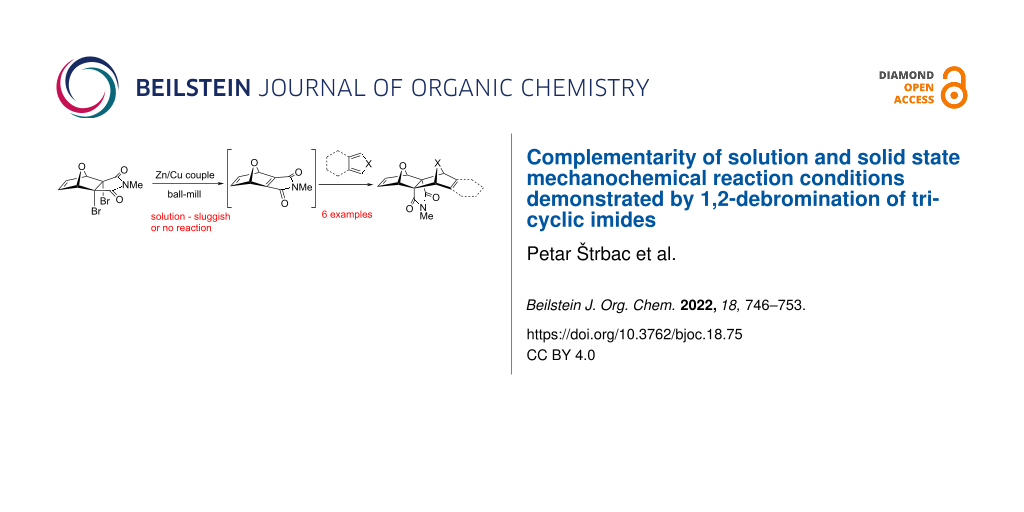
Abstract
The solution phase 1,2-debromination of polycyclic imides using the Zn/Ag couple was successfully transferred to solid state mechanochemical conditions. The Zn/Ag couple was replaced by the Zn/Cu couple which was prepared without any metal activation by in situ ball milling of zinc and copper dusts. The advantage of the ball milling process is that the whole procedure is operationally very simplified. The reactive alkene generated was trapped in situ by several dienes and the respective Diels−Alder cycloadducts were obtained. It was demonstrated that mechanochemical milling offers complementary conditions to solution (thermal) reaction by allowing chemical transformations to proceed which were not possible in solution and vice versa.

Graphical Abstract
Introduction
The complementarity of reaction conditions [1-3] where the reaction takes place under some, but not under other conditions, or where a chemical reaction proceeds in a different way or mechanism is a useful feature in synthetic organic chemistry. Advantageously, more difficult substrates or limitations of the conditions can be overcome by the change of the reaction methods. One of the emerging synthetic methods is mechanochemistry [4-7], a greener alternative to carry out synthesis which complements heating, irradiation and electrochemistry as methods of chemical activation [8]. Based upon our experience in applications of this method to organic synthesis [9-12], we recognized its potential for the adjustment of conditions in zinc-mediated debromination reactions.
Highly reactive dienophiles such as polycyclic molecules given in Figure 1 are interesting reactive intermediates which could be applied in the Diels−Alder reactions of less reactive or thermally susceptible dienes. Often, these are generated in situ and trapped with dienes in a single pot, such as 7-oxanorbornadiene imides 1–3. For instance, a synthetic methodology for the preparation of 1–3 was developed by Warrener and co-workers by a Zn/Ag couple debromination [13-15]. However, this methodology has some disadvantages, such as tedious preparation of the catalyst, the use of dry solvent and expensive silver acetate as well as side-reactions with solvent.
![[1860-5397-18-75-1]](/bjoc/content/figures/1860-5397-18-75-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
An alternative method for the synthesis 7-oxanorbornadiene-2,3-anhydrides and imides is via retro Diels−Alder reaction employing the flash vacuum pyrolysis (FVP) technique [16]. However, the FVP also has disadvantages such as limited scope of functionalities which can withstand harsh conditions (temperature) and the inability to control the elimination process [17].
The objective of this work was to establish whether the 1,2-debromination with the Zn/Ag couple could be carried out under solvent-free conditions in a ball mill and whether the tedious Zn/Ag couple preparation procedure [18] could be simplified by in situ generation of the catalyst. Moreover, in the debromination of norbornene imide 11, the expected Diels−Alder adduct with furan was not obtained, but compound 12 incorporating a tetrahydrofuran ring at position 2, presumably by radical reaction (Figure 2) [19,20]. We envisaged that the absence of solvent under mechanochemical conditions should prevent the formation of products from tetrahydrofuran and therefore allow cycloaddition to take place.
![[1860-5397-18-75-2]](/bjoc/content/figures/1860-5397-18-75-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Dibromide substrates and product 12.
Figure 2: Dibromide substrates and product 12.
Results and Discussion
Reaction optimization
Anthracene addition to dibromide 10 (Scheme 1) was used as the model reaction. Along with cycloadduct 14, three known side-products were obtained: endo-product 15 (hydrogenolysed inverted 10), N-methylphthalimide (16) and 17 (hydrogenolysed 10). Their ratio varied with the reaction conditions (Supporting Information, Table S1) and results of the optimizations are collected in Table 1.
![[1860-5397-18-75-i1]](/bjoc/content/inline/1860-5397-18-75-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Mechanochemical reaction of 10 with anthracene.
Scheme 1: Mechanochemical reaction of 10 with anthracene.
Table 1: Optimization of reaction conditions for reaction of 10 with anthracene.a
| Entry | Catalyst | Additives | Time [h] | Conversionb | Yield 14 [%]c |
| 1 | Zn/Ag couple | 0.5 | 81 | 2 | |
| 2 | Zn/Ag couple | NaCl | 0.5 | quant | 8 |
| 3 | Zn/Ag couple | LAG THF | 0.5 | quant | 58 |
| 4 | Zn/Ag couple | LAG THF, NaCl | 0.5 | 96 | 33 |
| 5 | Zn dust, Ag wire | 0.5 | NRd | ||
| 6 | Zn dust, Ag wire | LAG THF | 0.5 | 33 | 27 |
| 7 | Zn dust, Ag wire | LAG MeOH | 1 | quant | 64 |
| 8 | Zn dust, Ag wire | LAG MeCN | 1 | 88 | 63 |
| 9 | Zn activ., Ag wire | LAG THF, NaCl | 0.5 | quant | 56 |
| 10 | Zn dust, silvergal | 0,5 | NR | ||
| 11 | Zn dust, silvergal | LAG THF | 1 | 50 | 40 |
| 12 | Zn activated | 0,5 | NR | ||
| 13 | Zn dust | 0,5 | NR | ||
| 14 | Zn dust | LAG THF | 0,5 | 12 | 4 |
| 15 | Zn dust, Cu dust | LAG THF | 0,5 | 55 | 50 |
| 16 | Zn dust, Cu dust | LAG THF | 0.75 | 97 | 64 |
| 17 | Zn dust, Cu dust | LAG THF ZnBr2 | 1 | quant | 67 |
| 18 | Zn dust, Cu dust | LAG THFe | 1 | 66 | 48 |
| 19 | Zn dust, Cu dust | LAG THFe | 2 | quant | 76 (42) |
| Reactions in solution | Solvent | ||||
| 20 | Zn/Ag couple | dry THF, Ar | 1 | quant | 86 |
| 21 | Zn dust, Cu dust | dry THF | 1 | NR | |
| 22 | Zn dust, Cu dust | THF, ultrasound | 1.5 | NR | |
aRetsch MM400, 30 Hz, stainless steel 10 mL, one 12 mm SS ball; dibromide (50 mg); anthracene (132 mg, 5 equiv); reducing agent/catalyst (75 mg); silvergal = Ag/Cu powder 70% Ag; LAG THF η = 0.66 μL·mg−1; bNMR analysis; cNMR yields, isolated yield in parentheses; dNR = no reaction; eLAG THF η = 0.33 μL·mg−1.
The results of the optimization experiments showed that the solution reaction conditions could be transferred to mechanochemical conditions without significant loss of reactivity and identical side-products were formed. Initial experiments were performed with the Zn/Ag couple prepared by a usual procedure from Zn and silver acetate. Several simplifications of the Zn/Ag couple preparation were tested and showed that simple milling with Zn dust and Ag dust or wire can be also applied (Table 1, entries 5–8) [21]. Further improvement in the procedure was the replacement of the Ag dust with Cu dust. This combination of metals worked well, and the best conditions were obtained with the addition of a small amount of THF (liquid assisted grinding, LAG) [22], η = 0.5 μL·mg−1 (Table 1, entry 19). In contrast, the solution reaction catalyzed by Zn/Cu dust was totally ineffective (Table 1, entry 21), even with the agitation by ultrasound (Table 1, entry 22). The ratios of side-products vary depending on the reaction conditions (Table S1, Supporting Information File 1). The endo-product 15 was dominant in the neat grinding experiment (Table 1, entry 1), whereas phthalimide 16 dominates when NaCl was employed as grinding auxiliary (Table 1, entries 2 and 4). The addition of ZnBr2 (which is formed in the reaction and postulated that it could facilitate the oxa-ring opening of 15 to 16) [13] did not notably increase the amount of phthalimide, indicating that rather deoxygenation leading to 16 is facilitated by the Zn/Cu couple [23]. When LAG THF reactions were carried out without anthracene, 15 was major product, whereas 16 is the major product in LAG MeOH milling (Supporting Information File 1, Table S1).
Scope of the reaction
With the optimized conditions established, the scope of the reaction and its synthetic utility were investigated employing various dienes such as furan (18), 1,3-diphenylisobenzofuran (24) (DPIBF) and substituted anthracenes 31, 36 and 39 (Figure 3). Exclusive norbornene exo-π selectivity [24] was observed in all cycloadddition reactions.
![[1860-5397-18-75-3]](/bjoc/content/figures/1860-5397-18-75-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Scope of the Zn/Cu reaction with dibromide 10 (dienes are colored in red).
Figure 3: Scope of the Zn/Cu reaction with dibromide 10 (dienes are colored in red).
Selected five-membered dienes were subjected to established the Zn/Cu mechanochemical conditions (Figure 3). The furan reaction under ball milling conditions led to a mixture of exo,exo- and exo,endo-cycloadducts 19 and 20 in a 0.8:1 ratio. This is in contrast to classical conditions with the Zn/Ag couple in THF, where the ratio is different (0.6:1), and slightly more in favor of the unsymmetrical adduct 20. On the other hand, the reaction of cyclopentadiene (21) provided the linear exo,exo-cycloadduct 22 as the major product, together with the 2:1 adduct 23. In this reaction, identical stereospecificity was obtained by employment of the Zn/Ag couple in THF [19].
Linear exo,exo-product 25 was obtained exclusively in the reaction with DPIBF 24, which is in accordance to the stereospecificity of cycloadditions reported by Sasaki [25] where the linear adduct is greatly preferred over bent. An interesting feature of the 1H NMR spectrum is the very low-field position of the phenyl protons (8.15 ppm), not common for DPIBF adducts with 7-oxanorbornenes (usually below 8 ppm) [26,27]. Double adduct 26 was detected as minor product (evidenced by the lack of olefinic and presence of the endo protons at 2.64 ppm), whereas the DPIBF sideproduct in this reaction is 1,2-dibenzoylbenzene (27). The mechanism for the formation of 27 from DPIBF is not elucidated, but we confirmed that the reaction does not proceed by milling of DPIBF with Zn/Cu dust alone, indicating that the presence of the dibromide substrate is essential. Product 27 was also found in the reaction of bicyclo[2.2.2] dibromide 42 (Scheme 3), which suggests that the mechanism may involve radical anion intermediates. This result further supports a single electron transfer (SET) and radical anion mechanism which was postulated earlier for the Zn/Ag debromination reaction and was supported by a CH3OD trapping experiment [13]. Thus, DPIBF in this reaction acts both as Diels−Alder trap reagent for reactive alkene [28] as well as radical anion quencher [29,30].
The N,N-Boc-protected N-amidinylpyrrole 28 [31] and 1-guanidinoanthracene (29) [10,32] have functional groups which were not tolerated under the debromination conditions and intractable mixtures of products were obtained. Carbomethoxypyrrole (30) was less susceptible to the reaction conditions, but unreactive. In these reactions, N-methylphthalimide (16) was obtained as the major product.
Interestingly, when 9,10-diphenylanthracene (31) was subjected to milling with 10, the expected cycloadduct was not detected and 31 has remained unchanged, indicating its lower reactivity in comparison to anthracene (presumably due to steric reasons [33], the presence of phenyl substituents at reacting carbons). Instead, small amounts of another cycloadduct were obtained. It was found that this is the product arising from anthraquinone (32), which was present as an impurity in 31. Independent milling of 10 with anthraquinone afforded dihydroxy cycloadduct 33 (in 35% yield) indicating that in the reaction conditions of the anthraquinone ↔ 9,10-dihydroxyanthracene (DHA) equilibrium is shifted towards DHA [34,35]. To prove this premise, anthraquinone alone was ball milled, however, unreacted material was recovered and the formation of 9,10-dihydroxyanthracene was not spectroscopically detected. An analogous hydroxy adduct 35 was produced in the reaction with anthrone (34). When the reaction of anthraquinone was carried in THF solution (reflux, 1 h), dibromide 10 remained unchanged. However, a small amount of 33 was formed in refluxing THF by the use of the Zn/Ag couple in the case of anthraquinone. These results indicate that the Zn/Cu catalyst in solid state is much more effective than Zn/Cu or Zn/Ag in THF solution, and that ball milling offers different reaction outcomes and as such complements solution chemistry.
The reaction with 2,3-dicarbomethoxyanthracene (36) afforded two isomeric cycloadducts 37 and 38 in a 1:1.3 ratio (inseparable mixture, 32% overall yield). The minor isomer has a linear structure with carbomethoxy groups in the equatorial plane as depicted for 37, whereas in the major isomer 38 the carbomethoxy groups are positioned in axial plane of the molecule. The bent structure of 38 was established by 1H NMR analysis and comparison with product 10. The most indicative signals are of N-methyl groups, which are at an almost identical position for the bent isomer 38 (2.36 ppm), as in products 10 and 33 (2.38 and 2.37 ppm, respectively). The chemical shift of the NMe group in linear product 36 is affected by anthracene carbomethoxy 2,3-substituents and is shifted towards lower magnetic field (2.42 ppm). Furthermore, the highest lying aromatic multiplet of 38 is similarly positioned as for 10 (7.06 and 7.08 ppm, respectively), whereas the chemical shift of the methyl ester groups in 38 is closer to the starting anthracene 36 (3.87 ppm and 3.85 ppm in 38 and 37 vs 3.97 ppm in 36). When 2,3-anthracene anhydride (39) was subjected to ball milling, a complex mixture of products was obtained, indicating that the anhydride functionality is not compatible to the reaction conditions.
Product 22 could be also obtained by cycloaddition parity reversal principle [16,36], employing norbornene dibromide 11 and furan (18). Ball milling reaction without THF (neat grinding) provided 22 as the major product, together with a minor amount of dehalogenated product 40 (Scheme 2). When THF was added for LAG, milling again gave 22, but it was accompanied with a larger amount of 40 and some side-product 12. A control LAG milling experiment without furan led to 40 and a significant amount of 12. These experiments emphasize the complementarity of ball milling conditions with solution chemistry (in solution, only THF side-product 12 was obtained). Interestingly, small amounts of THF (used for LAG) were not detrimental for the reaction outcome in ball mill.
![[1860-5397-18-75-i2]](/bjoc/content/inline/1860-5397-18-75-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Mechanochemical reaction of 11 with furan.
Scheme 2: Mechanochemical reaction of 11 with furan.
The bicyclo[2.2.2] system
Besides the bicyclo[2.2.1] system (7-oxa or 7-methano), the reactivity of the bicyclo[2.2.2] moiety was investigated. Dibromide 42 was prepared by Diels−Alder reaction of anthracene (13) and 2,3-dibromo-N-methylmaleimide (41) (Scheme 3). Heating of the reactants at 180 °C for 10 min provided the required cycloadduct 42 (98% yield). When 42 was subjected to Zn/Cu debromination in a ball mill in the presence of anthracene (conditions a), imide 44 was the major product accompanied by a small amount of janusene imide derivative 45 (7:1 ratio). The formation of the intermediate alkene 43 was observed spectroscopically only in the milling reaction of 42 alone (an indicative 1H NMR signal of bicyclo[2.2.1] moiety at 5.28 ppm). Cycloreversion side-reaction in mechanochemical conditions [37] was noticed for dibromide 42, which led to mixtures consisting some janusene 45. Thus, milling of 42 with Zn/Cu without anthracene (13) (conditions b) provided a mixture of 43/44/45/anthracene in a 1:5:1:2 ratio) and milling of 42 alone without metal dust (conditions c) gave anthracene. Furthermore, alkene trapping with DPIBF provided 1:1 cycloadduct 46 as a single isomer (possessing linear structure as indicated by 1H NMR chemical shift of methyl group at 1.96 ppm). There is a large up-field shift of two aromatic protons of the anthracene moiety (6.31 ppm) arising from the magnetic shielding of phenyl groups. Analogously to the reaction of DPIBF with 10 (Figure 3), 1,2-dibenzoylbenzene (27) was found in the reaction mixture (Scheme 3). Starting from 42 in identical mechanochemical conditions, furan cycloadduct 14 was not formed, just 43 and 44 (6:1 ratio) and anthracene (13) were obtained. Our results demonstrate that the bicyclo[2.2.2] system is less reactive in comparison to the 7-oxabicyclo[2.2.1] system. Ball milling did not provide any cycloadduct even in the presence of 20-fold molar excess of furan and the alternative synthetic route (cycloaddition parity reversal) [16,17] for 14 was not viable.
![[1860-5397-18-75-i3]](/bjoc/content/inline/1860-5397-18-75-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reactivity of bicyclo[2.2.2] dibromide 42 with dienes.
Scheme 3: Reactivity of bicyclo[2.2.2] dibromide 42 with dienes.
Conclusion
The 1,2-debromination reactions employing the Zn/Ag couple could be effectively carried out in a ball mill, with advantageous employment of the Zn/Cu couple prepared in situ, avoiding the use of dry solvent precautions and tedious Zn/Ag couple preparation, in a simple procedure. It is an example of organic reactions in solution that could be transferred to greener conditions. The reaction works in some instances when classical conditions failed, and as such it offers a complementary synthetic approach towards required molecules.
Supporting Information
| Supporting Information File 1: Details of experimental procedures and characterization data of selected compounds. | ||
| Format: PDF | Size: 1.8 MB | Download |
References
-
McAtee, R. C.; McClain, E. J.; Stephenson, C. R. J. Trends Chem. 2019, 1, 111–125. doi:10.1016/j.trechm.2019.01.008
Return to citation in text: [1] -
Sambiagio, C.; Noël, T. Trends Chem. 2020, 2, 92–106. doi:10.1016/j.trechm.2019.09.003
Return to citation in text: [1] -
Wallace, S.; Balskus, E. P. Curr. Opin. Biotechnol. 2014, 30, 1–8. doi:10.1016/j.copbio.2014.03.006
Return to citation in text: [1] -
James, S. L.; Adams, C. J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K. D. M.; Hyett, G.; Jones, W.; Krebs, A.; Mack, J.; Maini, L.; Orpen, A. G.; Parkin, I. P.; Shearouse, W. C.; Steed, J. W.; Waddell, D. C. Chem. Soc. Rev. 2012, 41, 413–447. doi:10.1039/c1cs15171a
Return to citation in text: [1] -
Wang, G.-W. Chem. Soc. Rev. 2013, 42, 7668–7700. doi:10.1039/c3cs35526h
Return to citation in text: [1] -
Stolle, A.; Ranu, B., Eds. Ball Milling Towards Green Synthesis: Applications, Projects, Challenges; Green Chemistry Series, Vol. 31; Royal Society of Chemistry: Cambridge, UK, 2015. doi:10.1039/9781782621980
Return to citation in text: [1] -
Margetić, D.; Štrukil, V. Mechanochemical Organic Synthesis; Elsevier: Amsterdam, Netherlands, 2016. doi:10.1016/c2014-0-01621-8
Return to citation in text: [1] -
Howard, J. L.; Cao, Q.; Browne, D. L. Chem. Sci. 2018, 9, 3080–3094. doi:10.1039/c7sc05371a
Return to citation in text: [1] -
Đud, M.; Briš, A.; Jušinski, I.; Gracin, D.; Margetić, D. Beilstein J. Org. Chem. 2019, 15, 1313–1320. doi:10.3762/bjoc.15.130
Return to citation in text: [1] -
Đud, M.; Glasovac, Z.; Margetić, D. Tetrahedron 2019, 75, 109–115. doi:10.1016/j.tet.2018.11.038
Return to citation in text: [1] [2] -
Portada, T.; Margetić, D.; Štrukil, V. Molecules 2018, 23, 3163. doi:10.3390/molecules23123163
Return to citation in text: [1] -
Briš, A.; Đud, M.; Margetić, D. Beilstein J. Org. Chem. 2017, 13, 1745–1752. doi:10.3762/bjoc.13.169
Return to citation in text: [1] -
Warrener, R. N.; Maksimovic, L. Tetrahedron Lett. 1994, 35, 2389–2392. doi:10.1016/0040-4039(94)85227-8
Return to citation in text: [1] [2] [3] -
Warrener, R. N.; Maksimovic, L.; Butler, D. N. J. Chem. Soc., Chem. Commun. 1994, 1831–1832. doi:10.1039/c39940001831
Return to citation in text: [1] -
Warrener, R. N.; Pitt, I. G.; Russell, R. A. Aust. J. Chem. 1991, 44, 1293–1305. doi:10.1071/ch9911293
Return to citation in text: [1] -
Margetić, D.; Warrener, R. N.; Sun, G.; Butler, D. N. Tetrahedron 2007, 63, 4338–4346. doi:10.1016/j.tet.2007.03.009
Return to citation in text: [1] [2] [3] -
Margetić, D.; Butler, D. N.; Warrener, R. N.; Murata, Y. Tetrahedron 2011, 67, 1580–1588. doi:10.1016/j.tet.2010.12.032
Return to citation in text: [1] [2] -
Clark, R. D.; Heathcock, C. H. J. Org. Chem. 1976, 41, 636–643. doi:10.1021/jo00866a012
Return to citation in text: [1] -
Warrener, R. N.; Elsey, G. M.; Maksimovic, L.; Johnston, M. R.; Kennard, C. H. L. Tetrahedron Lett. 1995, 36, 7753–7756. doi:10.1016/0040-4039(95)01617-q
Return to citation in text: [1] [2] -
Matsuo, Y.; Zhang, Y.; Nakamura, E. Org. Lett. 2008, 10, 1251–1254. doi:10.1021/ol800143b
Return to citation in text: [1] -
Cao, Q.; Stark, R. T.; Fallis, I. A.; Browne, D. L. ChemSusChem 2019, 12, 2554–2557. doi:10.1002/cssc.201900886
Return to citation in text: [1] -
Friščić, T.; Trask, A. V.; Jones, W.; Motherwell, W. D. S. Angew. Chem., Int. Ed. 2006, 45, 7546–7550. doi:10.1002/anie.200603235
Return to citation in text: [1] -
Yide, X.; Qingqing, G.; Naizheng, H. Huaxue Xuebao 1983, 41, 934–938.
Return to citation in text: [1] -
Margetić, D.; Warrener, R. N. Croat. Chem. Acta 2003, 76, 357–363.
Return to citation in text: [1] -
Sasaki, T.; Kanematsu, K.; Iizuka, K. Heterocycles 1975, 3, 109–112. doi:10.3987/r-1975-02-0109
Return to citation in text: [1] -
Eda, S.; Eguchi, F.; Haneda, H.; Hamura, T. Chem. Commun. 2015, 51, 5963–5966. doi:10.1039/c5cc00077g
Return to citation in text: [1] -
Blatter, K.; Schlüter, A.-D. Chem. Ber. 1989, 122, 1351–1356. doi:10.1002/cber.19891220719
Return to citation in text: [1] -
Marchand, A. P.; Namboothiri, I. N. N.; Ganguly, B.; Watson, W. H.; Bodige, S. G. Tetrahedron Lett. 1999, 40, 5105–5109. doi:10.1016/s0040-4039(99)00888-6
Return to citation in text: [1] -
Ohyashiki, T.; Nunomura, M.; Katoh, T. Biochim. Biophys. Acta, Biomembr. 1999, 1421, 131–139. doi:10.1016/s0005-2736(99)00119-4
Return to citation in text: [1] -
Żamojć, K.; Zdrowowicz, M.; Rudnicki-Velasquez, P. B.; Krzymiński, K.; Zaborowski, B.; Niedziałkowski, P.; Jacewicz, D.; Chmurzyński, L. Free Radical Res. 2017, 51, 38–46. doi:10.1080/10715762.2016.1262541
Return to citation in text: [1] -
Parr, B. T.; Economou, C.; Herzon, S. B. Nature 2015, 525, 507–510. doi:10.1038/nature14902
Return to citation in text: [1] -
Antol, I.; Glasovac, Z.; Murata, Y.; Hashikawa, Y.; Margetić, D. ChemistrySelect 2022, 7, e202200633. doi:10.1002/slct.202200633
Return to citation in text: [1] -
Huynh, V. N.; Leitner, M.; Bhattacharyya, A.; Uhlstein, L.; Kreitmeier, P.; Sakrausky, P.; Rehbein, J.; Reiser, O. Commun. Chem. 2020, 3, 158. doi:10.1038/s42004-020-00407-9
Return to citation in text: [1] -
Koerner, M.; Rickborn, B. J. Org. Chem. 1989, 54, 6–9. doi:10.1021/jo00262a003
Return to citation in text: [1] -
Koerner, M.; Rickborn, B. J. Org. Chem. 1990, 55, 2662–2672. doi:10.1021/jo00296a024
Return to citation in text: [1] -
Butler, D. N.; Margetić, D.; O'Neill, P. J. C.; Warrener, R. N. Synlett 2000, 98–100. doi:10.1055/s-2000-6445
Return to citation in text: [1] -
Murata, Y.; Kato, N.; Fujiwara, K.; Komatsu, K. J. Org. Chem. 1999, 64, 3483–3488. doi:10.1021/jo990013z
Return to citation in text: [1]
| 1. | McAtee, R. C.; McClain, E. J.; Stephenson, C. R. J. Trends Chem. 2019, 1, 111–125. doi:10.1016/j.trechm.2019.01.008 |
| 2. | Sambiagio, C.; Noël, T. Trends Chem. 2020, 2, 92–106. doi:10.1016/j.trechm.2019.09.003 |
| 3. | Wallace, S.; Balskus, E. P. Curr. Opin. Biotechnol. 2014, 30, 1–8. doi:10.1016/j.copbio.2014.03.006 |
| 13. | Warrener, R. N.; Maksimovic, L. Tetrahedron Lett. 1994, 35, 2389–2392. doi:10.1016/0040-4039(94)85227-8 |
| 14. | Warrener, R. N.; Maksimovic, L.; Butler, D. N. J. Chem. Soc., Chem. Commun. 1994, 1831–1832. doi:10.1039/c39940001831 |
| 15. | Warrener, R. N.; Pitt, I. G.; Russell, R. A. Aust. J. Chem. 1991, 44, 1293–1305. doi:10.1071/ch9911293 |
| 19. | Warrener, R. N.; Elsey, G. M.; Maksimovic, L.; Johnston, M. R.; Kennard, C. H. L. Tetrahedron Lett. 1995, 36, 7753–7756. doi:10.1016/0040-4039(95)01617-q |
| 9. | Đud, M.; Briš, A.; Jušinski, I.; Gracin, D.; Margetić, D. Beilstein J. Org. Chem. 2019, 15, 1313–1320. doi:10.3762/bjoc.15.130 |
| 10. | Đud, M.; Glasovac, Z.; Margetić, D. Tetrahedron 2019, 75, 109–115. doi:10.1016/j.tet.2018.11.038 |
| 11. | Portada, T.; Margetić, D.; Štrukil, V. Molecules 2018, 23, 3163. doi:10.3390/molecules23123163 |
| 12. | Briš, A.; Đud, M.; Margetić, D. Beilstein J. Org. Chem. 2017, 13, 1745–1752. doi:10.3762/bjoc.13.169 |
| 25. | Sasaki, T.; Kanematsu, K.; Iizuka, K. Heterocycles 1975, 3, 109–112. doi:10.3987/r-1975-02-0109 |
| 8. | Howard, J. L.; Cao, Q.; Browne, D. L. Chem. Sci. 2018, 9, 3080–3094. doi:10.1039/c7sc05371a |
| 4. | James, S. L.; Adams, C. J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K. D. M.; Hyett, G.; Jones, W.; Krebs, A.; Mack, J.; Maini, L.; Orpen, A. G.; Parkin, I. P.; Shearouse, W. C.; Steed, J. W.; Waddell, D. C. Chem. Soc. Rev. 2012, 41, 413–447. doi:10.1039/c1cs15171a |
| 5. | Wang, G.-W. Chem. Soc. Rev. 2013, 42, 7668–7700. doi:10.1039/c3cs35526h |
| 6. | Stolle, A.; Ranu, B., Eds. Ball Milling Towards Green Synthesis: Applications, Projects, Challenges; Green Chemistry Series, Vol. 31; Royal Society of Chemistry: Cambridge, UK, 2015. doi:10.1039/9781782621980 |
| 7. | Margetić, D.; Štrukil, V. Mechanochemical Organic Synthesis; Elsevier: Amsterdam, Netherlands, 2016. doi:10.1016/c2014-0-01621-8 |
| 19. | Warrener, R. N.; Elsey, G. M.; Maksimovic, L.; Johnston, M. R.; Kennard, C. H. L. Tetrahedron Lett. 1995, 36, 7753–7756. doi:10.1016/0040-4039(95)01617-q |
| 20. | Matsuo, Y.; Zhang, Y.; Nakamura, E. Org. Lett. 2008, 10, 1251–1254. doi:10.1021/ol800143b |
| 22. | Friščić, T.; Trask, A. V.; Jones, W.; Motherwell, W. D. S. Angew. Chem., Int. Ed. 2006, 45, 7546–7550. doi:10.1002/anie.200603235 |
| 18. | Clark, R. D.; Heathcock, C. H. J. Org. Chem. 1976, 41, 636–643. doi:10.1021/jo00866a012 |
| 13. | Warrener, R. N.; Maksimovic, L. Tetrahedron Lett. 1994, 35, 2389–2392. doi:10.1016/0040-4039(94)85227-8 |
| 17. | Margetić, D.; Butler, D. N.; Warrener, R. N.; Murata, Y. Tetrahedron 2011, 67, 1580–1588. doi:10.1016/j.tet.2010.12.032 |
| 16. | Margetić, D.; Warrener, R. N.; Sun, G.; Butler, D. N. Tetrahedron 2007, 63, 4338–4346. doi:10.1016/j.tet.2007.03.009 |
| 21. | Cao, Q.; Stark, R. T.; Fallis, I. A.; Browne, D. L. ChemSusChem 2019, 12, 2554–2557. doi:10.1002/cssc.201900886 |
| 28. | Marchand, A. P.; Namboothiri, I. N. N.; Ganguly, B.; Watson, W. H.; Bodige, S. G. Tetrahedron Lett. 1999, 40, 5105–5109. doi:10.1016/s0040-4039(99)00888-6 |
| 26. | Eda, S.; Eguchi, F.; Haneda, H.; Hamura, T. Chem. Commun. 2015, 51, 5963–5966. doi:10.1039/c5cc00077g |
| 27. | Blatter, K.; Schlüter, A.-D. Chem. Ber. 1989, 122, 1351–1356. doi:10.1002/cber.19891220719 |
| 13. | Warrener, R. N.; Maksimovic, L. Tetrahedron Lett. 1994, 35, 2389–2392. doi:10.1016/0040-4039(94)85227-8 |
| 37. | Murata, Y.; Kato, N.; Fujiwara, K.; Komatsu, K. J. Org. Chem. 1999, 64, 3483–3488. doi:10.1021/jo990013z |
| 16. | Margetić, D.; Warrener, R. N.; Sun, G.; Butler, D. N. Tetrahedron 2007, 63, 4338–4346. doi:10.1016/j.tet.2007.03.009 |
| 17. | Margetić, D.; Butler, D. N.; Warrener, R. N.; Murata, Y. Tetrahedron 2011, 67, 1580–1588. doi:10.1016/j.tet.2010.12.032 |
| 34. | Koerner, M.; Rickborn, B. J. Org. Chem. 1989, 54, 6–9. doi:10.1021/jo00262a003 |
| 35. | Koerner, M.; Rickborn, B. J. Org. Chem. 1990, 55, 2662–2672. doi:10.1021/jo00296a024 |
| 16. | Margetić, D.; Warrener, R. N.; Sun, G.; Butler, D. N. Tetrahedron 2007, 63, 4338–4346. doi:10.1016/j.tet.2007.03.009 |
| 36. | Butler, D. N.; Margetić, D.; O'Neill, P. J. C.; Warrener, R. N. Synlett 2000, 98–100. doi:10.1055/s-2000-6445 |
| 10. | Đud, M.; Glasovac, Z.; Margetić, D. Tetrahedron 2019, 75, 109–115. doi:10.1016/j.tet.2018.11.038 |
| 32. | Antol, I.; Glasovac, Z.; Murata, Y.; Hashikawa, Y.; Margetić, D. ChemistrySelect 2022, 7, e202200633. doi:10.1002/slct.202200633 |
| 33. | Huynh, V. N.; Leitner, M.; Bhattacharyya, A.; Uhlstein, L.; Kreitmeier, P.; Sakrausky, P.; Rehbein, J.; Reiser, O. Commun. Chem. 2020, 3, 158. doi:10.1038/s42004-020-00407-9 |
| 29. | Ohyashiki, T.; Nunomura, M.; Katoh, T. Biochim. Biophys. Acta, Biomembr. 1999, 1421, 131–139. doi:10.1016/s0005-2736(99)00119-4 |
| 30. | Żamojć, K.; Zdrowowicz, M.; Rudnicki-Velasquez, P. B.; Krzymiński, K.; Zaborowski, B.; Niedziałkowski, P.; Jacewicz, D.; Chmurzyński, L. Free Radical Res. 2017, 51, 38–46. doi:10.1080/10715762.2016.1262541 |
| 31. | Parr, B. T.; Economou, C.; Herzon, S. B. Nature 2015, 525, 507–510. doi:10.1038/nature14902 |
© 2022 Štrbac and Margetić; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.