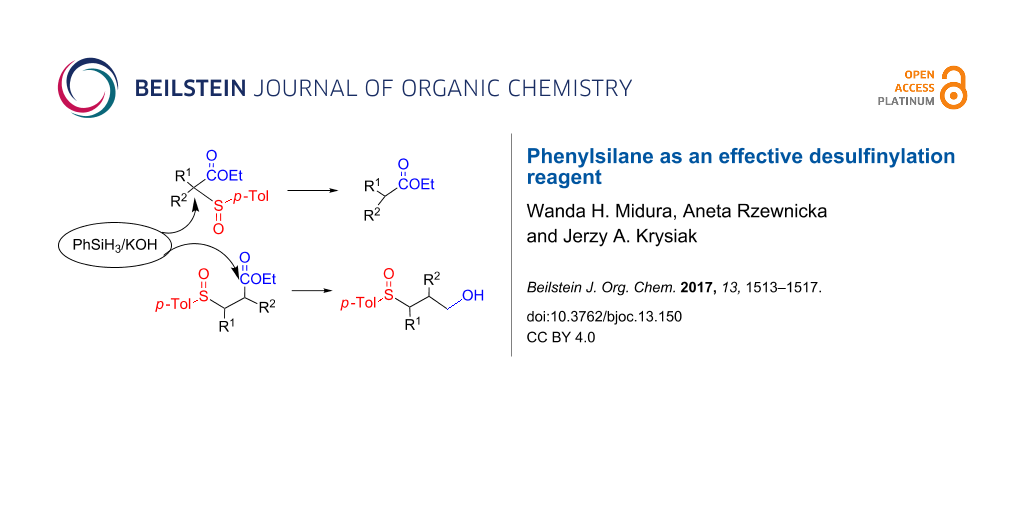
Abstract
The reduction using phenylsilane in a KOH-catalyzed system was applied successfully to the conversion of sulfinyl-substituted cyclopropylcarboxylates into the corresponding alcohols. The presence of sulfinyl substituents in the α-position to the carboxylate group caused a desulfinylation product formation with full regio- and stereoselectivity, instead of a carbonyl group reduction. Investigations performed on different α-sulfinylcarbonyl compounds revealed that phenylsilane treatment constitutes a regiospecific method for the desulfinylation of a-sulfinylesters; for corresponding ketones the reaction course depends on the character of the carbonyl group.

Graphical Abstract
Introduction
The conversion of carboxylic acid derivatives to the corresponding alcohols is one of the fundamental processes in organic synthesis. Over the past decades, many reagents and conditions for this transformation have been reported. Apart from traditional hydride reducing agents like lithium aluminum hydride and sodium borohydride [1,2], different modifications were applied, using transition metal salts as catalysts or additives to change or enhance the properties of these reagents [3-8]. Hydrogenation is another convenient method for the reduction of carbonyl compounds, although low selectivities have sometimes been observed and the reaction usually required drastic conditions [9]. Also, the catalytic hydrosilylation of carbonyl moieties has become an important transformation as an alternative reduction methodology [10-14]. Besides the well-known metal-catalyzed hydrosilylation, transition metals in the presence of Brønsted or Lewis acids were used for the reduction. Recently base-activated silanes were also used for this purpose [15-17].
Results and Discussion
Our recent investigations were connected with the synthesis and further conversion of multifunctional cyclopropanes, where the carboxylate moiety was present in close proximity to sulfinyl and phosphoryl substituents [18-20]. A selective and effective method of the independent conversion of each of these substituents is therefore a subject of our interest. To extend the investigations on the reduction of esters using a reagent with tolerance for other functional groups present in the structure, we applied a hydrosilylation methodology (Scheme 1). Various cyclopropyl phosphonates were subjected to the reaction with an excess of phenylsilane under neat reaction conditions in the presence of a catalytic amount of KOH. It appeared that results of this reaction strongly depended on the relative position of particular substituents [21].
![[1860-5397-13-150-i1]](/bjoc/content/inline/1860-5397-13-150-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Different behaviour of cyclopropylphosphonates in the reaction with phenylsilane.
Scheme 1: Different behaviour of cyclopropylphosphonates in the reaction with phenylsilane.
For compounds 1 and 2 the treatment with phenylsilane caused, as expected, in a reduction of the ester group. In both cyclopropanes the carboxylate moiety and the sulfinyl group were in vicinal relation, but in 1, formed in the reaction of vinyl phosphoryl sulfoxide with EDSA [22], the ester group was easily accessed, since the bulky phosphoryl group was also in vicinal position to the sulfoxide substituent. Compounds 2 and 3 were obtained by us earlier [23] in diastereomerically pure form as key intermediates in our approach to constrained phosphonic acids by the reaction of phosphoryl acrylates with (S)-dimethylsulfonium(p-tolylsulfinyl)methylide. The carboxylate moiety was geminal to the phosphoryl substituent in these compounds. In the case of cyclopropane 3 the reduction did not occur, since the bulky tert-butyl substituent effectively protected the carboxylate group. Hence, the starting material was recovered.
The next cyclopropane 4, which contained the sulfinyl substituent in geminal position to the ester group, was subjected to a reaction with phenylsilane using the procedure described above. In this case, to our surprise, no alcohol was detected, but the desulfinylation process occurred exclusively.
Cyclopropane 4 was formed by carboxylation of 3 with ethyl chloroformate as a continuation of our studies on asymmetric cyclopropanation of vinyl phosphonates using (S)-dimethylsulfonium(p-tolylsulfinyl)methylide and its further application. It was formed as a single diastereomer with full stereoselectivity. To determine the relative configuration of the obtained structure, in the absence of an α-proton at the sulfinyl group which was conclusive in our former studies, we benefited from 13C NMR analysis. The small value of coupling constant 3JP-C = 4 Hz indicated a trans relationship between the carboxylate and the phosphoryl substituent.
Desulfinylation of cyclopropane 4 under conditions used by us before, i.e., by treatment with isopropylmagnesium chloride, gave a mixture of diastereomers 7a and 7b. It was probably due to epimerization on carbon C2 of the carbanion, formed after attack of the Grignard reagent on sulfur. To our satisfaction, utilization of the silane procedure led to the single diastereomer 7a. In this case determination of the relative configuration as trans was based on a high coupling constant 3JP-H = 17.0 Hz, which evidenced retention of configuration during the desulfinylation process (Scheme 2).
![[1860-5397-13-150-i2]](/bjoc/content/inline/1860-5397-13-150-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis and desulfinylation of 4.
Scheme 2: Synthesis and desulfinylation of 4.
The reaction conducted under solvent-free conditions with a large excess of silyl reagent (procedure 1) occurred with full conversion of the cyclopropanes, although some problems with the isolation of the product due to an excess of silane induced a modification of the reduction procedure. The gain of using THF as a solvent [17] (procedure 2) let us decrease the amount of phenylsilane used in the reaction medium, which resulted in a high yield of the corresponding cyclopropane 7a.
The obtained results demonstrated that the presence of a sulfinyl substituent in α position to the carboxylate completely changed the direction of the silane attack on the structure, so this reaction could be considered as a new regiospecific method of a desulfinylation. In order to examine the scope of observed desulfinylation different carbonyl compounds with sulfinyl substituents were synthetized and subjected to the reaction with phenylsilane. Reactions of α-sulfinylcarboxylates 9 and 10 with phenylsilane evidenced that the desulfinylation process occurred also in the case of acyclic sulfoxides (Scheme 3).
![[1860-5397-13-150-i3]](/bjoc/content/inline/1860-5397-13-150-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction of acyclic sulfoxides with phenylsilane. Reagents and conditions: (a) BuLi, THF, −70 °C, p-TolS(O)Men; (b) PhSiH3, THF, rt, KOH (5 mol %); (c) MeI, 50% KOH, CH2Cl2.
Scheme 3: Reaction of acyclic sulfoxides with phenylsilane. Reagents and conditions: (a) BuLi, THF, −70 °C, p...
α-Sulfinylphenylacetate 9 was formed as a mixture of diastereomers difficult to separate. This mixture, without separation, was subjected to the reaction with phenylsilane utilizing the procedure described above and afforded phenylacetate 8 as the only product. Methylation of 9 afforded product 10 with full stereoselectivity as the only diastereomer although the configuration of this center was not determined. Also in this case treatment with PhSiH3/KOH caused a desulfinylation process leading to ester 11 [24]. Unfortunately in this case the stereogenic center at the carbon undergoes epimerization under these reaction conditions and 11 is obtained in racemic form.
To study the scope and limitation of the silane reactivity and its selectivity we checked the influence of the ketone function on the behaviour of the compounds with sulfur and carbonyl substituents in geminal position. This relation turned out to be more complex and was depended on the ketone structure (Table 1).
Table 1: Reaction of compounds with sulfur and carbonyl substituents in geminal relation with PhSiH3/KOH.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-150-i4.svg?max-width=637&scale=1.0)
|
||||
| Entry | Ketone | Reaction conditions | Product of reduction | Product of desulfinylation |
|---|---|---|---|---|
| 1 | 12 |
5 equiv PhSiH3, 0.1 equiv KOH,
0.5 h, rt, 20% conv. |
not detected |
13/14
2:1 |
| 2 | 12 |
5 equiv PhSiH3, 0.1 equiv KOH,
5 h, rt, 100% conv. |
not detected |
13/14
2:1 |
| 3 | 15 |
5 equiv PhSiH3, 0.1 equiv KOH,
5 h, rt, 100% conv. |
not detected | 17 |
| 4 | 16 |
5 equiv PhSiH3, 0.1 equiv KOH,
5 h, rt, 100% conv. |
12 | not detected |
| 5 | 19 |
5 equiv PhSiH3, 0.1 equiv KOH,
5 h, rt, 100% conv. |
21 | not detected |
| 6 | 20 |
5 equiv PhSiH3, 0.1 equiv KOH,
5 h, rt, 100% conv. |
22 | not detected |
Introduction of an acyl moiety by substitution of 3 with acetic anhydride led to compound 12, which was subjected to the reaction with PhSiH3/KOH. Also in this case desulfinylation of cyclopropane took place leading to 13 as a major product, however, accompanied by the corresponding alcohol 14, formed as a result of the subsequent reduction of the carbonyl group (Table 1, entries 1 and 2). Oxidation of the crude mixture by stirring with chromium trioxide and pyridine allowed to obtain desulfinylated ketone 13 in a reasonable yield.
For acyclic structures the reaction course dramatically depended on the character of ketone. In the case of aliphatic ketone 15, silane treatment gave desulfinylated product 17 as the only product (Table 1, entry 3). However, the presence of a more electrophilic aryl ketone group in 19 caused that the reduction of carbonyl group took place, leading to alcohol 21 without desulfinylation (Table 1, entry 5).
For comparison, for analogous carbonyl structures with sulfide substituents, under these conditions, reduction of the carbonyl group occurred leading to the corresponding alcohols (18 [25], 22) as the only products in high yield. It means that attack on sulfur is not connected with the presence of a C–S bond but a sulfinyl substituent is required.
In a former research performed by Fernandes and Romão [26], an α-sulfinyl-substituted ester (methyl(phenylsulfinyl)acetate) was treated by phenylsilane, but using a silane/MoO2Cl2 system as a catalyst. Under those conditions both functional groups were reduced, leading to the corresponding α-phenylsulfenyl alcohol. The desulfinylation process of compounds of this type by phenylsilane is the first observation. It is unknown until now in literature. One could assume that the active species, usually postulated in the fluoride- and base-catalyzed hydrosilylation [27-30], in the presented case is additionally chelated by both polar carbonyl substituents. It facilitates hydride anion formation, which attacks the most electrophilic center of a particular structure.
Conclusion
A phenylsilane/KOH system was successfully applied as a reducing agent for conversion of different cyclopropylcarboxylates to the corresponding alcohols. However, our study revealed that the presence of a sulfinyl substituent in the α-position to the carboxylate moiety totally changes the direction of the reaction, leading to the corresponding ester deprived of the sulfinyl group. The desulfinylation process of compounds of this type by phenylsilane was observed for the first time, so far unknown in the literature. This approach increases the number of selective desulfinylation methods which can be performed under mild conditions [31-33]. The reactivity of α-sulfinylketones with phenylsilane is less regioselective and the direction of the reaction strongly depends on the character of the ketone.
Supporting Information
| Supporting Information File 1: Experimental and analytical data and NMR spectra. | ||
| Format: PDF | Size: 1.0 MB | Download |
References
-
Seyden-Penne, J. Reductions by the Alumino- and Borohydrides in Organic Synthesis, 2nd ed.; Wiley: New York, 1997.
Return to citation in text: [1] -
Gribble, G. W. Chem. Soc. Rev. 1998, 27, 395–404. doi:10.1039/a827395z
Return to citation in text: [1] -
Ganem, B.; Osby, J. O. Chem. Rev. 1988, 86, 763–780. doi:10.1021/cr00075a003
Return to citation in text: [1] -
Kanth, J. V. B.; Periasamy, M. J. Org. Chem. 1991, 56, 5964–5965. doi:10.1021/jo00020a052
Return to citation in text: [1] -
Narasimhan, S.; Madhavan, S.; Prasad, K. G. J. Org. Chem. 1995, 60, 5314–5315. doi:10.1021/jo00121a060
Return to citation in text: [1] -
Reding, M. T.; Buchwald, S. L. J. Org. Chem. 1995, 60, 7884–7890. doi:10.1021/jo00129a031
Return to citation in text: [1] -
Jagdale, A. R.; Paraskar, A. S.; Sudalai, A. Synthesis 2009, 660–664. doi:10.1055/s-0028-1083353
Return to citation in text: [1] -
Morales-Serna, J. A.; García-Rios, E.; Bernal, J.; Paleo, E.; Gaviño, R.; Cárdenas, J. Synthesis 2011, 1375–1382. doi:10.1055/s-0030-1259988
Return to citation in text: [1] -
Pouilloux, Y.; Autin, F.; Barrault, J. Catal. Today 2000, 63, 87–100. doi:10.1016/S0920-5861(00)00448-X
Return to citation in text: [1] -
Marciniec, B.; Maciejewski, H.; Pietraszuk, C.; Pawluć, P. Hydrosilylation. In A Comprehensive Review on Recent Advances; Marciniec, B., Ed.; Springer, 2009; pp 290–326.
Return to citation in text: [1] -
Marciniec, B. Coord. Chem. Rev. 2005, 249, 2374–2390. doi:10.1016/j.ccr.2005.02.025
Return to citation in text: [1] -
Addis, D.; Das, S.; Junge, K.; Beller, M. Angew. Chem., Int. Ed. 2011, 50, 6004–6011. doi:10.1002/anie.201100145
Return to citation in text: [1] -
Marciniec, B.; Gulinsky, J.; Urbaniak, W.; Kornetka, Z. W. In Comprehensive Handbook on Hydrosilylation; Marciniec, B., Ed.; Pergamon: Oxford, 1992.
Return to citation in text: [1] -
Ojima, I. In The Chemistry of Organic Silicon Compounds; Patai, S.; Rappoport, Z., Eds.; Wiley: Chichester, 1989; Vol. 1.
Return to citation in text: [1] -
Fedorov, A.; Toutov, A. A.; Swisher, N. A.; Grubbs, R. H. Chem. Sci. 2013, 4, 1640–1645. doi:10.1039/c3sc22256j
Return to citation in text: [1] -
Fernández-Salas, J. A.; Manzini, S.; Nolan, S. P. Chem. Commun. 2013, 49, 9758–9760. doi:10.1039/c3cc45930f
Return to citation in text: [1] -
Revunova, K.; Nikonov, G. I. Chem. – Eur. J. 2014, 20, 839–845. doi:10.1002/chem.201302728
Return to citation in text: [1] [2] -
Midura, W. H.; Cypryk, M.; Rzewnicka, A.; Krysiak, J. A.; Sieroń, L. Eur. J. Org. Chem. 2016, 2064–2074. doi:10.1002/ejoc.201600111
Return to citation in text: [1] -
Midura, W. H.; Krysiak, J.; Rzewnicka, A.; Supeł, A.; Łyżwa, P.; Ewas, A. M. Tetrahedron 2013, 69, 730–737. doi:10.1016/j.tet.2012.10.085
Return to citation in text: [1] -
Krysiak, J.; Midura, W. H.; Wieczorek, W.; Sieroń, L.; Mikołajczyk, M. Tetrahedron: Asymmetry 2010, 21, 1486–1493. doi:10.1016/j.tetasy.2010.04.051
Return to citation in text: [1] -
Midura, W. H.; Rzewnicka, A.; Krysiak, J. A. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 180–181. doi:10.1080/10426507.2016.1250763
Preliminary results of this work were presented ISOCS-27, Jena, 2016.
Return to citation in text: [1] -
Midura, W. H.; Krysiak, J.; Mikołajczyk, M. Tetrahedron: Asymmetry 2003, 14, 1245–1249. doi:10.1016/S0957-4166(03)00211-8
Return to citation in text: [1] -
Midura, W. H.; Rzewnicka, A. Tetrahedron: Asymmetry 2013, 24, 937–941. doi:10.1016/j.tetasy.2013.05.024
Return to citation in text: [1] -
Yamashita, T.; Yasueda, H.; Miyauchi, Y.; Nakamura, N. Bull. Chem. Soc. Jpn. 1977, 50, 1532–1534. doi:10.1246/bcsj.50.1532
Return to citation in text: [1] -
Sun, J.; Yang, M.; Yuan, F.; Jia, X.; Pan, Y.; Zhu, C. Adv. Synth. Catal. 2009, 351, 920–930. doi:10.1002/adsc.200800767
Return to citation in text: [1] -
Fernandes, A. C.; Romão, C. C. J. Mol. Catal. A: Chem. 2006, 253, 96–98. doi:10.1016/j.molcata.2006.03.006
Return to citation in text: [1] -
Lawrence, N. J.; Drew, M. D.; Bushell, S. M. J. Chem. Soc., Perkin Trans. 1 1999, 3381–3391. doi:10.1039/a903662h
Return to citation in text: [1] -
Kobayashi, Y.; Takahisa, E.; Nakano, M.; Watatani, K. Tetrahedron 1997, 53, 1627–1634. doi:10.1016/S0040-4020(96)01092-7
Return to citation in text: [1] -
Hojo, M.; Murakami, C.; Fujii, A.; Hosomi, A. Tetrahedron Lett. 1999, 40, 911–914. doi:10.1016/S0040-4039(98)02480-0
Return to citation in text: [1] -
Hojo, M.; Fujii, A.; Murakami, C.; Aihara, H.; Hosomi, A. M. Tetrahedron Lett. 1995, 36, 571–574. doi:10.1016/0040-4039(94)02255-A
Return to citation in text: [1] -
Capozzi, M. A. M.; Cardellicchio, C.; Naso, F.; Tortorella, P. J. Org. Chem. 2000, 65, 2843–2846. doi:10.1021/jo991912q
Return to citation in text: [1] -
García Ruano, J. L.; Marzo, L.; Marcos, V.; Alvarado, C.; Alemán, J. Chem. – Eur. J. 2012, 18, 9775–9779. doi:10.1002/chem.201201607
Return to citation in text: [1] -
Frutos, M.; Ortuño, M. A.; Lledos, A.; Viso, A.; Fernández de la Pradilla, R.; de la Torre, M. C.; Sierra, M. A.; Gornitzka, H.; Hemmert, C. Org. Lett. 2017, 19, 822–825. doi:10.1021/acs.orglett.6b03808
Return to citation in text: [1]
| 1. | Seyden-Penne, J. Reductions by the Alumino- and Borohydrides in Organic Synthesis, 2nd ed.; Wiley: New York, 1997. |
| 2. | Gribble, G. W. Chem. Soc. Rev. 1998, 27, 395–404. doi:10.1039/a827395z |
| 15. | Fedorov, A.; Toutov, A. A.; Swisher, N. A.; Grubbs, R. H. Chem. Sci. 2013, 4, 1640–1645. doi:10.1039/c3sc22256j |
| 16. | Fernández-Salas, J. A.; Manzini, S.; Nolan, S. P. Chem. Commun. 2013, 49, 9758–9760. doi:10.1039/c3cc45930f |
| 17. | Revunova, K.; Nikonov, G. I. Chem. – Eur. J. 2014, 20, 839–845. doi:10.1002/chem.201302728 |
| 31. | Capozzi, M. A. M.; Cardellicchio, C.; Naso, F.; Tortorella, P. J. Org. Chem. 2000, 65, 2843–2846. doi:10.1021/jo991912q |
| 32. | García Ruano, J. L.; Marzo, L.; Marcos, V.; Alvarado, C.; Alemán, J. Chem. – Eur. J. 2012, 18, 9775–9779. doi:10.1002/chem.201201607 |
| 33. | Frutos, M.; Ortuño, M. A.; Lledos, A.; Viso, A.; Fernández de la Pradilla, R.; de la Torre, M. C.; Sierra, M. A.; Gornitzka, H.; Hemmert, C. Org. Lett. 2017, 19, 822–825. doi:10.1021/acs.orglett.6b03808 |
| 10. | Marciniec, B.; Maciejewski, H.; Pietraszuk, C.; Pawluć, P. Hydrosilylation. In A Comprehensive Review on Recent Advances; Marciniec, B., Ed.; Springer, 2009; pp 290–326. |
| 11. | Marciniec, B. Coord. Chem. Rev. 2005, 249, 2374–2390. doi:10.1016/j.ccr.2005.02.025 |
| 12. | Addis, D.; Das, S.; Junge, K.; Beller, M. Angew. Chem., Int. Ed. 2011, 50, 6004–6011. doi:10.1002/anie.201100145 |
| 13. | Marciniec, B.; Gulinsky, J.; Urbaniak, W.; Kornetka, Z. W. In Comprehensive Handbook on Hydrosilylation; Marciniec, B., Ed.; Pergamon: Oxford, 1992. |
| 14. | Ojima, I. In The Chemistry of Organic Silicon Compounds; Patai, S.; Rappoport, Z., Eds.; Wiley: Chichester, 1989; Vol. 1. |
| 9. | Pouilloux, Y.; Autin, F.; Barrault, J. Catal. Today 2000, 63, 87–100. doi:10.1016/S0920-5861(00)00448-X |
| 26. | Fernandes, A. C.; Romão, C. C. J. Mol. Catal. A: Chem. 2006, 253, 96–98. doi:10.1016/j.molcata.2006.03.006 |
| 3. | Ganem, B.; Osby, J. O. Chem. Rev. 1988, 86, 763–780. doi:10.1021/cr00075a003 |
| 4. | Kanth, J. V. B.; Periasamy, M. J. Org. Chem. 1991, 56, 5964–5965. doi:10.1021/jo00020a052 |
| 5. | Narasimhan, S.; Madhavan, S.; Prasad, K. G. J. Org. Chem. 1995, 60, 5314–5315. doi:10.1021/jo00121a060 |
| 6. | Reding, M. T.; Buchwald, S. L. J. Org. Chem. 1995, 60, 7884–7890. doi:10.1021/jo00129a031 |
| 7. | Jagdale, A. R.; Paraskar, A. S.; Sudalai, A. Synthesis 2009, 660–664. doi:10.1055/s-0028-1083353 |
| 8. | Morales-Serna, J. A.; García-Rios, E.; Bernal, J.; Paleo, E.; Gaviño, R.; Cárdenas, J. Synthesis 2011, 1375–1382. doi:10.1055/s-0030-1259988 |
| 27. | Lawrence, N. J.; Drew, M. D.; Bushell, S. M. J. Chem. Soc., Perkin Trans. 1 1999, 3381–3391. doi:10.1039/a903662h |
| 28. | Kobayashi, Y.; Takahisa, E.; Nakano, M.; Watatani, K. Tetrahedron 1997, 53, 1627–1634. doi:10.1016/S0040-4020(96)01092-7 |
| 29. | Hojo, M.; Murakami, C.; Fujii, A.; Hosomi, A. Tetrahedron Lett. 1999, 40, 911–914. doi:10.1016/S0040-4039(98)02480-0 |
| 30. | Hojo, M.; Fujii, A.; Murakami, C.; Aihara, H.; Hosomi, A. M. Tetrahedron Lett. 1995, 36, 571–574. doi:10.1016/0040-4039(94)02255-A |
| 23. | Midura, W. H.; Rzewnicka, A. Tetrahedron: Asymmetry 2013, 24, 937–941. doi:10.1016/j.tetasy.2013.05.024 |
| 24. | Yamashita, T.; Yasueda, H.; Miyauchi, Y.; Nakamura, N. Bull. Chem. Soc. Jpn. 1977, 50, 1532–1534. doi:10.1246/bcsj.50.1532 |
| 22. | Midura, W. H.; Krysiak, J.; Mikołajczyk, M. Tetrahedron: Asymmetry 2003, 14, 1245–1249. doi:10.1016/S0957-4166(03)00211-8 |
| 25. | Sun, J.; Yang, M.; Yuan, F.; Jia, X.; Pan, Y.; Zhu, C. Adv. Synth. Catal. 2009, 351, 920–930. doi:10.1002/adsc.200800767 |
| 21. |
Midura, W. H.; Rzewnicka, A.; Krysiak, J. A. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 180–181. doi:10.1080/10426507.2016.1250763
Preliminary results of this work were presented ISOCS-27, Jena, 2016. |
| 18. | Midura, W. H.; Cypryk, M.; Rzewnicka, A.; Krysiak, J. A.; Sieroń, L. Eur. J. Org. Chem. 2016, 2064–2074. doi:10.1002/ejoc.201600111 |
| 19. | Midura, W. H.; Krysiak, J.; Rzewnicka, A.; Supeł, A.; Łyżwa, P.; Ewas, A. M. Tetrahedron 2013, 69, 730–737. doi:10.1016/j.tet.2012.10.085 |
| 20. | Krysiak, J.; Midura, W. H.; Wieczorek, W.; Sieroń, L.; Mikołajczyk, M. Tetrahedron: Asymmetry 2010, 21, 1486–1493. doi:10.1016/j.tetasy.2010.04.051 |
| 17. | Revunova, K.; Nikonov, G. I. Chem. – Eur. J. 2014, 20, 839–845. doi:10.1002/chem.201302728 |
© 2017 Midura et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)