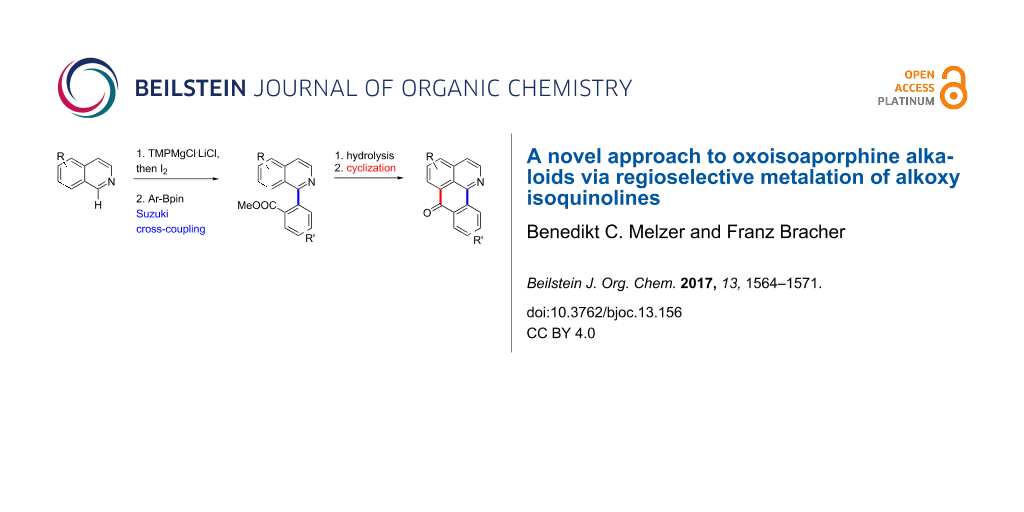
Abstract
Oxoisoaporphine alkaloids are conveniently prepared via direct ring metalation of alkoxy-substituted isoquinolines at C-1, followed by reaction with iodine. Subsequent Suzuki cross-coupling of the resulting 1-iodoisoquinolines to methyl 2-(isoquinolin-1-yl)benzoates and intramolecular acylation of the corresponding carboxylic acids with Eaton’s reagent afforded five alkaloids of the oxoisoaporphine type. The yield of the cyclization step strongly depends on the electrophilic properties of ring B. An alternative cyclization protocol via directed remote metalation of ester and amide intermediates was investigated thoroughly, but found to be not feasible. Two of the alkaloids showed strong cytotoxicity against the HL-60 tumor cell line.

Graphical Abstract
Introduction
Benzylisoquinoline alkaloids represent a very large group of plant secondary metabolites, which includes about 2,500 known structures. Besides simple benzylisoquinolines, more complex tetracyclic ring systems like aporphines, protoberberines, cularines, pavines, as well as pentacyclic morphinane-type alkaloids belong to this class. Biosynthetically, all of these chemotypes are derived from the 1-benzyltetrahydroisoquinoline (S)-norcoclaurine. The structures, biosynthesis and pharmacology of benzylisoquinoline alkaloids have been reviewed recently by Hagel and Facchini [1].
The subgroup of aporphinoid alkaloids [2] consists of the aporphines and oxoaporphines (e.g., liriodenine (1)), as well as several truncated chemotypes like aristolactams and azafluoranthenes, and is of considerable pharmacological interest due to the significant cytotoxicity of numerous representatives [3]. A unique subclass of the aporphinoid alkaloids are the oxoisoaporphines (7H-dibenzo[de,h]quinolin-7-ones, e.g., menisporphine, (2)), which at the first glance appear to be not derived from 1-benzyltetrahydroisoquinoline intermediates, albeit a hypothesis of Kunitomo postulates a biosynthesis from a benzylisoquinoline precursor involving a rearrangement (Figure 1) [4].
![[1860-5397-13-156-1]](/bjoc/content/figures/1860-5397-13-156-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Prominent oxoaporphine and oxoisoaporphine alkaloids: liriodenine (1), menisporphine (2), dauriporphine (3), 6-O-demethylmenisporphine (4), dauriporphinoline (5), and bianfugecine (6).
Figure 1: Prominent oxoaporphine and oxoisoaporphine alkaloids: liriodenine (1), menisporphine (2), dauriporp...
Menisporphine (2), first isolated from Menispermum dauricum DC [4], shows antiangiogenic activity. Some, partly synthetic, oxoisoaporphine-like analogues were found to have strong DNA binding affinity and therefore high cytotoxicity [5] as well as antiplasmodial activity [6]. Besides menisporphine (2), the related oxoisoaporphine alkaloids dauriporphine (3), 6-O-demethylmenisporphine (4), dauriporphinoline (5) and bianfugecine (6) were isolated from Menispermum dauricum DC, among a few other plants [1,7,8].
A number of approaches to the oxoaporphine ring system has been published, typically involving the synthesis of 1-benzyl/1-benzoylisoquinoline intermediates, followed by cyclization under intramolecular biaryl synthesis, utilizing either photochemical [9-13], radical [14] or Pd-catalyzed [15,16] reactions.
In contrast, only minute work has been published on the synthesis of oxoisoaporphines. The approaches are mainly based on Kunitomo’s total syntheses of menisporphine (2) [7] and dauriporphine (3) [17], which start with the synthesis of 1-(2-bromoaryl)isoquinolines using Bischler–Napieralski chemistry, followed by tedious replacement of the bromine substituent by cyanide (for modern variants, see ref. [18,19]), and subsequent conversion to a carboxylate and Friedel–Crafts-type cyclization using polyphosphoric acid (Scheme 1). In the course of this cyclization, the methoxy group at C-6 is typically hydrolized to the phenol, subsequent O-methylation gives the 6-methoxy derivatives menisporphine (2) and dauriporphine (3). Recently, Zhang et al. [20] described an alternative approach starting from an isoquinoline bearing an ester group at C-8. In a photoredox-catalyzed direct C–H arylation a 4-methoxyphenyl residue from a methoxyphenyldiazonium salt was introduced at C-1, and after ester hydrolysis intramolecular Friedel–Crafts acylation afforded menisporphine (2).
In continuation of our recent work on the synthesis of polycyclic aromatic alkaloids like pyridoacridine, benzylisoquinoline-type and oxoaporphine alkaloids using direct ring metalations of heterocycles as vital step [13,21] we intended to develop a novel and more flexible access to oxoisoaporphine alkaloids. Again we were inspired by Knochel’s reports on the direct metalation of isoquinoline [22] and 6,7-dimethoxyisoquinoline [23] as well as by our own results for the metalation of various alkoxyisoquinolines [13] at C-1 with the Knochel–Hauser base TMPMgCl·LiCl. Transmetalation of the intermediate organomagnesium species with ZnCl2 should lead to at C-1 zincated isoquinolines, which could undergo Negishi cross-coupling reactions with appropriately substituted methyl 2-bromobenzoates to give methyl 2-(isoquinolin-1-yl)benzoates. These would again open an access to oxoisoaporphine alkaloids via intramolecular acylation following Kunitomo’s strategy. Main advantage of this approach should be that the laborious introduction of the carboxy residue at a late stage of the total synthesis is circumvented. Alternatively, quenching of the 1-magnesiated alkoxyisoquinolines with iodine should lead to 1-iodoisoquinolines, which were expected to be versatile substrates for Suzuki cross-coupling reactions with appropriate phenylboronic acids to gain methyl 2-(isoquinolin-1-yl)benzoates as the central intermediates (Scheme 1).
![[1860-5397-13-156-i1]](/bjoc/content/inline/1860-5397-13-156-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Previously reported [7,17] and new approach to oxoisoaporphine alkaloids.
Scheme 1: Previously reported [7,17] and new approach to oxoisoaporphine alkaloids.
Results and Discussion
As we reported previously, metalation of 6,7-dimethoxyisoquinoline (7a) and 5,6,7-trimethoxyisoquinoline (7b) with 1.5 equivalents of the Knochel–Hauser base (TMPMgCl∙LiCl) exclusively occurs at C-1. Trapping with appropriate benzaldehydes opened an access to benzylisoquinoline and oxoaporphine-type alkaloids [13]. Here we describe the extension of this approach to the synthesis of oxoisoaporphines.
By using our modification (1.5 equivalents of TMPMgCl∙LiCl over 4 h at room temperature) [13] of Knochel’s [23] protocol for the metalation of isoquinolines, the alkoxy-substituted isoquinolines 7a,b were metalated at C-1, as shown before, and this method could conveniently be extended to 6-methoxyisoquinoline (7c). Unfortunately, transmetalation with ZnCl2 at 0 °C followed by palladium-catalyzed (5 mol % Pd(dba)2/ 10 mol % P(2-furyl)3 or 1 mol % Pd2(dba)3/2 mol % RuPhos) Negishi cross-coupling reaction with methyl 2-bromo-5-methoxybenzoate did not lead to the expected methyl 2-(isoquinolin-1-yl)benzoates 10 after up to 72 h at room temperature or 60 °C [22]. Starting materials were recovered almost quantitatively. However, quenching with iodine gave the 1-iodoisoquinolines 8a [23], 8b, and 8c in good yields (53–67%, Scheme 2).
![[1860-5397-13-156-i2]](/bjoc/content/inline/1860-5397-13-156-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of iodinated isoquinolines 8a–c from alkoxy-substituted isoquinolines 7a–c.
Scheme 2: Synthesis of iodinated isoquinolines 8a–c from alkoxy-substituted isoquinolines 7a–c.
For the introduction of ring D of the oxoisoaporphine alkaloids one common building block, (4-methoxy-2-(methoxycarbonyl)phenyl)boronic acid pinacol ester (9) [24], could be applied, since the alkaloids of interest all bear the methoxy group at C-9. Suzuki cross-coupling reaction of the iodinated isoquinolines 8a–c with this boronate under Pd(PPh3)4 catalysis gave the desired 1-arylisoquinolines 10a–c in moderate to good isolated yields (65–77%, Scheme 3). Up to 20% of the starting materials 8a–c were typically recovered, but all attempts to achieve complete conversion (longer reaction times, other catalysts and bases) were in vain.
![[1860-5397-13-156-i3]](/bjoc/content/inline/1860-5397-13-156-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of methyl 2-(isoquinolin-1-yl)benzoates 10a–c from 1-iodoisoquinolines 8a–c.
Scheme 3: Synthesis of methyl 2-(isoquinolin-1-yl)benzoates 10a–c from 1-iodoisoquinolines 8a–c.
The esters 10a–c are valuable intermediates for the further conversion to the target oxoisoaporphines, and in previous protocols [7,17] the esters were typically converted into the corresponding carboxylic acids, and then cyclized using polyphosphoric acid. Previously we found that esters can directly be subjected to this type of intramolecular Friedel–Crafts-type acylations, and trifluoromethanesulfonic acid [25] is superior to polyphosphoric acid [26,27] for the synthesis of polycyclic ketones. Unfortunately, direct cyclization of esters 10a–c using this reagent failed completely, despite numerous variations of the reaction conditions. As an alternative reagent for the acylation of arenes with esters Eaton’s reagent (phosphorus pentoxide, 7.7 wt % in methanesulfonic acid) has been described in the literature [28,29]. Eaton’s reagent has advantages over polyphosphoric acid as it is an easy to handle, non-viscous and freely measurable liquid, and therefore more suitable for small-scale synthesis. However, attempted direct cyclization of ester 10a with Eaton’s reagent to menisporphine (2) failed. Consequently, we had to come back to Kunitomo’s approach [7,17] via carboxylate intermediates. Carboxylic acid 11a was conveniently obtained by hydrolysis of the methyl ester 10a with concentrated hydrochloric acid. The crude carboxylic acid 11a underwent cyclization to 6-O-demethylmenisporphine (4) with Eaton’s reagent in 45% yield over both steps (Scheme 4). In accordance with Kunitomo’s observations [7,17], we also observed demethylation of the methoxy group at C-6. Dauriporphinoline (5) was synthesized in 58% yield using the same protocol starting from the methyl ester 10b, once again under hydrolysis of the 6-methoxy group.
![[1860-5397-13-156-i4]](/bjoc/content/inline/1860-5397-13-156-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of the alkaloids 6-O-demethylmenisporphine (4), dauriporphinoline (5), and bianfugecine (6).
Scheme 4: Synthesis of the alkaloids 6-O-demethylmenisporphine (4), dauriporphinoline (5), and bianfugecine (6...
In order to explore the scope of this new cyclization methodology, we further investigated the synthesis of the alkaloid bianfugecine (6, Scheme 4), which contains only one methoxy group at ring B. Until now, only one partial synthesis of this alkaloid is described in the literature. Kunitomo et al. [30] obtained alkaloid 6 by hydrogenolytic demethoxylation of alkaloid menisporphine (2) with H2/PtO2 in acetic acid. Unfortunately, upon cyclization of the carboxylic acid 11c, obtained by acidic hydrolysis of ester 10c, with Eaton’s reagent alkaloid 6 could be isolated in only very poor yield (7%). Other established cyclization methods starting from methyl ester 10c were tested in order to increase the yield. As described above, attempted direct intramolecular Friedel–Crafts-type cyclization of 10c catalyzed by trifluoromethanesulfonic acid [25] failed completely. Also, generation of an acid chloride from 11c with thionyl chloride, followed by the reaction with AlCl3 did not even give traces of bianfugecine (6).
Taken all of these results together, intramolecular Friedel–Crafts-type cyclization aimed at the construction of the oxoisoaporphine scaffold is a very challenging conversion, and the electronic properties of the ring to be attacked (ring B) strongly determine the outcome of this reaction.
Inspired by a report from the Snieckus group [31] on the application of the directed remote metalation (DreM) methodology for generation of carbanionic Friedel–Crafts equivalents (generating acridones from diarylamine carboxamides), we converted methyl ester 10c into the corresponding diethyl amide 12. For this purpose, ester 10c was reacted in a Weinreb amidation [32] with a mixture of trimethylaluminium and diethylamine to give the amide 12 in 31% yield (Scheme 5). The diethyl amide moiety was designated to promote a directed remote metalation by lithium diisopropylamide (LDA) at C-8 of the isoquinoline ring, which should be followed by an intramolecular trapping of the amide group to give the oxoisoaporphine bianfugecine (6, Scheme 5). However, only starting material 12 was recovered from this reaction.
![[1860-5397-13-156-i5]](/bjoc/content/inline/1860-5397-13-156-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Attempted synthesis of bianfugecine (6) via directed remote metalation and subsequent trapping of the carboxamide group.
Scheme 5: Attempted synthesis of bianfugecine (6) via directed remote metalation and subsequent trapping of t...
A D2O quenching experiment after the metalation period (4 equiv LDA, 1 h, 25 °C) was performed in order to identify the position(s) of ring metalation of 12. In recovered (about 80%) educt, deuterium incorporation (about 40%; calculated from integrals of the 1H NMR spectrum) was observed exclusively at C-6 of the benzamide moiety (Scheme 6). It is known for decades [33] that cooperative effects of directing groups lead to directed ortho-metalation (DoM) at their common ortho-position (C-6 in this case, located ortho to both the amide and the methoxy group). Nevertheless, under reversible metalation conditions, typically existing when amide bases like LDA are employed, kinetically controlled DoM can be followed by an equilibration to give a thermodynamically controlled DreM product [34]. This has been demonstrated by Tilly et al. [35] in the synthesis of fluorenones by treatment of N,N-dialkyl biphenyl-2-carboxamides with LDA. In our case, however, initial metalation at C-6 of the benzamide moiety was not followed by an equilibration giving the desired 8’-metalated intermediate (which in turn should be trapped by the amide group to give the tetracyclic ketone 6). Probably, the DreM at C-8’ is prevented, since the amide moiety forms a chelate with a lithium ion and N-2 of the isoquinoline moiety, thus is kept away from C-8’. In contrast, we found in previous investigations on the synthesis of the pyridoacridone alkaloid demethyldeoxyamphimendine [21], that an ester group located at a comparable position (suitable for forming a chelate in cooperation with a pyridine nitrogen), can very well promote a directed remote metalation.
![[1860-5397-13-156-i6]](/bjoc/content/inline/1860-5397-13-156-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Outcome of a D2O quenching experiment after metalation of amide 12.
Scheme 6: Outcome of a D2O quenching experiment after metalation of amide 12.
To test our hypothesis, we performed two more D2O quenching experiments with naphthalene analogues of esters 10 and amide 12. The methyl ester analogue 15 was obtained by Suzuki cross-coupling of naphthalene-1-boronic acid (13) and methyl 2-bromo-5-methoxybenzoate (14) in 68% yield. Methyl ester 15 was converted into the corresponding diethyl amide 16 in 58% yield by Weinreb amidation following the protocol described above for the synthesis of 12 (Scheme 7).
![[1860-5397-13-156-i7]](/bjoc/content/inline/1860-5397-13-156-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of 1-arylnaphthalene analogues 15 and 16.
Scheme 7: Synthesis of 1-arylnaphthalene analogues 15 and 16.
Incubation of methyl ester 15 with LDA (4 equiv LDA, 25 °C) over a period of 1 h, followed by D2O quenching resulted in complete decomposition. In contrast, metalation of amide 16 under the same conditions and quenching with D2O led to the formation of the benzo[c]fluoren-7-one 17 in 38% yield. In recovered (about 20%) educt 16, deuterium incorporation (about 20%; calculated from integrals of the 1H NMR spectrum) was exclusively observed at C-6 of the benzamide moiety (Scheme 8). These data show no indication for a DreM at the peri-position C-8’, since neither a 8’-deuterated arylnaphthalene nor a benzo[de]anthracen-7-one was detected. This observation is in accordance with results from the Snieckus group [36] on the remote metalation/cyclization of a 2-(1-naphthyl)benzamide, which gave exclusively benzo[c]fluorenone (analogue of 17 lacking the methoxy group), and not the benzo[de]anthracen-7-one. This clearly indicates that remote metalation, if any, occurs only in the ortho-position to the benzamide residue (C-2 of the naphthalene ring system), but not in the peri-position (C-8 of the naphthalene). Consequently, further attempts to initialize the cyclization of arylisoquinolines 10c and 12 to the oxoisoaporphine alkaloid bianfugecine (6, Scheme 5) by a DreM did not appear to be promising.
![[1860-5397-13-156-i8]](/bjoc/content/inline/1860-5397-13-156-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Outcome of a D2O quenching experiment after metalation of amide 16 with LDA.
Scheme 8: Outcome of a D2O quenching experiment after metalation of amide 16 with LDA.
Finally, the alkaloids 6-O-demethylmenisporphine (4) and dauriporphinoline (5) were converted into their corresponding 6-methoxy derivatives, the oxoisoaporphine alkaloids menisporphine (2) and dauriporphine (3). Using Kunitomo’s [7,17] protocol (methyl iodide in the presence of silver oxide), phenols 4 and 5 were converted into the alkaloids 2 and 3 in 33 and 49% yields (Scheme 9). In accordance to Kunitomo’s observations, the isomeric methoxy compounds 18 and 19 were obtained as byproducts in significant yields (Scheme 9). In order to find a more convenient methylation protocol, we explored diazomethane, but no conversion was observed at all.
![[1860-5397-13-156-i9]](/bjoc/content/inline/1860-5397-13-156-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Synthesis of the alkaloids menisporphine (2) and dauriporphine (3) by O-methylation of the alkaloids 6-O-demethylmenisporphine (4) and dauriporphinoline (5).
Scheme 9: Synthesis of the alkaloids menisporphine (2) and dauriporphine (3) by O-methylation of the alkaloid...
Finally, all of the synthesized alkaloids were tested for their cytotoxic potential in a MTT assay on the HL-60 cell line. Except for menisporphine (2) and bianfugecine (6, both IC50 values > 50 µM), all of the described oxoisoaporphine alkaloids and the isomers 18 and 19 showed significant cytotoxic activity. IC50 values of alkaloid 3 and of compounds 18 and 19 are ranging from 3–6 µM. Very strong cytotoxicity was determined for 6-O-demethylmenisporphine (4) with an IC50 value of 0.06 µM and for dauriporphinoline (5) with 0.23 µM. So, at least in the HL-60 cell line, oxoisoaporphines bearing a free hydroxy group at C-6 show outstanding cytotoxicity. These data are in accordance with previously reported results [37].
Conclusion
In conclusion, we worked out a novel approach for the synthesis of oxoisoaporphine alkaloids by direct ring metalation of alkoxy isoquinolines at C-1, followed by the reaction with iodine as central step. Subsequent Suzuki cross-coupling to methyl 2-(isoquinolin-1-yl)benzoates and intramolecular acylation with Eaton’s reagent afforded five alkaloids of the oxoisoaporphine type. Significant cytotoxicity was found for oxoisoaporphines bearing a free 6-hydroxy group.
Experimental
For experimental procedures and copies of 1H and 13C NMR spectra of all compounds see Supporting Information File 1.
Supporting Information
| Supporting Information File 1: Experimental procedures and copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 2.5 MB | Download |
References
-
Hagel, J. M.; Facchini, P. J. Plant Cell Physiol. 2013, 54, 647–672. doi:10.1093/pcp/pct020
Return to citation in text: [1] [2] -
Shamma, M.; Guinaudeau, H. Nat. Prod. Rep. 1984, 1, 201–207. doi:10.1039/np9840100201
Return to citation in text: [1] -
Stévigny, C.; Bailly, C.; Quetin-Leclercq, J. Curr. Med. Chem. - Anti-Cancer Agents 2005, 5, 173–182. doi:10.2174/1568011053174864
Return to citation in text: [1] -
Kunitomo, J.; Satoh, M. Chem. Pharm. Bull. 1982, 30, 2659–2660. doi:10.1248/cpb.30.2659
Return to citation in text: [1] [2] -
Tang, H.; Wang, X.-D.; Wie, Y.-B.; Huang, S.-L.; Huang, Z.-S.; Tan, J.-H.; An, L.-K.; Wu, J.-Y.; Chan, A. S.-C.; Gu, L.-Q. Eur. J. Med. Chem. 2008, 43, 973–980. doi:10.1016/j.ejmech.2007.07.004
Return to citation in text: [1] -
Castro-Castillo, V.; Suárez-Rozas, C.; Pabón, A.; Pérez, E. G.; Cassels, B. K.; Blair, S. Bioorg. Med. Chem. Lett. 2013, 23, 327–329. doi:10.1016/j.bmcl.2012.10.092
Return to citation in text: [1] -
Kunitomo, J.; Satoh, M.; Shingu, T. Tetrahedron 1983, 39, 3261–3265. doi:10.1016/S0040-4020(01)91573-X
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Okamoto, Y.; Tanaka, S.; Kitayama, K.; Isomoto, M.; Masaishi, M.; Yanagawa, H.; Kunitomo, J.-I. Yakugaku Zasshi 1971, 91, 684–687. doi:10.1248/yakushi1947.91.6_684
Return to citation in text: [1] -
Kupchan, S. M.; Moniot, J. L.; Kanojia, R. M.; O'Brien, J. B. J. Org. Chem. 1971, 36, 2413–2418. doi:10.1021/jo00816a007
Return to citation in text: [1] -
Kupchan, S. M.; O’Brien, P. F. J. Chem. Soc., Chem. Commun. 1973, 915–916. doi:10.1039/c39730000915
Return to citation in text: [1] -
Castedo, L.; Saá, J. M.; Suau, R.; Villaverde, C. Heterocycles 1980, 14, 1131–1134. doi:10.3987/R-1980-08-1131
Return to citation in text: [1] -
Kessar, S. V.; Gupta, Y. P.; Yadav, V. S.; Narula, M.; Mohammed, T. Tetrahedron Lett. 1980, 21, 3307–3308. doi:10.1016/S0040-4039(00)78675-8
Return to citation in text: [1] -
Melzer, B.; Bracher, F. Org. Biomol. Chem. 2015, 13, 7664–7672. doi:10.1039/C5OB00926J
Return to citation in text: [1] [2] [3] [4] [5] -
Orito, K.; Uchiito, S.; Satoh, Y.; Tatsuzawa, T.; Harada, R.; Tokuda, M. Org. Lett. 2000, 2, 307–310. doi:10.1021/ol990360v
Return to citation in text: [1] -
Cuny, G. D. Tetrahedron Lett. 2004, 45, 5167–5170. doi:10.1016/j.tetlet.2004.04.194
Return to citation in text: [1] -
Chaudhary, S.; Pecic, S.; LeGendre, O.; Harding, W. W. Tetrahedron Lett. 2009, 50, 2437–2439. doi:10.1016/j.tetlet.2009.03.029
Return to citation in text: [1] -
Kunitomo, J.; Kaede, S.; Satoh, M. Chem. Pharm. Bull. 1985, 33, 2778–2782. doi:10.1248/cpb.33.2778
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Jia, X.; Yang, D.; Zhang, S.; Cheng, J. Org. Lett. 2009, 11, 4716–4719. doi:10.1021/ol9017529
Return to citation in text: [1] -
Chaitanya, M.; Yadagiri, D.; Anbarasan, P. Org. Lett. 2013, 15, 4960–4963. doi:10.1021/ol402201c
Return to citation in text: [1] -
Zhang, J.; Chen, J.; Zhang, X.; Lei, X. J. Org. Chem. 2014, 79, 10682–10688. doi:10.1021/jo5020432
Return to citation in text: [1] -
Melzer, B.; Plodek, A.; Bracher, F. J. Org. Chem. 2014, 79, 7239–7242. doi:10.1021/jo501312d
Return to citation in text: [1] [2] -
Kraskovskiy, A.; Kraskovskaya, V.; Knochel, P. Angew. Chem., Int. Ed. 2006, 45, 2958–2961. doi:10.1002/anie.200504024
Return to citation in text: [1] [2] -
Metzger, A.; Schade, M. A.; Knochel, P. Org. Lett. 2008, 10, 1107–1110. doi:10.1021/ol7030697
Return to citation in text: [1] [2] [3] -
Krätzschmar, F.; Kaßel, M.; Delony, D.; Breder, A. Chem. – Eur. J. 2015, 21, 7030–7034. doi:10.1002/chem.201406290
Return to citation in text: [1] -
Plodek, A.; Raeder, S.; Bracher, F. Tetrahedron 2013, 69, 9857–9864. doi:10.1016/j.tet.2013.08.085
Return to citation in text: [1] [2] -
Bracher, F. Arch. Pharm. 1989, 322, 293–294. doi:10.1002/ardp.19893220511
Return to citation in text: [1] -
Mink, K.; Bracher, F. Arch. Pharm. 2007, 340, 429–433. doi:10.1002/ardp.200700064
Return to citation in text: [1] -
Dorow, R. L.; Herrinton, P. M.; Hohler, R. A.; Maloney, M. T.; Mauragis, M. A.; McGhee, W. E.; Moeslein, J. A.; Strohbach, J. W.; Veley, M. F. Org. Process Res. Dev. 2006, 10, 493–499. doi:10.1021/op050251y
Return to citation in text: [1] -
Zewge, D.; Chen, C.-y.; Deer, C.; Dormer, P. G.; Hughes, D. L. J. Org. Chem. 2007, 72, 4276–4279. doi:10.1021/jo070181o
Return to citation in text: [1] -
Kunitomo, J.-i.; Miyata, Y. Heterocycles 1986, 24, 437–440. doi:10.3987/R-1986-02-0437
Return to citation in text: [1] -
MacNeil, S. L.; Gray, M.; Gusev, D. G.; Briggs, L. E.; Snieckus, V. J. Org. Chem. 2008, 73, 9710–9719. doi:10.1021/jo801856n
Return to citation in text: [1] -
Levin, J. I.; Turos, E.; Weinreb, S. M. Synth. Commun. 1982, 12, 989–993. doi:10.1080/00397918208061938
Return to citation in text: [1] -
Snieckus, V. Chem. Rev. 1990, 90, 879–933. doi:10.1021/cr00104a001
Return to citation in text: [1] -
Tilly, D.; Magolan, J.; Mortier, J. Chem. – Eur. J. 2012, 18, 3804–3820. doi:10.1002/chem.201103920
Return to citation in text: [1] -
Tilly, D.; Fu, J.-m.; Zhao, B.-p.; Alessi, M.; Castanet, A. S.; Snieckus, V.; Mortier, J. Org. Lett. 2010, 12, 68–71. doi:10.1021/ol902268h
Return to citation in text: [1] -
Fu, J. M.; Zhao, B. P.; Sharp, M. J.; Snieckus, V. J. Org. Chem. 1991, 56, 1683–1685. doi:10.1021/jo00005a004
Return to citation in text: [1] -
Cheng, J.-J.; Tsai, T.-H.; Lin, L.-C. Planta Med. 2012, 78, 1873–1877. doi:10.1055/s-0032-1327785
Return to citation in text: [1]
| 25. | Plodek, A.; Raeder, S.; Bracher, F. Tetrahedron 2013, 69, 9857–9864. doi:10.1016/j.tet.2013.08.085 |
| 26. | Bracher, F. Arch. Pharm. 1989, 322, 293–294. doi:10.1002/ardp.19893220511 |
| 27. | Mink, K.; Bracher, F. Arch. Pharm. 2007, 340, 429–433. doi:10.1002/ardp.200700064 |
| 28. | Dorow, R. L.; Herrinton, P. M.; Hohler, R. A.; Maloney, M. T.; Mauragis, M. A.; McGhee, W. E.; Moeslein, J. A.; Strohbach, J. W.; Veley, M. F. Org. Process Res. Dev. 2006, 10, 493–499. doi:10.1021/op050251y |
| 29. | Zewge, D.; Chen, C.-y.; Deer, C.; Dormer, P. G.; Hughes, D. L. J. Org. Chem. 2007, 72, 4276–4279. doi:10.1021/jo070181o |
| 1. | Hagel, J. M.; Facchini, P. J. Plant Cell Physiol. 2013, 54, 647–672. doi:10.1093/pcp/pct020 |
| 4. | Kunitomo, J.; Satoh, M. Chem. Pharm. Bull. 1982, 30, 2659–2660. doi:10.1248/cpb.30.2659 |
| 20. | Zhang, J.; Chen, J.; Zhang, X.; Lei, X. J. Org. Chem. 2014, 79, 10682–10688. doi:10.1021/jo5020432 |
| 4. | Kunitomo, J.; Satoh, M. Chem. Pharm. Bull. 1982, 30, 2659–2660. doi:10.1248/cpb.30.2659 |
| 13. | Melzer, B.; Bracher, F. Org. Biomol. Chem. 2015, 13, 7664–7672. doi:10.1039/C5OB00926J |
| 21. | Melzer, B.; Plodek, A.; Bracher, F. J. Org. Chem. 2014, 79, 7239–7242. doi:10.1021/jo501312d |
| 34. | Tilly, D.; Magolan, J.; Mortier, J. Chem. – Eur. J. 2012, 18, 3804–3820. doi:10.1002/chem.201103920 |
| 3. | Stévigny, C.; Bailly, C.; Quetin-Leclercq, J. Curr. Med. Chem. - Anti-Cancer Agents 2005, 5, 173–182. doi:10.2174/1568011053174864 |
| 17. | Kunitomo, J.; Kaede, S.; Satoh, M. Chem. Pharm. Bull. 1985, 33, 2778–2782. doi:10.1248/cpb.33.2778 |
| 31. | MacNeil, S. L.; Gray, M.; Gusev, D. G.; Briggs, L. E.; Snieckus, V. J. Org. Chem. 2008, 73, 9710–9719. doi:10.1021/jo801856n |
| 2. | Shamma, M.; Guinaudeau, H. Nat. Prod. Rep. 1984, 1, 201–207. doi:10.1039/np9840100201 |
| 18. | Jia, X.; Yang, D.; Zhang, S.; Cheng, J. Org. Lett. 2009, 11, 4716–4719. doi:10.1021/ol9017529 |
| 19. | Chaitanya, M.; Yadagiri, D.; Anbarasan, P. Org. Lett. 2013, 15, 4960–4963. doi:10.1021/ol402201c |
| 32. | Levin, J. I.; Turos, E.; Weinreb, S. M. Synth. Commun. 1982, 12, 989–993. doi:10.1080/00397918208061938 |
| 9. | Kupchan, S. M.; Moniot, J. L.; Kanojia, R. M.; O'Brien, J. B. J. Org. Chem. 1971, 36, 2413–2418. doi:10.1021/jo00816a007 |
| 10. | Kupchan, S. M.; O’Brien, P. F. J. Chem. Soc., Chem. Commun. 1973, 915–916. doi:10.1039/c39730000915 |
| 11. | Castedo, L.; Saá, J. M.; Suau, R.; Villaverde, C. Heterocycles 1980, 14, 1131–1134. doi:10.3987/R-1980-08-1131 |
| 12. | Kessar, S. V.; Gupta, Y. P.; Yadav, V. S.; Narula, M.; Mohammed, T. Tetrahedron Lett. 1980, 21, 3307–3308. doi:10.1016/S0040-4039(00)78675-8 |
| 13. | Melzer, B.; Bracher, F. Org. Biomol. Chem. 2015, 13, 7664–7672. doi:10.1039/C5OB00926J |
| 15. | Cuny, G. D. Tetrahedron Lett. 2004, 45, 5167–5170. doi:10.1016/j.tetlet.2004.04.194 |
| 16. | Chaudhary, S.; Pecic, S.; LeGendre, O.; Harding, W. W. Tetrahedron Lett. 2009, 50, 2437–2439. doi:10.1016/j.tetlet.2009.03.029 |
| 30. | Kunitomo, J.-i.; Miyata, Y. Heterocycles 1986, 24, 437–440. doi:10.3987/R-1986-02-0437 |
| 1. | Hagel, J. M.; Facchini, P. J. Plant Cell Physiol. 2013, 54, 647–672. doi:10.1093/pcp/pct020 |
| 7. | Kunitomo, J.; Satoh, M.; Shingu, T. Tetrahedron 1983, 39, 3261–3265. doi:10.1016/S0040-4020(01)91573-X |
| 8. | Okamoto, Y.; Tanaka, S.; Kitayama, K.; Isomoto, M.; Masaishi, M.; Yanagawa, H.; Kunitomo, J.-I. Yakugaku Zasshi 1971, 91, 684–687. doi:10.1248/yakushi1947.91.6_684 |
| 7. | Kunitomo, J.; Satoh, M.; Shingu, T. Tetrahedron 1983, 39, 3261–3265. doi:10.1016/S0040-4020(01)91573-X |
| 25. | Plodek, A.; Raeder, S.; Bracher, F. Tetrahedron 2013, 69, 9857–9864. doi:10.1016/j.tet.2013.08.085 |
| 6. | Castro-Castillo, V.; Suárez-Rozas, C.; Pabón, A.; Pérez, E. G.; Cassels, B. K.; Blair, S. Bioorg. Med. Chem. Lett. 2013, 23, 327–329. doi:10.1016/j.bmcl.2012.10.092 |
| 7. | Kunitomo, J.; Satoh, M.; Shingu, T. Tetrahedron 1983, 39, 3261–3265. doi:10.1016/S0040-4020(01)91573-X |
| 17. | Kunitomo, J.; Kaede, S.; Satoh, M. Chem. Pharm. Bull. 1985, 33, 2778–2782. doi:10.1248/cpb.33.2778 |
| 5. | Tang, H.; Wang, X.-D.; Wie, Y.-B.; Huang, S.-L.; Huang, Z.-S.; Tan, J.-H.; An, L.-K.; Wu, J.-Y.; Chan, A. S.-C.; Gu, L.-Q. Eur. J. Med. Chem. 2008, 43, 973–980. doi:10.1016/j.ejmech.2007.07.004 |
| 14. | Orito, K.; Uchiito, S.; Satoh, Y.; Tatsuzawa, T.; Harada, R.; Tokuda, M. Org. Lett. 2000, 2, 307–310. doi:10.1021/ol990360v |
| 7. | Kunitomo, J.; Satoh, M.; Shingu, T. Tetrahedron 1983, 39, 3261–3265. doi:10.1016/S0040-4020(01)91573-X |
| 17. | Kunitomo, J.; Kaede, S.; Satoh, M. Chem. Pharm. Bull. 1985, 33, 2778–2782. doi:10.1248/cpb.33.2778 |
| 13. | Melzer, B.; Bracher, F. Org. Biomol. Chem. 2015, 13, 7664–7672. doi:10.1039/C5OB00926J |
| 22. | Kraskovskiy, A.; Kraskovskaya, V.; Knochel, P. Angew. Chem., Int. Ed. 2006, 45, 2958–2961. doi:10.1002/anie.200504024 |
| 35. | Tilly, D.; Fu, J.-m.; Zhao, B.-p.; Alessi, M.; Castanet, A. S.; Snieckus, V.; Mortier, J. Org. Lett. 2010, 12, 68–71. doi:10.1021/ol902268h |
| 23. | Metzger, A.; Schade, M. A.; Knochel, P. Org. Lett. 2008, 10, 1107–1110. doi:10.1021/ol7030697 |
| 21. | Melzer, B.; Plodek, A.; Bracher, F. J. Org. Chem. 2014, 79, 7239–7242. doi:10.1021/jo501312d |
| 36. | Fu, J. M.; Zhao, B. P.; Sharp, M. J.; Snieckus, V. J. Org. Chem. 1991, 56, 1683–1685. doi:10.1021/jo00005a004 |
| 24. | Krätzschmar, F.; Kaßel, M.; Delony, D.; Breder, A. Chem. – Eur. J. 2015, 21, 7030–7034. doi:10.1002/chem.201406290 |
| 7. | Kunitomo, J.; Satoh, M.; Shingu, T. Tetrahedron 1983, 39, 3261–3265. doi:10.1016/S0040-4020(01)91573-X |
| 17. | Kunitomo, J.; Kaede, S.; Satoh, M. Chem. Pharm. Bull. 1985, 33, 2778–2782. doi:10.1248/cpb.33.2778 |
| 22. | Kraskovskiy, A.; Kraskovskaya, V.; Knochel, P. Angew. Chem., Int. Ed. 2006, 45, 2958–2961. doi:10.1002/anie.200504024 |
| 23. | Metzger, A.; Schade, M. A.; Knochel, P. Org. Lett. 2008, 10, 1107–1110. doi:10.1021/ol7030697 |
| 13. | Melzer, B.; Bracher, F. Org. Biomol. Chem. 2015, 13, 7664–7672. doi:10.1039/C5OB00926J |
| 23. | Metzger, A.; Schade, M. A.; Knochel, P. Org. Lett. 2008, 10, 1107–1110. doi:10.1021/ol7030697 |
| 7. | Kunitomo, J.; Satoh, M.; Shingu, T. Tetrahedron 1983, 39, 3261–3265. doi:10.1016/S0040-4020(01)91573-X |
| 17. | Kunitomo, J.; Kaede, S.; Satoh, M. Chem. Pharm. Bull. 1985, 33, 2778–2782. doi:10.1248/cpb.33.2778 |
| 7. | Kunitomo, J.; Satoh, M.; Shingu, T. Tetrahedron 1983, 39, 3261–3265. doi:10.1016/S0040-4020(01)91573-X |
| 17. | Kunitomo, J.; Kaede, S.; Satoh, M. Chem. Pharm. Bull. 1985, 33, 2778–2782. doi:10.1248/cpb.33.2778 |
| 13. | Melzer, B.; Bracher, F. Org. Biomol. Chem. 2015, 13, 7664–7672. doi:10.1039/C5OB00926J |
| 37. | Cheng, J.-J.; Tsai, T.-H.; Lin, L.-C. Planta Med. 2012, 78, 1873–1877. doi:10.1055/s-0032-1327785 |
© 2017 Melzer and Bracher; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)