J. Med. Chem. 1988, 31, 2235. doi:10.1021/jm00120a002 |
| 40. | Prostoba, Y.; Coelho, P. J.; Pina, J.; Seixas de Melo, J. J. Photochem. Photobiol., A: Chem. 2010, 216, 59. doi:10.1016/j.jphotochem.2010.09.006 |
| 26. | Golubev, P.; Bakulina, O.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2016, 3969. doi:10.1002/ejoc.201600656 |
| 32. | Sarnpitak, P.; Krasavin, M. Tetrahedron Lett. 2014, 55, 2299. doi:10.1016/j.tetlet.2014.02.099 |
| 30. | Ling, T.; Rivas, F. Tetrahedron 2016, 72, 6729. doi:10.1016/j.tet.2016.09.002 |
| 31. | Luo, W.-K.; Shi, X.; Zhou, W.; Yang, L. Org. Lett. 2016, 18, 2036. doi:10.1021/acs.orglett.6b00646 |
| 26. | Golubev, P.; Bakulina, O.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2016, 3969. doi:10.1002/ejoc.201600656 |
| 29. | Lovering, F.; Bikker, J.; Humblet, C. J. Med. Chem. 2009, 52, 6752. doi:10.1021/jm901241e |
| 27. | Estévez, V.; Kloeters, L.; Kwietniewska, N.; Vicente-García, E.; Ruijter, E.; Orru, R. V. A. Synlett 2017, 28, 376. doi:10.1055/s-0036-1588084 |
| 28. | Golubev, P.; Krasavin, M. Eur. J. Org. Chem. 2017, 1740. doi:10.1002/ejoc.201700152 |
© 2017 Bakulina et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)