Abstract
Spiropyrans bearing an N-alkylcarboxylate tether are a common structure in dynamic, photoactive materials and serve as colourimetric/fluorimetric cation receptors. In this study, we describe an efficient synthesis of spiropyrans with 2–12 carbon atom alkylcarboxylate substituents, and a systematic analysis of their interactions with metal cations using 1H NMR and UV-visible spectroscopy. All N-alkylcarboxyspiropyrans in this study displayed a strong preference for binding divalent metal cations and a modest increase in M2+ binding affinity correlated with increased alkycarboxylate tether length.

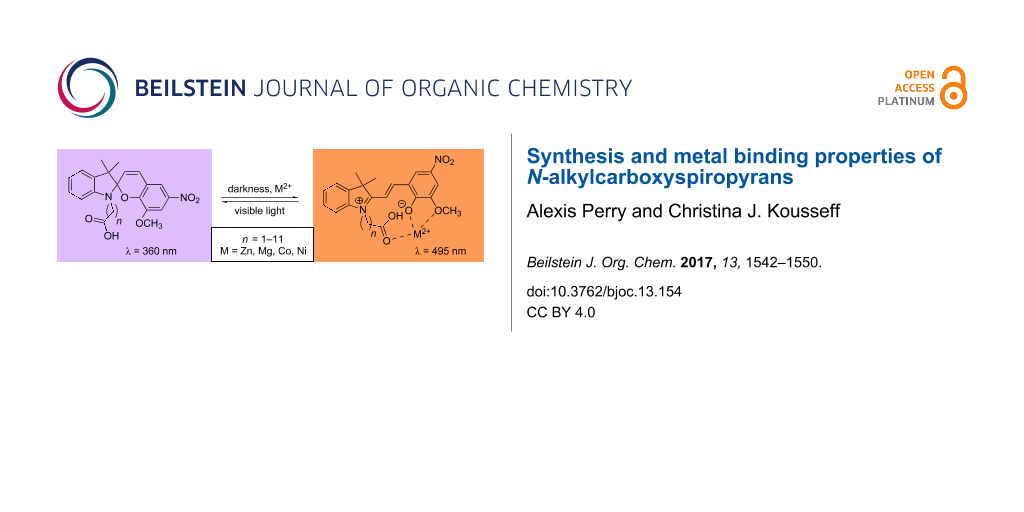
Graphical Abstract
Introduction
Spiropyrans are a class of spiro-fused indolochromene (e.g., C4SP, Scheme 1) which exist in photo-controlled equilibrium with their zwitterionic, fully-conjugated merocyanine isomer [1] (e.g., C4MC, Scheme 1). The controllable nature of this isomerisation and the dramatic differences in physical and chemical properties displayed by the isomeric forms, coupled with the facile elaboration of the spiropyran core, have made spiropyran–merocyanine systems a common motif in molecular switch and sensing applications [2]. In this respect, the difference in optical properties between colourless, non-fluorescent spiropyran and coloured, fluorescent merocyanine has been extensively exploited [2].
Spiropyrans bearing an N-alkylcarboxylate tether are photo-reversible colourimetric/fluorimetric receptors for metal cations [3-5] and amino acids [6], and serve as convenient building blocks in the synthesis of dynamic materials [7] (Figure 1). In this latter role, N-alkylcarboxyspiropyrans have been tagged (via ester or amide linkage) to carbon nanotubes (e.g., for photocontrolled Zn2+ delivery in biological media [8]) and gold electrodes (for photoswitching the bioelectrocatalytic cascade of cytochrome c/cytochrome oxidase [9]), and attachment to nanoparticles has enabled photomodulation of nanoparticle fluorescence [10,11]. Similarly, tagged N-alkylcarboxyspiropyrans have provided the basis for photoactive biopolymers, e.g., polypeptides with photocontrolled folding [12], light-activated enzymes [13], light-enhanced affinity chromatography using spiropyran-modified agarose gel [14,15] and the creation of light-responsive nanopores through modification of natural channel proteins [16]. Furthermore, addition of an N-alkylcarboxylate moiety is a common tactic used to enhance water solubility of spiropyran derivatives [6].
![[1860-5397-13-154-1]](/bjoc/content/figures/1860-5397-13-154-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: General uses of N-alkylcarboxyspiropyrans.
Figure 1: General uses of N-alkylcarboxyspiropyrans.
Spiropyran–metal cation binding occurs as a result of stabilisation of the zwitterionic merocyanine isomer via phenoxide–metal complexation [17] (Figure 1). Commonly, merocyanines undergo photoreversion to their corresponding spiropyran under visible light irradiation and metal complexation is usually achieved either in darkness or under UV irradiation. Greater stabilisation of merocyanine–metal cation complexes is often achieved through the addition of extra ligation sites and this effect is particularly pronounced in structures bearing an 8′-OMe substituent with respect to binding divalent metal cations [18] (Scheme 1). The extension of this basic bidentate ligand with further substituents has been employed to generate structures with bespoke binding characteristics (e.g., metal ion specificity, control of complex stoichiometry, greater binding affinity) [17]. Typifying this approach, Natali et al. synthesised butanoate-tagged spiropyran C4SP and demonstrated its high affinity for Zn2+ and Cu2+ through the involvement of the N-alkylcarboxylate in metal binding [3] (Scheme 1).
![[1860-5397-13-154-i1]](/bjoc/content/inline/1860-5397-13-154-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: C4SP–C4MC spiropyran-merocyanine equilibrium and M2+ binding.
Scheme 1: C4SP–C4MC spiropyran-merocyanine equilibrium and M2+ binding.
Given the evident importance and widespread application of N-alkylcarboxyspiropyrans as building blocks in the synthesis of photoactive materials – and as interesting molecules in themselves – we describe herein an efficient synthesis of N-alkylcarboxyspiropyrans (analogous to C4SP) with alkyl chain lengths from C2 to C12, and a systematic analysis of their metal-binding properties across a range of metal cations. It was envisaged that spiropyrans bearing incrementally-spaced carboxylate groups on conformationally flexible tethers would present trends in their metal-binding characteristics and that such information would have relevance to the application of N-alkylcarboxyspiropyrans generally.
Results and Discussion
Synthesis
Commonly, the synthesis of N-alkylcarboxyspiropyrans (e.g., C4SP) involves N-alkylation of 2,3,3-trimethylindolenine (1) with a bromoalkanoic acid or ester 2, isolation of the resulting indolium salt 3 or treatment with base to generate the exo-methylene enamine 4, then condensation with the appropriate salicylaldehyde 5 (and ester hydrolysis if required) (Scheme 2).
![[1860-5397-13-154-i2]](/bjoc/content/inline/1860-5397-13-154-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: General synthesis of N-alkylcarboxyspiropyrans.
Scheme 2: General synthesis of N-alkylcarboxyspiropyrans.
Initially, we followed the conditions employed by Natali et al. in the three-step synthesis of butyric acid derivative C4SP, which involved: (i) alkylation of 2,3,3-trimethylindolenine (1) with ethyl 4-bromobutyrate (2; R = Et, n = 3) in chloroform; (ii) condensation of the resulting indolium salt 3 with 3-methoxy-5-nitrosalicylaldehyde (5) in ethanol; (iii) basic ester hydrolysis [3] (Scheme 2). Unfortunately, in our hands the alkylation was extremely sluggish, and both this step and the final ester hydrolysis were low yielding. Consequently, we pursued a more efficient approach, reacting trimethylindolenine (1) directly with bromoalkanoic acids 2a–g, obviating the requirement for ester hydrolysis (Table 1). N-Alkylation of trimethylindolenine (1) with bromoalkanoic acids/esters is often slow and the solvent choice is crucial in identifying viable, robust conditions. Reported protocols have employed chloroform [3], acetonitrile [19], acetone [20], nitromethane [21], 1,2-dichlorobenzene [22], toluene [23], reaction in the absence of solvent [24] or use of microwave irradiation [25]. We found that refluxing acetonitrile was relatively effective, if slow (completion in ca. 60 h), and that a more rapid process occurred if the solvent was allowed to evaporate during the course of the reaction (completion in ca. 20 h). The reaction in the absence of solvent was less effective, perhaps due to inefficient stirring of the small reaction volume (ca. 0.3 mL) or the absence of polar aprotic solvent-mediated acceleration of this SN2 process (Table 1, compare entries 6, 7 and 8).
Table 1: Synthesis of spiropyrans C2–C12SP.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-154-i5.svg?max-width=637&scale=1.0)
|
||||||
| Entry | n | Bromoacid |
Alkylation
conditionsa |
Condensation
conditionsb |
Product |
Yield
from 1 |
| 1 | 1 | 2a | method A | – | C2SP | 0% |
| 2 | 1 | 2a | method Bc | method Cc | C2SP | 21% |
| 3 | 2 | 2b | method A | method C | C3SP | 81% |
| 4 | 3 | 2c | method A | – | C4SP | 0% |
| 5 | 3 | 2cd | method A | method D | C4SP | 38% |
| 6 | 5 | 2d | method B | method C | C6SP | 52% |
| 7 | 5 | 2d | method A | method C | C6SP | 71% |
| 8 | 5 | 2d | method Ae | method C | C6SP | 48% |
| 9 | 7 | 2e | method A | method C | C8SP | 60% |
| 10 | 9 | 2f | method A | method C | C10SP | 82% |
| 11 | 11 | 2g | method A | method C | C12SP | 67% |
aMethod A: Solution of bromoacid (1 equiv) and 2,3,3-trimethylindolenine (1 equiv) in acetonitrile (0.6 M) heated at 80 °C without a condenser for 20 h; Method B: Solution of bromoacid (1 equiv) and 2,3,3-trimethylindolenine (1 equiv) in acetonitrile (0.6 M) heated at reflux for 72 h. bMethod C: Solution of crude indolium salt (1 equiv), salicylaldehyde 5 (1 equiv) and piperidine (1 equiv) in MEK (50 mM) heated at reflux for 20 h; Method D: (i) Solution of crude indolium salt (1 equiv) and salicylaldehyde 5 (1 equiv) in ethanol (0.1 M) heated at reflux for 20 h then (ii) solution of crude spiropyran stirred in 2:1 THF/NaOH (50 mM). cConducted at room temperature. dReaction used ethyl 4-bromobutyrate. eConducted without solvent.
The relative instability of indolium salts is well-documented [26] and our attempts to purify compounds 3 by silica gel chromatography or recrystallisation were unsuccessful. In light of this, and given that our alkylation reactions were relatively clean, crude indolium salts 3 were then condensed with 3-methoxy-5-nitrosalicylaldehyde (5) to give the required spiropyrans CnSP (Table 1). Commonly, this has been achieved through simple reflux in ethanol; however, this transformation proved relatively ineffective for alkylcarboxyindolium salts 3 and consequently, we applied alternative conditions using MEK and piperidine [27]. This two-step approach was used to synthesise a range of N-alkylcarboxyspiropyrans derived from bromoalkanoic acids of varying chain length in two-step yields from 60–82% (Table 1, entries 3, 7, and 9–11).
This protocol was ineffective for the synthesis of C2SP (n = 1) and C4SP (n = 3). In the former case, the intermediate N-ethanoate indolium salt 3a underwent thermal decarboxylation to give N-methylindolium salt 7, in a similar manner to that previously reported [28]. Presumably, this reaction proceeds via azomethine ylid 6 (Scheme 3); analogous indolium ylids have been used synthetically in 1,3-dipolar cycloadditions [29] and mechanistic studies have been published on the related decarboxylation of pyridinium 2-carboxylates [30]. Fortunately, α-bromocarbonyls such as bromoacetic acid are excellent SN2 electrophiles, and N-alkylation of 2,3,3-trimethylindolenine (1) with bromoacetic acid (2a) was successful, if somewhat sluggish, at room temperature. Reaction of the crude indolium salt 3a with 3-methoxy-5-nitrosalicylaldehyde (5) was also conducted at room temperature and generated the ethanoic acid substituted spiropyran C2SP, albeit in modest yield (Table 1, entry 2).
![[1860-5397-13-154-i3]](/bjoc/content/inline/1860-5397-13-154-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Decarboxylation of N-ethanoic acid indolium salt 3a.
Scheme 3: Decarboxylation of N-ethanoic acid indolium salt 3a.
Our attempts to synthesise C4SP using our two-step protocol were undermined by the ineffective alkylation of trimethylindolenine (1) with 4-bromobutyric acid (2c). Under our alkylation conditions, intramolecular lactonisation of 4-bromobutyric acid to γ-butyrolactone (8) was more rapid than N-alkylation and no indolium product was observed (Scheme 4). Consequently, to obtain C4SP, we employed an optimised version of the three-step procedure of Natali et al. [3], wherein lactonisation is avoided through the use of ethyl 4-bromobutyrate, and basic ester hydrolysis is required as a final step (Table 1, entry 5).
![[1860-5397-13-154-i4]](/bjoc/content/inline/1860-5397-13-154-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Lactonisation of 4-bromobutyric acid 2c.
Scheme 4: Lactonisation of 4-bromobutyric acid 2c.
Binding studies
Analysis of spiropyran–merocyanine equilibria with respect to metal binding has frequently employed UV-visible spectroscopy. Merocyanine–metal cation complexes absorb strongly in the visible range (often 450–600 nm), giving rapid access to clear, quantitative data at low concentrations and without interference from paramagnetic transition metal cations (a feature which causes significant problems with NMR analysis). Conversely, use of bound merocyanine–metal cation absorbance intensity as the sole metric for quantification of metal binding of different metal cations should be undertaken with caution. Direct comparison of absorbance intensity measurements (as a surrogate for concentration) is only valid if the molar extinction coefficient, ε, remains constant for all merocyanine complexes of different metals, and indeed for different merocyanine complexes of the same metal. We thus sought initial validation of UV–vis data by direct comparison with data from 1H NMR, derived from merocyanine complexes of two non-paramagnetic metal cations, Mg2+ and Zn2+. 1H NMR allows rapid assessment of spiropyran:merocyanine via measurement and comparison of integrals, and hence enables calculation of [merocyanine]. In turn, [merocyanine] can be applied to quantify the corresponding UV data. Related approaches have employed IR spectroscopy [31] and partial least squares regression analysis [32] as adjuncts to UV–vis spectroscopy in the analysis of merocyanine binding across ranges of transition metal cations.
We prepared solutions of C2SP–C12SP in CD3CN to which were added aqueous solutions of either Zn(NO3)2·6H2O or Mg(NO3)2·6H2O. These samples were allowed to equilibrate in darkness at room temperature overnight and were then analysed by 1H NMR spectroscopy. Following this, the samples were diluted, allowed to re-equilibrate in darkness, and then analysed by UV–vis spectroscopy. Similar Zn2+ and Mg2+ samples were prepared with N-methyl spiropyran 9 (Figure 2, prepared according to a reported procedure [33]), which served as a carboxylate-free control.
![[1860-5397-13-154-2]](/bjoc/content/figures/1860-5397-13-154-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
The results from these experiments, and example spectra, are shown in Figure 3 and Figure 4, respectively, from which two trends are apparent: (i) ε remains relatively constant for a given compound, irrespective of divalent metal cation; (ii) ε decreases slightly with increasing chain length. Unexpectedly, 1H NMR analysis of C3SP in the presence of Zn2+ and Mg2+ produced complex and intractable spectra and it would appear that these metal salts are able to promote degradation of this compound. Irrespective of the precise fate of C3SP under these conditions, without NMR evidence of merocyanine formation, this compound was necessarily omitted from this study.
![[1860-5397-13-154-3]](/bjoc/content/figures/1860-5397-13-154-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Example spectra illustrating binding studies of spiropyrans with M2+. (a) 1H NMR spectrum of C10SP (10 mM) and Zn(NO3)2·6H2O (2 mM) in CD3CN–H2O (99.9% v/v) after 18 h in darkness. Peaks corresponding to H nuclei within the spiropyran isomer are labelled; aromatic peaks from the merocyanine–Zn2+ complex are broad and unassigned. SP:MC was estimated by comparison of integral values from SP 3′-H and MC (N+CH2)/2 (as shown in red). (b) UV-visible absorbance spectrum of C10SP (0.1 mM) and Zn(NO3)2·6H2O (0.02 mM) in CD3CN/CH3CN–H2O (99.9% v/v) with MC–Zn2+ prominent at 495 nm. [C10MC–Zn2+], derived from (a), was used to calculate of ε for this absorbance.
Figure 3: Example spectra illustrating binding studies of spiropyrans with M2+. (a) 1H NMR spectrum of C10SP ...
![[1860-5397-13-154-4]](/bjoc/content/figures/1860-5397-13-154-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: ε for MC–M2+ complexes of C2SP–C12SP and 9: (left) with Zn2+; (right) with Mg2+. Values for ε were calculated by application of 1H NMR-derived values for [MC–M2+] to UV–vis spectroscopy-derived absorbance intensities of MC–M2+ (see Figure 3).
Figure 4: ε for MC–M2+ complexes of C2SP–C12SP and 9: (left) with Zn2+; (right) with Mg2+. Values for ε were ...
On the basis that the nature of the metal cation appeared to have minimal influence upon the merocyanine extinction coefficient, we were able to use UV–vis spectroscopy to rapidly assess the impact of binding various metal cations upon compounds C2SP–C12SP and 9 by applying the NMR-derived ε values (average of ε for MC–Zn2+ and MC–Mg2+ for each compound) to calculate approximate [merocyanine] from merocyanine absorbance intensity (Figure 5). Although this is based on the analysis of only two metal cations, studies using dynamic modelling have displayed similar, limited impact of different metal cations upon merocyanine ε values [34] so we have cautious confidence in the validity of this approach. All compounds, including control compound 9, showed selectivity for M2+ over M+ which follows the pattern previously observed for merocyanines bearing an 8′-OMe substituent [18]. Broadly speaking, the effects upon metal binding of incremental increase in alkylcarboxylate chain length were subtle. The general trends were: (i) compounds bearing longer tethers produced higher concentrations of MC–M2+ complex per unit metal ion (presumably on the basis that carboxylates on longer tethers with greater conformational flexibility are able to interact more effectively with cations, despite their inherent entropic penalty [35]); (ii) affinity for metals was in the order Zn2+ > Mg2+ > Co2+ > Ni2+. Binding to Ni2+ was considerably less effective than for other divalent metal cations and this was particularly pronounced for non-carboxylate 9, wherein 9–Ni2+ complexation did not exceed background [merocyanine]. Merocyanine complexation of Cu2+ was also investigated; however, the absorbance wavelength for MC–Cu2+ (≈400 nm) overlapped with that of the larger spiropyran absorbance (≈370 nm) and we were unable to extract meaningful data from this. This is consistent with related studies [3] and, furthermore, spiropyran-based Cu2+ detection can be complicated by Cu2+-catalysed spiropyran dimerisation [36]. In the light of the results from Natali et al. [3] – in which evidence for involvement of a carboxylate ligand in metal binding was presented but no comparison was made against a control – it is reassuring to note that incorporation of such a ligand into the merocyanine structure did increase the affinity for Zn2+, Mg2+ and Co2+ over that seen in the non-carboxylate control compound 9.
![[1860-5397-13-154-5]](/bjoc/content/figures/1860-5397-13-154-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: [MC] for compounds C2SP–C12SP and 9 in the presence of various metal cations. Solutions of spiropyrans (0.1 mM, 1 equiv) and metal nitrates (0.04 mM, 0.4 equiv) in CH3CN–H2O (99.9% v/v) were kept in darkness for 18 h then analysed by UV–vis spectroscopy. Values for MC absorbance intensity were converted into [MC] using appropriate ε values.
Figure 5: [MC] for compounds C2SP–C12SP and 9 in the presence of various metal cations. Solutions of spiropyr...
It is apparent from these data that the length of the carboxylate tether can also affect the spiropyran–merocyanine equilibrium in the absence of metal cations (Figure 5 and Figure 6). All compounds tested existed as mixtures of spiropyran and merocyanine isomers at 0.1 mM in acetonitrile in darkness. For shorter-chain compounds C2SP and C4SP, and control compound 9, this resulted in an approximate 2:8 MC:SP ratio. Longer chain carboxylates C6SP–C12SP, however, showed higher merocyanine concentrations and these data appear to indicate a sharp threshold between 4-C and 6-C carboxylates. Correspondingly, we synthesised 5-C carboxylate C5SP (using our standard protocol; 77%) and assessed its MC:SP equilibrium behaviour under similar conditions. In this case, [C5MC] resembled [merocyanine] values for the shorter chain compounds and reinforced the idea of a threshold distance required for carboxylate-mediated merocyanine stabilisation. It is as yet unclear how a long carboxylate tether might stabilise a merocyanine structure whilst structures bearing a superficially similar tether do not. To probe involvement of the carboxylate/carboxylic acid moiety (e.g., in forming inter- or intramolecular hydrogen bonds) we prepared C6 ester derivative 10 (Figure 7) as a direct point of comparison with C6SP. In the absence of a hydrogen bond donor group, the concentration of 10MC at equilibrium is approximately half of that observed for C6MC under identical conditions. On the other hand, this value for [10MC] is considerably higher than that recorded with shorter chain carboxylates. It would appear, therefore, that tether length is the defining factor in merocyanine stabilisation and that only beyond a threshold distance does hydrogen bond donation of the carboxylic acid become important. This is a key point in designing functional materials based on N-alkylcarboxyspiropyrans.
![[1860-5397-13-154-6]](/bjoc/content/figures/1860-5397-13-154-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: [MC] for spiropyrans C2SP–C12SP, 9 and 10 (0.1 mM) in CH3CN–H2O (99.9% v/v). Samples were kept in darkness for 18 h then analysed by UV–vis spectroscopy. Values for MC absorbance intensity were converted into [MC] using appropriate ε values.
Figure 6: [MC] for spiropyrans C2SP–C12SP, 9 and 10 (0.1 mM) in CH3CN–H2O (99.9% v/v). Samples were kept in d...
![[1860-5397-13-154-7]](/bjoc/content/figures/1860-5397-13-154-7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, we have developed a high-yielding, two-step synthesis of N-alkylcarboxyspiropyrans via N-alkylation of trimethylindolenine with bromoalkanoic acids, then condensation of the resulting indolium salts with methoxynitrosalicylaldehyde. This protocol is generally effective and enabled the synthesis of a range of spiropyrans bearing different length alkylcarboxylate tethers; however, it is ineffective with bromoacetic acid and bromobutyric acid, where decarboxylation and intramolecular lactonisation respectively compete with N-alkylation. In these cases, we have developed alternative, though lower-yielding procedures.
N-Alkylcarboxyspiropyrans can function as colourimetric/fluorimetric receptors for metal cations via complexation of the merocyanine isomer. Consequently, we have assessed the metal binding behaviour of spiropyrans bearing N-acetic acid through to N-dodecanoic acid tethers by 1H NMR and UV–vis spectroscopy. All compounds tested displayed a strong preference for divalent over monovalent metal cations and a modest increase in M2+ binding affinity correlated with increasing alkycarboxylate tether length.
This paper details a clear, effective protocol for the synthesis of N-alkylcarboxyspiropyrans and a thorough analysis of the effect of metal cations upon their spiropyran–merocyanine equilibria. Consequently, the results of this study will impact upon the design and synthesis of dynamic functional materials based on spiropyran–merocyanine units, with particular relevance to cases where such materials are used in the presence of metal cations (e.g., in biological media).
Supporting Information
| Supporting Information File 1: Full experimental details. | ||
| Format: PDF | Size: 189.5 KB | Download |
References
-
Fischer, E.; Hirshberg, Y. J. Chem. Soc. 1952, 11, 4522–4524.
Return to citation in text: [1] -
Lukyanov, B. S.; Lukyanova, M. B. Chem. Heterocycl. Compd. 2005, 41, 281–311. doi:10.1007/s10593-005-0148-x
Return to citation in text: [1] [2] -
Natali, M.; Aakeröy, C.; Desper, J.; Giordani, S. Dalton Trans. 2010, 39, 8269–8277. doi:10.1039/c0dt00242a
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Krikun, V. M.; Sadimenko, L. P.; Voloshina, E. N.; Voloshin, N. A. Russ. J. Gen. Chem. 2009, 79, 1191–1196. doi:10.1134/S1070363209060279
Return to citation in text: [1] -
Wojtyk, J. T. C.; Kazmaier, P. M.; Buncel, E. Chem. Mater. 2001, 13, 2547–2551. doi:10.1021/cm010038q
Return to citation in text: [1] -
Liu, Y.; Fan, M.; Zhang, S.; Sheng, X.; Yao, J. New J. Chem. 2007, 31, 1878–1881. doi:10.1039/b713247f
Return to citation in text: [1] [2] -
Klajn, R. Chem. Soc. Rev. 2014, 43, 148–184. doi:10.1039/C3CS60181A
Return to citation in text: [1] -
Del Canto, E.; Natali, M.; Movia, D.; Giordani, S. Phys. Chem. Chem. Phys. 2012, 14, 6034–6043. doi:10.1039/c2cp40275k
Return to citation in text: [1] -
Lion-Dagan, M.; Katz, E.; Willner, I. J. Chem. Soc., Chem. Commun. 1994, 2741–2742. doi:10.1039/c39940002741
Return to citation in text: [1] -
Medintz, I. L.; Trammell, S. A.; Mattoussi, H.; Mauro, J. M. J. Am. Chem. Soc. 2004, 126, 30–31. doi:10.1021/ja037970h
Return to citation in text: [1] -
Tomasulo, M.; Yildiz, I.; Raymo, F. M. Aust. J. Chem. 2006, 59, 175–178. doi:10.1071/CH05332
Return to citation in text: [1] -
Ciardelli, F.; Fabbri, D.; Pieroni, O.; Fissi, A. J. Am. Chem. Soc. 1989, 111, 3470–3472. doi:10.1021/ja00191a076
Return to citation in text: [1] -
Ito, Y.; Sugimura, N.; Kwon, O. H.; Imanishi, Y. Nat. Biotechnol. 1999, 17, 73–75. doi:10.1038/5250
Return to citation in text: [1] -
Karube, I.; Ishimori, Y.; Suzuki, S.; Sato, T. Biotechnol. Bioeng. 1978, 20, 1775–1783. doi:10.1002/bit.260201107
Return to citation in text: [1] -
Karube, I.; Ishimori, Y.; Suzuki, S. Anal. Biochem. 1978, 86, 100–106. doi:10.1016/0003-2697(78)90322-6
Return to citation in text: [1] -
Koçer, A.; Walko, M.; Meijberg, W.; Feringa, B. L. Science 2005, 309, 755–758. doi:10.1126/science.1114760
Return to citation in text: [1] -
Paramonov, S. V.; Lokshin, V.; Fedorova, O. A. J. Photochem. Photobiol., C 2011, 12, 209–236. doi:10.1016/j.jphotochemrev.2011.09.001
Return to citation in text: [1] [2] -
Roxburgh, C. J.; Sammes, P. G.; Abdullah, A. Eur. J. Inorg. Chem. 2008, 4951–4960. doi:10.1002/ejic.200800614
Return to citation in text: [1] [2] -
Lopalco, M.; Koini, E. N.; Cho, J. K.; Bradley, M. Org. Biomol. Chem. 2009, 7, 856–859. doi:10.1039/b820719b
Return to citation in text: [1] -
Pertusati, F.; Menger, F. M. J. Org. Chem. 2008, 73, 2939–2942. doi:10.1021/jo7027294
Return to citation in text: [1] -
Kvach, M. V.; Ustinov, A. V.; Stepanova, I. A.; Malakhov, A. D.; Skorobogatyi, M. V.; Shmanai, V. V.; Korshun, V. A. Eur. J. Org. Chem. 2008, 2107–2117. doi:10.1002/ejoc.200701190
Return to citation in text: [1] -
Kiyose, K.; Hanaoka, K.; Oushiki, D.; Nakamura, T.; Kajimura, M.; Suematsu, M.; Nishimatsu, H.; Yamane, T.; Terai, T.; Hirata, Y.; Nagano, T. J. Am. Chem. Soc. 2010, 132, 15846–15848. doi:10.1021/ja105937q
Return to citation in text: [1] -
An, H.-W.; Qiao, S.-L.; Hou, C.-Y.; Lin, Y.-X.; Li, L.-L.; Xie, H.-Y.; Wang, Y.; Wang, L.; Wang, H. Chem. Commun. 2015, 51, 13488–13491. doi:10.1039/C5CC05395A
Return to citation in text: [1] -
Zhang, Y.; Xiao, L.; Popovic, K.; Xie, X.; Chordia, M. D.; Chung, L. W. K.; Williams, M. B.; Yue, W.; Pan, D. Bioorg. Med. Chem. Lett. 2013, 23, 6350–6354. doi:10.1016/j.bmcl.2013.09.074
Return to citation in text: [1] -
Altevogt, A.; Flehr, R.; Gehne, S.; Kumke, M. U.; Bannwarth, W. Helv. Chim. Acta 2012, 95, 543–555. doi:10.1002/hlca.201100460
Return to citation in text: [1] -
Owens, E. A.; Bruschi, N.; Tawney, J. G.; Henary, M. Dyes Pigm. 2015, 113, 27–37. doi:10.1016/j.dyepig.2014.07.035
Return to citation in text: [1] -
Chen, J.; Zeng, F.; Wu, S.; Chen, Q.; Tong, Z. Chem. – Eur. J. 2008, 14, 4851–4860. doi:10.1002/chem.200701994
Return to citation in text: [1] -
Wang, B.; Fan, J.; Sun, S.; Wang, L.; Song, B.; Peng, X. Dyes Pigm. 2010, 85, 43–50. doi:10.1016/j.dyepig.2009.10.002
Return to citation in text: [1] -
Fishwick, C. W. G.; Jones, A. D.; Mitchell, M. B.; Eggleston, D. S.; Baures, P. W. Synlett 1990, 359–361. doi:10.1055/s-1990-21095
Return to citation in text: [1] -
Dunn, G. E.; Lee, G. K. J.; Thimm, H. Can. J. Chem. 1972, 50, 3017–3027. doi:10.1139/v72-480
Return to citation in text: [1] -
Fries, K. H.; Driskell, J. D.; Samanta, S.; Locklin, J. Anal. Chem. 2010, 82, 3306–3314. doi:10.1021/ac1001004
Return to citation in text: [1] -
Fries, K. H.; Driskell, J. D.; Sheppard, G. R.; Locklin, J. Langmuir 2011, 27, 12253–12260. doi:10.1021/la202344w
Return to citation in text: [1] -
Roxburgh, C. J.; Sammes, P. G.; Abdullah, A. Dyes Pigm. 2011, 90, 146–162. doi:10.1016/j.dyepig.2010.10.006
Return to citation in text: [1] -
Zakharova, M. I.; Coudret, C.; Pimienta, V.; Micheau, J. C.; Delbaere, S.; Vermeersch, G.; Metelitsa, A. V.; Voloshin, N.; Minkin, V. I. Photochem. Photobiol. Sci. 2010, 9, 199–207. doi:10.1039/b9pp00112c
Return to citation in text: [1] -
Kumar, S.; Chau, C.; Chau, G.; McCurdy, A. Tetrahedron 2008, 64, 7097–7105. doi:10.1016/j.tet.2008.05.083
Return to citation in text: [1] -
Natali, M.; Giordani, S. Org. Biomol. Chem. 2012, 10, 1162–1171. doi:10.1039/C1OB06375H
Return to citation in text: [1]
| 26. | Owens, E. A.; Bruschi, N.; Tawney, J. G.; Henary, M. Dyes Pigm. 2015, 113, 27–37. doi:10.1016/j.dyepig.2014.07.035 |
| 27. | Chen, J.; Zeng, F.; Wu, S.; Chen, Q.; Tong, Z. Chem. – Eur. J. 2008, 14, 4851–4860. doi:10.1002/chem.200701994 |
| 28. | Wang, B.; Fan, J.; Sun, S.; Wang, L.; Song, B.; Peng, X. Dyes Pigm. 2010, 85, 43–50. doi:10.1016/j.dyepig.2009.10.002 |
| 6. | Liu, Y.; Fan, M.; Zhang, S.; Sheng, X.; Yao, J. New J. Chem. 2007, 31, 1878–1881. doi:10.1039/b713247f |
| 17. | Paramonov, S. V.; Lokshin, V.; Fedorova, O. A. J. Photochem. Photobiol., C 2011, 12, 209–236. doi:10.1016/j.jphotochemrev.2011.09.001 |
| 34. | Zakharova, M. I.; Coudret, C.; Pimienta, V.; Micheau, J. C.; Delbaere, S.; Vermeersch, G.; Metelitsa, A. V.; Voloshin, N.; Minkin, V. I. Photochem. Photobiol. Sci. 2010, 9, 199–207. doi:10.1039/b9pp00112c |
| 3. | Natali, M.; Aakeröy, C.; Desper, J.; Giordani, S. Dalton Trans. 2010, 39, 8269–8277. doi:10.1039/c0dt00242a |
| 4. | Krikun, V. M.; Sadimenko, L. P.; Voloshina, E. N.; Voloshin, N. A. Russ. J. Gen. Chem. 2009, 79, 1191–1196. doi:10.1134/S1070363209060279 |
| 5. | Wojtyk, J. T. C.; Kazmaier, P. M.; Buncel, E. Chem. Mater. 2001, 13, 2547–2551. doi:10.1021/cm010038q |
| 18. | Roxburgh, C. J.; Sammes, P. G.; Abdullah, A. Eur. J. Inorg. Chem. 2008, 4951–4960. doi:10.1002/ejic.200800614 |
| 18. | Roxburgh, C. J.; Sammes, P. G.; Abdullah, A. Eur. J. Inorg. Chem. 2008, 4951–4960. doi:10.1002/ejic.200800614 |
| 2. | Lukyanov, B. S.; Lukyanova, M. B. Chem. Heterocycl. Compd. 2005, 41, 281–311. doi:10.1007/s10593-005-0148-x |
| 16. | Koçer, A.; Walko, M.; Meijberg, W.; Feringa, B. L. Science 2005, 309, 755–758. doi:10.1126/science.1114760 |
| 32. | Fries, K. H.; Driskell, J. D.; Sheppard, G. R.; Locklin, J. Langmuir 2011, 27, 12253–12260. doi:10.1021/la202344w |
| 2. | Lukyanov, B. S.; Lukyanova, M. B. Chem. Heterocycl. Compd. 2005, 41, 281–311. doi:10.1007/s10593-005-0148-x |
| 6. | Liu, Y.; Fan, M.; Zhang, S.; Sheng, X.; Yao, J. New J. Chem. 2007, 31, 1878–1881. doi:10.1039/b713247f |
| 33. | Roxburgh, C. J.; Sammes, P. G.; Abdullah, A. Dyes Pigm. 2011, 90, 146–162. doi:10.1016/j.dyepig.2010.10.006 |
| 10. | Medintz, I. L.; Trammell, S. A.; Mattoussi, H.; Mauro, J. M. J. Am. Chem. Soc. 2004, 126, 30–31. doi:10.1021/ja037970h |
| 11. | Tomasulo, M.; Yildiz, I.; Raymo, F. M. Aust. J. Chem. 2006, 59, 175–178. doi:10.1071/CH05332 |
| 13. | Ito, Y.; Sugimura, N.; Kwon, O. H.; Imanishi, Y. Nat. Biotechnol. 1999, 17, 73–75. doi:10.1038/5250 |
| 3. | Natali, M.; Aakeröy, C.; Desper, J.; Giordani, S. Dalton Trans. 2010, 39, 8269–8277. doi:10.1039/c0dt00242a |
| 9. | Lion-Dagan, M.; Katz, E.; Willner, I. J. Chem. Soc., Chem. Commun. 1994, 2741–2742. doi:10.1039/c39940002741 |
| 14. | Karube, I.; Ishimori, Y.; Suzuki, S.; Sato, T. Biotechnol. Bioeng. 1978, 20, 1775–1783. doi:10.1002/bit.260201107 |
| 15. | Karube, I.; Ishimori, Y.; Suzuki, S. Anal. Biochem. 1978, 86, 100–106. doi:10.1016/0003-2697(78)90322-6 |
| 31. | Fries, K. H.; Driskell, J. D.; Samanta, S.; Locklin, J. Anal. Chem. 2010, 82, 3306–3314. doi:10.1021/ac1001004 |
| 8. | Del Canto, E.; Natali, M.; Movia, D.; Giordani, S. Phys. Chem. Chem. Phys. 2012, 14, 6034–6043. doi:10.1039/c2cp40275k |
| 29. | Fishwick, C. W. G.; Jones, A. D.; Mitchell, M. B.; Eggleston, D. S.; Baures, P. W. Synlett 1990, 359–361. doi:10.1055/s-1990-21095 |
| 12. | Ciardelli, F.; Fabbri, D.; Pieroni, O.; Fissi, A. J. Am. Chem. Soc. 1989, 111, 3470–3472. doi:10.1021/ja00191a076 |
| 30. | Dunn, G. E.; Lee, G. K. J.; Thimm, H. Can. J. Chem. 1972, 50, 3017–3027. doi:10.1139/v72-480 |
| 3. | Natali, M.; Aakeröy, C.; Desper, J.; Giordani, S. Dalton Trans. 2010, 39, 8269–8277. doi:10.1039/c0dt00242a |
| 17. | Paramonov, S. V.; Lokshin, V.; Fedorova, O. A. J. Photochem. Photobiol., C 2011, 12, 209–236. doi:10.1016/j.jphotochemrev.2011.09.001 |
| 35. | Kumar, S.; Chau, C.; Chau, G.; McCurdy, A. Tetrahedron 2008, 64, 7097–7105. doi:10.1016/j.tet.2008.05.083 |
| 3. | Natali, M.; Aakeröy, C.; Desper, J.; Giordani, S. Dalton Trans. 2010, 39, 8269–8277. doi:10.1039/c0dt00242a |
| 3. | Natali, M.; Aakeröy, C.; Desper, J.; Giordani, S. Dalton Trans. 2010, 39, 8269–8277. doi:10.1039/c0dt00242a |
| 36. | Natali, M.; Giordani, S. Org. Biomol. Chem. 2012, 10, 1162–1171. doi:10.1039/C1OB06375H |
| 24. | Zhang, Y.; Xiao, L.; Popovic, K.; Xie, X.; Chordia, M. D.; Chung, L. W. K.; Williams, M. B.; Yue, W.; Pan, D. Bioorg. Med. Chem. Lett. 2013, 23, 6350–6354. doi:10.1016/j.bmcl.2013.09.074 |
| 25. | Altevogt, A.; Flehr, R.; Gehne, S.; Kumke, M. U.; Bannwarth, W. Helv. Chim. Acta 2012, 95, 543–555. doi:10.1002/hlca.201100460 |
| 22. | Kiyose, K.; Hanaoka, K.; Oushiki, D.; Nakamura, T.; Kajimura, M.; Suematsu, M.; Nishimatsu, H.; Yamane, T.; Terai, T.; Hirata, Y.; Nagano, T. J. Am. Chem. Soc. 2010, 132, 15846–15848. doi:10.1021/ja105937q |
| 23. | An, H.-W.; Qiao, S.-L.; Hou, C.-Y.; Lin, Y.-X.; Li, L.-L.; Xie, H.-Y.; Wang, Y.; Wang, L.; Wang, H. Chem. Commun. 2015, 51, 13488–13491. doi:10.1039/C5CC05395A |
| 20. | Pertusati, F.; Menger, F. M. J. Org. Chem. 2008, 73, 2939–2942. doi:10.1021/jo7027294 |
| 21. | Kvach, M. V.; Ustinov, A. V.; Stepanova, I. A.; Malakhov, A. D.; Skorobogatyi, M. V.; Shmanai, V. V.; Korshun, V. A. Eur. J. Org. Chem. 2008, 2107–2117. doi:10.1002/ejoc.200701190 |
| 3. | Natali, M.; Aakeröy, C.; Desper, J.; Giordani, S. Dalton Trans. 2010, 39, 8269–8277. doi:10.1039/c0dt00242a |
| 3. | Natali, M.; Aakeröy, C.; Desper, J.; Giordani, S. Dalton Trans. 2010, 39, 8269–8277. doi:10.1039/c0dt00242a |
| 19. | Lopalco, M.; Koini, E. N.; Cho, J. K.; Bradley, M. Org. Biomol. Chem. 2009, 7, 856–859. doi:10.1039/b820719b |
© 2017 Perry and Kousseff; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)