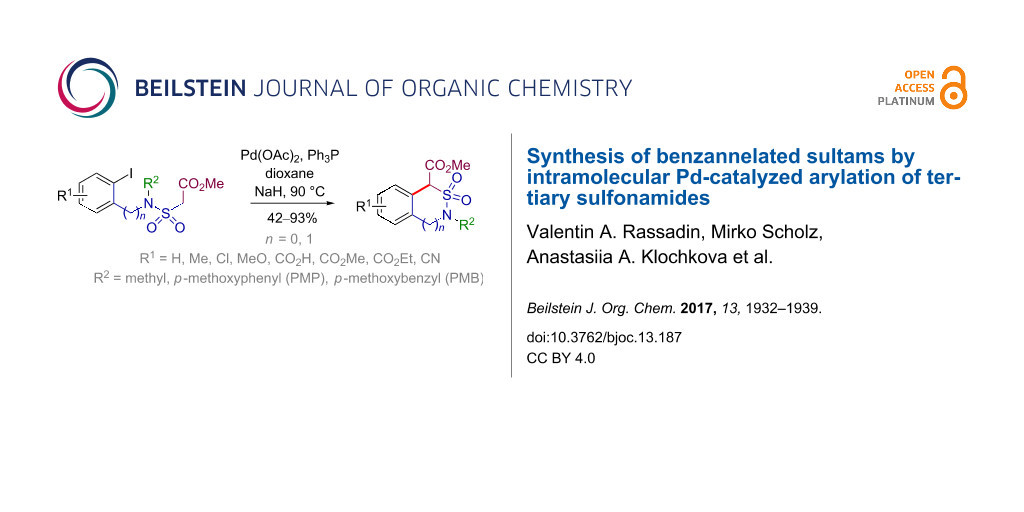
Abstract
A new and efficient approach to five- and six-membered benzannelated sultams by intramolecular C-arylation of tertiary 1-(methoxycarbonyl)methanesulfonamides under palladium catalysis is described. In case of the α-toluenesulfonamide derivative, an unexpected formation of a 2,3-diarylindole was observed under the same conditions.

Graphical Abstract
Introduction
The sulfonamide functional group stands out as one of the most important pharmacophores. At the same time, cyclic sulfonamides (sultams) have also received significant attention due to their biological activities and medicinal uses [1-7]. On the other hand, especially fused sultams are among the most commonly used therapeutic agents owing to the broad spectrum of their activities [8-13]. Up to now, sultams have been prepared employing Diels–Alder reactions, radical cyclizations, reductions of sulfonylimines, ring-closing metatheses, nucleophilic aromatic substitutions and Heck cyclizations [14-16].
Earlier on, our group has developed versatile syntheses of sultams based on the transformation of methanesulfonamides bearing an additional α-acceptor (EWG) group [17-20]. Thus, intermolecular cyclodialkylation of α-substituted methanesulfonamides, upon treatment with α,ω-dibromoalkanes and bases, provides a rather simple and efficient route to five-, six- and seven-membered sultams. Moreover, bridged sultams with a pyramidal nitrogen atom and a sulfur atom in the apex positions were prepared from the same kind of starting materials in good to excellent yields [20]. Quite recently, Xu et al. employed 1-diazo-1-ethoxycarbonylmethanesulfonamides for the synthesis of benzo-γ-sultams. The key step in the latter transformation was a rhodium octanoate-catalyzed intramolecular carbenoid insertion into an ortho CAr–H bond (Scheme 1) [21,22], which proceeded with good yields in most cases. However, with non-equivalent aryl ortho-positions in the starting diazo compounds, mixtures of regioisomers – virtually in 1:1 ratios – were always formed, which is a serious drawback of this approach. Another well-known method based on retrosynthetic disconnections at the same C–C bond employed intramolecular vicarious nucleophilic substitution of hydrogen in the substituted N-(3-nitrophenyl)chloromethylsulfonamides [23,24]. However, this method is limited to sultams bearing a strong electron-withdrawing group on the aromatic ring. A similar approach utilizes a nucleophilic aromatic substitution and leads to benzo-γ-sultams, yet this reaction requires harsh conditions and has a limited scope with only few examples [25,26]. More recently, Zard et al. have developed a sequence of lauroyl peroxide-catalyzed radical additions of xanthate to substituted N-aryl vinyl sulfonamides and subsequent intramolecular cyclization to yield benzo-annelated γ-sultams [27]. Quite interestingly, the obtained sultams were efficiently used for the preparation of ortho-functionalized anilines.
![[1860-5397-13-187-i1]](/bjoc/content/inline/1860-5397-13-187-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: A previous and a new approach to arene-annelated sultams.
Scheme 1: A previous and a new approach to arene-annelated sultams.
This contribution deals with a new access to ring-annelated sultams by an intramolecular Pd(0)-catalyzed arylation of tertiary sulfonamides bearing an additional C–H acidic center (Scheme 1). From several potentially suitable electron-withdrawing groups (EWG), e.g., alkoxycarbonyl, aryl, cyano, or trifluoromethyl, we have chosen methoxycarbonyl (CO2Me) and aryl (Ar), since the corresponding starting sulfonyl chlorides are the most easily accessible ones.
Results and Discussion
To begin with, methyl 2-[N-(2-iodophenyl)sulfamoyl]acetate (3a) was prepared from commercially available 2-iodoaniline (1a) and methyl (chlorosulfonyl)acetate (2a) according to a previously published protocol (Scheme 2) [18,19].
![[1860-5397-13-187-i2]](/bjoc/content/inline/1860-5397-13-187-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Pd-catalyzed cyclization of (2-iodophenyl)sulfonamides 3 and 5.
Scheme 2: Pd-catalyzed cyclization of (2-iodophenyl)sulfonamides 3 and 5.
The initial reaction conditions were chosen based on several reports on intermolecular arylations of “classic” weakly C–H acidic compounds under palladium catalysis [28-35]. Disappointingly, an attempted cyclization of 3a employing one of the established precatalyst cocktails – Pd(OAc)2, Ph3P, NaH in dioxane – virtually failed. The target product 4 could be detected in the reaction mixture by 1H NMR spectroscopy, but the conversion of the starting material was less than 20% after 4 days, and 4 could not be isolated in pure form (Scheme 2). A possible reason for this failure could be the presence of the N–H acidic fragment in 3a, and therefore the tertiary analogue 5 was prepared by N-alkylation of 3a with methyl iodide (Scheme 2). Unfortunately, this apparently simple transformation yielded a mixture of C-, N- and C,N-methylation products due to similar reactivities of the C- and N-nucleophilic centers in 3a. At best, the tertiary sulfonamide 5 was isolated in 32% yield. In contrast to the behavior of 3a, the tertiary analogue 5 underwent cyclization smoothly and rapidly (1 h) under the same conditions to furnish the benzannelated sultam 6 in 78% yield (Scheme 2).
Thus, in order to enable the preparation of arene-annelated tertiary sultams of type 6 along this route, an effective and versatile access to the N-alkylated precursors of type 5 had to be found. In this context, the 4-methoxybenzyl (PMB) was chosen as a possible protective group for the nitrogen atom, since it can subsequently be removed under acidic [36,37] as well as oxidative conditions [38,39]. In view of the regioselectivety problem in the alkylation of the sulfonamide 3a, the sequence of steps was reversed. Thus, 2-iodo-N-(4-methoxybenzyl)anilines 7a–g were prepared by reduction of the Schiff bases obtained from 2-iodoanilines 1a–g and 4-methoxybenzaldehyde with sodium cyanoborohydride in a mixture of glacial acetic acid and acetonitrile according to a previously published protocol (for more details see Supporting Information File 1) [40-42].
Surprisingly, the obtained 2-iodo-N-(4-methoxybenzyl)aniline (7a) turned out to be rather sluggish in its reaction with the sulfonyl chloride 2. Previously used conditions viz. carrying out the reaction in MeCN in the presence of pyridine at rt gave poor results [17-20]. Thus, in case of aniline 7a the desired sulfonamide 8a was obtained in only 7% yield. Employment of a stronger base such as triethylamine in various solvents (MeCN, DMF, CH2Cl2, THF) led to substantial decomposition of the sulfonyl chloride 2a and did not improve the yield of the target product. However, sulfonylation of 7a can be achieved with two equivalents of 2a in MeCN in the presence of pyridine at 60 °C for 2 days to give the sulfonamide 8a in an acceptable yield of 46%. Employing these conditions, methyl 2-[N-(2-iodo-4-methylphenyl)-N-(4-methoxybenzyl)sulfamoyl]acetate (8b) and methyl 2-[N-(4-chloro-2-iodophenyl)-N-(4-methoxybenzyl)sulfamoyl]acetate (8c) could be prepared in 52 and 62% yield, respectively (Scheme 3). In a recent paper Xu et al. report sulfonylation of sluggish anilines with sulfonyl chloride 2a should be performed in the presence of 2 equivalents of aniline without an additional base [21]. However, we figured out that the second equivalent of aniline could efficiently be replaced with an inexpensive weak base such as N,N-diethylaniline and under such conditions obtained the sulfonamides 8d–g in good yields (Scheme 3).
![[1860-5397-13-187-i3]](/bjoc/content/inline/1860-5397-13-187-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of 4-methoxybenzyl-protected methyl 2-(N-o-iodoarylsulfamoyl)acetates 8. Reagents and conditions A: MeO2CCH2SO2Cl (2a), Py, MeCN, 60 °C, 48 h; conditions B: MeO2CCH2SO2Cl (2a), PhNEt2, CH2Cl2, rt, 20 h; conditions C: EtO2CCH(Me)SO2Cl (2b), Py, MeCN, 60 °C, 48 h.
Scheme 3: Preparation of 4-methoxybenzyl-protected methyl 2-(N-o-iodoarylsulfamoyl)acetates 8. Reagents and c...
Next, the key step in the targeted synthesis, the intramolecular Pd-catalyzed arylation was optimized for the sulfonamide 8a (Table 1).
Table 1: Optimization of the reaction conditions for the Pd-catalyzed cyclization of the sulfonamide 8a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-187-i8.svg?max-width=637&scale=1.0)
|
||
| entry | base (equiv) | conditionsa (yield/conversion %)b |
|---|---|---|
| 1 | NaH (4.0) | 90 °C, 1 h (89/100) |
| 2 | t-BuOK (4.0) | 90 °C, 1 h (49/94) |
| 3 | MeONa (4.0) | 90 °C, 1 h (44/80) |
| 4 | K2CO3 (4.0) | 90 °C, 1 h (0/16) |
| 5c | K2CO3 (4.0) | 90 °C, 1 h (10/58) |
| 6 | NaH (2.5) | 90 °C, 1 h (48/52) |
| 7d | NaH (4.0) | 90 °C, 1 h (4/28) |
| 8 | NaH (4.0) | 90 °C, 30 min (98/100) |
| 9 | NaH (4.0) | 90 °C, 15 min (91/96) |
| 10 | NaH (4.0) | 25 °C, 1 h (0/10) |
aReactions were performed with 0.25 mmol of 8a. bYield and conversion were estimated on the basis of 1H NMR spectra (1,2-diphenylethane was used as an internal standard). cReaction performed in DMAA. d1 mol % and 4 mol % of Pd(OAc)2 and Ph3P, respectively, were used.
Initially the studied reaction was performed in dioxane at 90 °C employing 5 mol % of Pd(OAc)2, 20 mol % of Ph3P, and 4.0 equivalents of NaH. Although full conversion of 8a was achieved and the NMR-determined yield of sultam 10a was 89% (Table 1, entry 1), attempts were made to further improve the reaction conditions by testing different bases such as potassium tert-butoxide and sodium methoxide in dioxane and potassium carbonate in dioxane as well as in DMAA (Table 1, entries 2–5). Unfortunately, in all cases the yields of the target sultam 10a were poorer. Since NaH in dioxane had given the best results, various parameters of these conditions were tested. With a reduced amount of NaH (2.5 instead of 4.0 equiv) the conversion was lower and so was the yield. With a lower amount of the precatalyst (1 mol % of Pd(OAc)2 and 4 mol % of Ph3P) the sultam 10a was formed in an extremely low yield of 4% (Table 1, entry 7). Finally, when keeping the mixture at 90 °C for only 30 min instead of 1 h, a virtually quantitative transformation of 8a to the sultam 10a was achieved (Table 1, entries 8, 9). To demonstrate the scalability of this synthesis of the sultam 10a, the reaction was carried out with 7.1 g of 8a and just 2 mol % of Pd(OAc)2, to furnish an isolated yield of 4.3 g (83%) of the sultam 10a.
Under the optimized reaction conditions the whole series of sulfonamides 8b–g was converted to the corresponding sultams 10b–g in good to excellent yields (Scheme 4).
![[1860-5397-13-187-i4]](/bjoc/content/inline/1860-5397-13-187-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of arene-annelated sultams 10 by Pd-catalyzed intramolecular arylation of a C–H acidic methylene group.
Scheme 4: Synthesis of arene-annelated sultams 10 by Pd-catalyzed intramolecular arylation of a C–H acidic me...
No significant differences between the unsubstituted sulfonamide 8a, and its derivatives bearing weak electron-donating (8b) and strong electron-withdrawing (8d,e) groups in the para-position relative to the nitrogen atom were detected. However, methyl 2-(N-(5-cyano-2-iodophenyl)-N-(4-methoxybenzyl)sulfamoyl)acetate (8g) and methyl 2-(N-(2-iodo-5-methoxyphenyl)-N-(4-methoxybenzyl)sulfamoyl)acetate (8f) behaved slightly differently. Sulfonamide 8g was completely converted to the corresponding sultam 10g within 5 min (checked by TLC), whereas 8f underwent cyclization significantly more slowly than the unsubstituted analog 8a, and was fully converted after 3 h at 90 °C. When the esters 8d,e were treated with the precatalyst under the standard conditions in freshly distilled dioxane, the sultams 10d and 10e were isolated in 78 and 86% yield, respectively. However, when commercial dioxane was used, both sulfonamide 8d as well as 8e gave the same product, namely the benzoic acid derivative 10h in 65 and 71% yield, respectively. Apparently, the commercial dioxane contained a certain amount of water, so that the benzoates 10d,e underwent hydrolysis. Indeed, when the sultam 10e was treated with sodium hydride in freshly distilled dioxane followed by addition of a small amount of water, the acid 10h was isolated in an excellent yield of 93%. The relative stability of the methoxycarbonyl group at C-3 under these basic conditions must be due to its reduced electrophilicity in connection with the facile enolate formation at this position. The structures of all obtained compounds were assigned on the basis of 1H and 13C NMR spectra, as well as high resolution mass spectrometric data. The structure of the sultam 10c was unequivocally confirmed by a single-crystal X-ray diffraction study (Figure 1).
![[1860-5397-13-187-1]](/bjoc/content/figures/1860-5397-13-187-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of methyl 5-chloro-1-(4-methoxybenzyl)-1,3-dihydrobenzo[c]isothiazole-3-carboxylate-2,2-dioxide (10c) in the crystal [CCDC: 1520988].
Figure 1: Structure of methyl 5-chloro-1-(4-methoxybenzyl)-1,3-dihydrobenzo[c]isothiazole-3-carboxylate-2,2-d...
While the intramolecular arylation of the C–H acidic CH2 group in sulfamoylacetates 8a–g to yield 1,3-dihydrobenzo[c]isothiazole-2,2-dioxide derivatives 10a–h works well, the sulfonamide 9a with a tertiary C–H acidic group, which was prepared from the 2-iodoaniline derivative 7a and ethyl 2-(chlorosulfonyl)propanoate (2b) according to the protocol employed for the sulfonamides 8a–c, under the same conditions did not yield the corresponding cyclization product, but a complex and inseparable mixture.
To further test the scope and limitations of this cyclization with respect to other C–H acidifying groups (EWG) on the tether, the α-toluenesulfonamide 12 was prepared in two steps from o-iodoaniline (1a, Scheme 5). The subsequent sulfonylation of 1a with α-toluenesulfonyl chloride yielded a mixture of the sulfonamide 11 along with a side product resulting from twofold N-sulfonylation. Gratifyingly, subsequent treatment of the reaction mixture with a solution of sodium methoxide in methanol converted this side product to 11, so that the overall yield of the latter was 86%. Alkylation of 11 with 4-methoxybenzyl chloride in DMF in the presence of potassium carbonate according to a previously published protocol [17] gave the N-PMB-protected tertiary sulfonamide 12 in 89% yield. The attempted palladium-catalyzed cyclization of 12 under the conditions employed above (Scheme 4), yet at 70 °C did not give 13, but the unexpected 2,3-diarylindole 18 in 47% isolated yield (Scheme 5).
![[1860-5397-13-187-i5]](/bjoc/content/inline/1860-5397-13-187-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Palladium-catalyzed transformation of N-(2-iodophenyl)-N-(4-methoxybenzyl-benzylsulfonamide 12. Ar = 4-MeOC6H4; a) BnSO2Cl, NMM, CH2Cl2, rt, 0.5 h; b) MeONa, MeOH, reflux, 3 h; c) 4-MeOC6H4CH2Cl, K2CO3, DMF, 60 °C, 2 h; d) Pd(OAc)2, Ph3P, NaH, dioxane, 70 °C, 30 min.
Scheme 5: Palladium-catalyzed transformation of N-(2-iodophenyl)-N-(4-methoxybenzyl-benzylsulfonamide 12. Ar ...
The formation of 18 can best be rationalized assuming a ring opening of the initially formed anionic cyclization product 13 to furnish the aza-ortho-quinodimethane sulfonyl anion 14 [43]. The formation of 14 can also proceed as a cheletropic extrusion of SO2 from 13 followed by recombination of SO2 with the formed carbanionic center. Similar fragmentations of such 1,3-dihydro-2,1-benzothiazol-2,2-dioxides and related compounds are known, but usually require heating to above 200 °C to yield aza-ortho-quinodimethane derivatives [44], which can rearrange into the corresponding Schiff bases by sigmatropic [1,5]-hydrogen shift [45]. In the case of 13, the extrusion of SO2 must be drastically accelerated due to its carbanionic nature, so that it can occur at 70 °C. The resulting anionic aza-ortho-quinodimethane intermediate 14 then, by [1,5]-hydrogen shift gives the ortho-benzylaniline-derived Schiff base 15, which undergoes cyclization to give the indoline derivative 16, and this by subsequent elimination of SO2 and protonation yields 17. The latter, like many indolines [46-48] probably is susceptible to very rapid oxidation upon work-up in air to give the corresponding indole 18.
In order to test the applicability of the palladium-catalyzed intramolecular arylation of a sulfonamide with a C–H acidic group for the preparation of benzannelated six-membered sultams like 21, the appropriate precursor 20 was prepared in one step from the respective sulfonamide 19 and 2-iodobenzyl alcohol employing a Mitsunobu protocol (Scheme 6) [49].
![[1860-5397-13-187-i6]](/bjoc/content/inline/1860-5397-13-187-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Palladium-catalyzed intramolecular arylation to yield a benzannelated six-membered sultam 21. Ar = 4-MeOC6H4; a) 2-IC6H4CH2OH, Ph3P, DEAD, THF, rt, 12 h; b) Pd(OAc)2, Ph3P, NaH, dioxane, 90 °C, 20 h.
Scheme 6: Palladium-catalyzed intramolecular arylation to yield a benzannelated six-membered sultam 21. Ar = ...
Treatment of 20 with palladium acetate, Ph3P, and sodium hydride in dioxane at 90 °С for 20 h furnished the target sultam 21 in 49% yield. Apparently, the cyclization of 20 occurs significantly more slowly than those of the sulfonamides 8a–g, which is in accordance with the well-known fact that an intermediate six-membered palladacycle forms more rapidly than a seven-membered analogue in similar transformations.
In an attempt to remove the p-methoxybenzyl group (PMB) from the nitrogen, the methyl 1-(4-methoxybenzyl)-1,3-dihydrobenzo[c]isothiazole-3-carboxylate-2,2-dioxide (10a) was treated with cerium(IV) ammonium nitrate (CAN) according to a previously developed protocol [18,20]. Unexpectedly, this PMB resisted its removal, and the only formed product was the nitrooxy derivative 22, presumbaly via the corresponding benzyl-type free radical (Scheme 7). The structure of 22 was proven by single-crystal X-ray diffraction (Figure 2).
![[1860-5397-13-187-i7]](/bjoc/content/inline/1860-5397-13-187-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: An attempted and a successful removal of the PMB group from the sultam 10a.
Scheme 7: An attempted and a successful removal of the PMB group from the sultam 10a.
![[1860-5397-13-187-2]](/bjoc/content/figures/1860-5397-13-187-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Structure of methyl 1-(4-methoxybenzyl)-3-(nitrooxy)-1,3-dihydrobenzo[c]isothiazole-3-carboxylate-2,2-dioxide (22) in the crystal [CCDC: 1520989].
Figure 2: Structure of methyl 1-(4-methoxybenzyl)-3-(nitrooxy)-1,3-dihydrobenzo[c]isothiazole-3-carboxylate-2...
According to a publication by Metz et al., a N-(4-methoxybenzyl) group can be removed from sultams by treatment with trifluoroacetic acid [36]. Indeed, when 10a was treated with CF3CO2H in refluxing dichloromethane for 3 days, the deprotected sultam 23 was isolated in a good yield of 72% (Scheme 7).
Conclusion
In conclusion, the intramolecular palladium-catalyzed C-arylation of tertiary 1-(methoxycarbonyl)methanesulfonanilides can be considered as a viable approach to five- and six-membered benzannelated sultams. The 4-methoxybenzyl group is a suitable protective group for the sultam synthesis since it is stable under the reaction conditions and can be easily removed by acidolysis with trifluoroacetic acid.
Supporting Information
| Supporting Information File 1: Experimental procedure and analytical data. | ||
| Format: PDF | Size: 5.6 MB | Download |
Acknowledgements
V.A.R. gratefully acknowledges the support of the Scientific Council of the President of the Russian Federation (Grant MK-2382.2017.3). M. S. is indebted to the Studienstiftung des Deutschen Volkes for a scholarship. All physicochemical measurements were performed at the Center for Magnetic Resonance, Сenter for Chemical Analysis and Material Research, and Center for X-ray Diffraction Methods all at Saint Petersburg State University.
References
-
Abbaz, T.; Bendjeddou, A.; Gouasmia, A.; Bouchouk, D.; Boualleg, C.; Kaouachi, N.; Inguimbert, N.; Villemin, D. Lett. Org. Chem. 2014, 11, 59–63. doi:10.2174/157017861101140113162502
Return to citation in text: [1] -
Inagaki, M.; Tsuri, T.; Jyoyama, H.; Ono, T.; Yamada, K.; Kobayashi, M.; Hori, Y.; Arimura, A.; Yasui, K.; Ohno, K.; Kakudo, S.; Koizumi, K.; Suzuki, R.; Kato, M.; Kawai, S.; Matsumoto, S. J. Med. Chem. 2000, 43, 2040–2048. doi:10.1021/jm9906015
Return to citation in text: [1] -
Kolb, R.; Bach, N. C.; Sieber, S. A. Chem. Commun. 2014, 50, 427–429. doi:10.1039/C3CC46002A
Return to citation in text: [1] -
Kim, C.-Y.; Whittington, D. A.; Chang, J. S.; Liao, J.; May, J. A.; Christianson, D. W. J. Med. Chem. 2002, 45, 888–893. doi:10.1021/jm010163d
Return to citation in text: [1] -
Ali, A.; Reddy, G. S. K. K.; Cao, H.; Anjum, S. G.; Nalam, M. N. L.; Schiffer, C. A.; Rana, T. M. J. Med. Chem. 2006, 49, 7342–7356. doi:10.1021/jm060666p
Return to citation in text: [1] -
Smits, R. A.; Adami, M.; Istyastono, E. P.; Zuiderveld, O. P.; van Dam, C. M. E.; de Kanter, F. J. J.; Jongejan, A.; Coruzzi, G.; Leurs, R.; de Esch, I. J. P. J. Med. Chem. 2010, 53, 2390–2400. doi:10.1021/jm901379s
Return to citation in text: [1] -
Cherney, R. J.; Mo, R.; Meyer, D. T.; Hardman, K. D.; Liu, R.-Q.; Covington, M. B.; Qian, M.; Wasserman, Z. R.; Christ, D. D.; Trzaskos, J. M.; Newton, R. C.; Decicco, C. P. J. Med. Chem. 2004, 47, 2981–2983. doi:10.1021/jm049833g
Return to citation in text: [1] -
Wales, J. K.; Krees, S. V.; Grant, A. M.; Viktora, J. K.; Wolff, F. W. J. Pharmacol. Exp. Ther. 1968, 164, 421–432.
Return to citation in text: [1] -
Wells, G. J.; Tao, M.; Josef, K. A.; Bihovsky, R. J. Med. Chem. 2001, 44, 3488–3503. doi:10.1021/jm010178b
Return to citation in text: [1] -
Wang, Y.; Busch-Petersen, J.; Wang, F.; Ma, L.; Fu, W.; Kerns, J. K.; Jin, J.; Palovich, M. R.; Shen, J.-K.; Burman, M.; Foley, J. J.; Schmidt, D. B.; Hunsberger, G. E.; Sarau, H. M.; Widdowson, K. L. Bioorg. Med. Chem. Lett. 2007, 17, 3864–3867. doi:10.1016/j.bmcl.2007.05.011
Return to citation in text: [1] -
Combrink, K. D.; Gulgeze, H. B.; Thuring, J. W.; Yu, K.-L.; Civiello, R. L.; Zhang, Y.; Pearce, B. C.; Yin, Z.; Langley, D. R.; Kadow, K. F.; Cianci, C. W.; Li, Z.; Clarke, J.; Genovesi, E. V.; Medina, I.; Lamb, L.; Yang, Z.; Zadjura, L.; Krystal, M.; Meanwell, N. A. Bioorg. Med. Chem. Lett. 2007, 17, 4784–4790. doi:10.1016/j.bmcl.2007.06.065
Return to citation in text: [1] -
Cherney, R. J.; Duan, J. J.-W.; Voss, M. E.; Chen, L.; Wang, L.; Meyer, D. T.; Wasserman, Z. R.; Hardman, K. D.; Liu, R.-Q.; Covington, M. B.; Qian, M.; Mandlekar, S.; Christ, D. D.; Trzaskos, J. M.; Newton, R. C.; Magolda, R. L.; Wexler, R. R.; Decicco, C. P. J. Med. Chem. 2003, 46, 1811–1823. doi:10.1021/jm020475w
Return to citation in text: [1] -
Giannotti, D.; Viti, G.; Sbraci, P.; Pestellini, V.; Volterra, G.; Borsini, F.; Lecci, A.; Meli, A.; Dapporto, P.; Paoli, P. J. Med. Chem. 1991, 34, 1356–1362. doi:10.1021/jm00108a018
Return to citation in text: [1] -
Rassadin, V. A.; Grosheva, D. S.; Tomashevskii, A. A.; Sokolov, V. V. Chem. Heterocycl. Compd. 2013, 49, 39–65. doi:10.1007/s10593-013-1231-3
Return to citation in text: [1] -
Majumdar, K. C.; Mondal, S. Chem. Rev. 2011, 111, 7749–7773. doi:10.1021/cr1003776
Return to citation in text: [1] -
Szostak, M.; Aubé, J. Chem. Rev. 2013, 113, 5701–5765. doi:10.1021/cr4000144
Return to citation in text: [1] -
Rassadin, V. A.; Grosheva, D. S.; Arefeva, I. A.; Tomashevskiy, A. A.; Sokolov, V. V.; de Meijere, A. Eur. J. Org. Chem. 2012, 5028–5037. doi:10.1002/ejoc.201200670
Return to citation in text: [1] [2] [3] -
Rassadin, V. A.; Tomashevskiy, A. A.; Sokolov, V. V.; Ringe, A.; Magull, J.; de Meijere, A. Eur. J. Org. Chem. 2009, 2635–2641. doi:10.1002/ejoc.200900113
Return to citation in text: [1] [2] [3] [4] -
Rassadin, V. A.; Tomashevskii, A. A.; Sokolov, V. V.; Potekhin, A. A. Chem. Heterocycl. Compd. 2008, 44, 474–485. doi:10.1007/s10593-008-0066-9
Return to citation in text: [1] [2] [3] -
Rassadin, V. A.; Grosheva, D. S.; Tomashevskiy, A. A.; Sokolov, V. V.; Yufit, D. S.; Kozhushkov, S. I.; de Meijere, A. Eur. J. Org. Chem. 2010, 3481–3486. doi:10.1002/ejoc.201000345
Return to citation in text: [1] [2] [3] [4] -
Yang, Z.; Xu, J. Chem. Commun. 2014, 50, 3616–3618. doi:10.1039/C4CC00250D
Return to citation in text: [1] [2] -
Huang, P.; Yang, Z.; Xu, J. Tetrahedron 2017, 73, 3255–3265. doi:10.1016/j.tet.2017.04.056
Return to citation in text: [1] -
Makosza, M.; Wojciechowski, K. Tetrahedron Lett. 1984, 25, 4791–4792. doi:10.1016/S0040-4039(01)81520-3
Return to citation in text: [1] -
Wojciechowski, K.; Makosza, M. Synthesis 1992, 571–576. doi:10.1055/s-1992-34113
Return to citation in text: [1] -
Skorcz, J. A.; Suh, J. T.; Germershausen, R. L. J. Heterocycl. Chem. 1974, 11, 73–75. doi:10.1002/jhet.5570110116
Return to citation in text: [1] -
Kosinski, S.; Wojciechowski, K. Eur. J. Org. Chem. 2000, 1263–1270. doi:10.1002/1099-0690(200004)2000:7<1263::AID-EJOC1263>3.0.CO;2-E
Return to citation in text: [1] -
Moutrille, C.; Zard, S. Z. Tetrahedron Lett. 2004, 45, 4631–4634. doi:10.1016/j.tetlet.2004.04.111
Return to citation in text: [1] -
Grimm, J. B.; Katcher, M. H.; Witter, D. J.; Northrup, A. B. J. Org. Chem. 2007, 72, 8135–8138. doi:10.1021/jo701431j
Return to citation in text: [1] -
Zhou, G.; Ting, P. C.; Aslanian, R. G. Tetrahedron Lett. 2010, 51, 939–941. doi:10.1016/j.tetlet.2009.12.035
Return to citation in text: [1] -
Kashin, A. N.; Mitin, A. V.; Beletskaya, I. P.; Wife, R. Tetrahedron Lett. 2002, 43, 2539–2542. doi:10.1016/S0040-4039(02)00321-0
Return to citation in text: [1] -
Uno, M.; Seto, K.; Takahashi, S. J. Chem. Soc., Chem. Commun. 1984, 932–933. doi:10.1039/c39840000932
Return to citation in text: [1] -
Uno, M.; Seto, K.; Ueda, W.; Masuda, M.; Takahashi, S. Synthesis 1985, 506–508. doi:10.1055/s-1985-31250
Return to citation in text: [1] -
Sakamoto, T.; Kayoh, E.; Kondo, Y.; Yamanaka, H. Chem. Pharm. Bull. 1988, 36, 1664–1668. doi:10.1248/cpb.36.1664
Return to citation in text: [1] -
Sakamoto, T.; Kayoh, E.; Kondo, Y.; Yamanaka, H. Chem. Pharm. Bull. 1990, 38, 1513–1517. doi:10.1248/cpb.38.1513
Return to citation in text: [1] -
Jiao, Z.; Chee, K. W.; Zhou, J. J. Am. Chem. Soc. 2016, 138, 16240–16243. doi:10.1021/jacs.6b11610
Return to citation in text: [1] -
Rogachev, V. O.; Metz, P. ARKIVOC 2007, No. 5, 167–190.
Return to citation in text: [1] [2] -
Semple, J. E.; Wang, P. C.; Lysenko, Z.; Joullie, M. M. J. Am. Chem. Soc. 1980, 102, 7505–7510. doi:10.1021/ja00545a018
Return to citation in text: [1] -
Yamaura, M.; Suzuki, T.; Hashimoto, H.; Yoshimura, J.; Okamoto, T.; Shin, C.-g. Bull. Chem. Soc. Jpn. 1985, 58, 1413–1420. doi:10.1246/bcsj.58.1413
Return to citation in text: [1] -
Yoshimura, J.; Yamaura, M.; Suzuki, T.; Hashimoto, H. Chem. Lett. 1983, 1001–1002. doi:10.1246/cl.1983.1001
Return to citation in text: [1] -
Roman, D. S.; Takahashi, Y.; Charette, A. B. Org. Lett. 2011, 13, 3242–3245. doi:10.1021/ol201160s
Return to citation in text: [1] -
Zhang, H.; Hay, E. B.; Geib, S. J.; Curran, D. P. Beilstein J. Org. Chem. 2015, 11, 1649–1655. doi:10.3762/bjoc.11.181
Return to citation in text: [1] -
Grosheva, D. S.; Rassadin, V. A.; Sokolov, V. V. Eur. J. Org. Chem. 2015, 1355–1363. doi:10.1002/ejoc.201403416
Return to citation in text: [1] -
Mitin, A. V.; Kashin, A. N.; Beletskaya, I. P. J. Organomet. Chem. 2004, 689, 1085–1090. doi:10.1016/j.jorganchem.2003.12.039
Return to citation in text: [1] -
Wojciechowski, K. Eur. J. Org. Chem. 2001, 3587–3605. doi:10.1002/1099-0690(200110)2001:19<3587::AID-EJOC3587>3.0.CO;2-5
Return to citation in text: [1] -
Wojciechowski, K. Tetrahedron 1993, 49, 10017–10026. doi:10.1016/S0040-4020(01)80198-8
Return to citation in text: [1] -
Padwa, A.; Kappe, C. O.; Cochran, J. E.; Snyder, J. P. J. Org. Chem. 1997, 62, 2786–2797. doi:10.1021/jo962358c
Return to citation in text: [1] -
Samet, A. V.; Zakharov, E. P.; Semenov, V. V.; Buchanan, A. C.; Gakh, A. A. Synth. Commun. 2001, 31, 1441–1445. doi:10.1081/SCC-100104054
Return to citation in text: [1] -
Kappe, C. O.; Cochran, J. E.; Padwa, A. Tetrahedron Lett. 1995, 36, 9285–9288. doi:10.1016/0040-4039(95)02013-F
Return to citation in text: [1] -
Mitsunobu, O.; Yamada, M. Bull. Chem. Soc. Jpn. 1967, 40, 2380–2382. doi:10.1246/bcsj.40.2380
Return to citation in text: [1]
| 44. | Wojciechowski, K. Eur. J. Org. Chem. 2001, 3587–3605. doi:10.1002/1099-0690(200110)2001:19<3587::AID-EJOC3587>3.0.CO;2-5 |
| 17. | Rassadin, V. A.; Grosheva, D. S.; Arefeva, I. A.; Tomashevskiy, A. A.; Sokolov, V. V.; de Meijere, A. Eur. J. Org. Chem. 2012, 5028–5037. doi:10.1002/ejoc.201200670 |
| 43. | Mitin, A. V.; Kashin, A. N.; Beletskaya, I. P. J. Organomet. Chem. 2004, 689, 1085–1090. doi:10.1016/j.jorganchem.2003.12.039 |
| 1. | Abbaz, T.; Bendjeddou, A.; Gouasmia, A.; Bouchouk, D.; Boualleg, C.; Kaouachi, N.; Inguimbert, N.; Villemin, D. Lett. Org. Chem. 2014, 11, 59–63. doi:10.2174/157017861101140113162502 |
| 2. | Inagaki, M.; Tsuri, T.; Jyoyama, H.; Ono, T.; Yamada, K.; Kobayashi, M.; Hori, Y.; Arimura, A.; Yasui, K.; Ohno, K.; Kakudo, S.; Koizumi, K.; Suzuki, R.; Kato, M.; Kawai, S.; Matsumoto, S. J. Med. Chem. 2000, 43, 2040–2048. doi:10.1021/jm9906015 |
| 3. | Kolb, R.; Bach, N. C.; Sieber, S. A. Chem. Commun. 2014, 50, 427–429. doi:10.1039/C3CC46002A |
| 4. | Kim, C.-Y.; Whittington, D. A.; Chang, J. S.; Liao, J.; May, J. A.; Christianson, D. W. J. Med. Chem. 2002, 45, 888–893. doi:10.1021/jm010163d |
| 5. | Ali, A.; Reddy, G. S. K. K.; Cao, H.; Anjum, S. G.; Nalam, M. N. L.; Schiffer, C. A.; Rana, T. M. J. Med. Chem. 2006, 49, 7342–7356. doi:10.1021/jm060666p |
| 6. | Smits, R. A.; Adami, M.; Istyastono, E. P.; Zuiderveld, O. P.; van Dam, C. M. E.; de Kanter, F. J. J.; Jongejan, A.; Coruzzi, G.; Leurs, R.; de Esch, I. J. P. J. Med. Chem. 2010, 53, 2390–2400. doi:10.1021/jm901379s |
| 7. | Cherney, R. J.; Mo, R.; Meyer, D. T.; Hardman, K. D.; Liu, R.-Q.; Covington, M. B.; Qian, M.; Wasserman, Z. R.; Christ, D. D.; Trzaskos, J. M.; Newton, R. C.; Decicco, C. P. J. Med. Chem. 2004, 47, 2981–2983. doi:10.1021/jm049833g |
| 20. | Rassadin, V. A.; Grosheva, D. S.; Tomashevskiy, A. A.; Sokolov, V. V.; Yufit, D. S.; Kozhushkov, S. I.; de Meijere, A. Eur. J. Org. Chem. 2010, 3481–3486. doi:10.1002/ejoc.201000345 |
| 17. | Rassadin, V. A.; Grosheva, D. S.; Arefeva, I. A.; Tomashevskiy, A. A.; Sokolov, V. V.; de Meijere, A. Eur. J. Org. Chem. 2012, 5028–5037. doi:10.1002/ejoc.201200670 |
| 18. | Rassadin, V. A.; Tomashevskiy, A. A.; Sokolov, V. V.; Ringe, A.; Magull, J.; de Meijere, A. Eur. J. Org. Chem. 2009, 2635–2641. doi:10.1002/ejoc.200900113 |
| 19. | Rassadin, V. A.; Tomashevskii, A. A.; Sokolov, V. V.; Potekhin, A. A. Chem. Heterocycl. Compd. 2008, 44, 474–485. doi:10.1007/s10593-008-0066-9 |
| 20. | Rassadin, V. A.; Grosheva, D. S.; Tomashevskiy, A. A.; Sokolov, V. V.; Yufit, D. S.; Kozhushkov, S. I.; de Meijere, A. Eur. J. Org. Chem. 2010, 3481–3486. doi:10.1002/ejoc.201000345 |
| 17. | Rassadin, V. A.; Grosheva, D. S.; Arefeva, I. A.; Tomashevskiy, A. A.; Sokolov, V. V.; de Meijere, A. Eur. J. Org. Chem. 2012, 5028–5037. doi:10.1002/ejoc.201200670 |
| 18. | Rassadin, V. A.; Tomashevskiy, A. A.; Sokolov, V. V.; Ringe, A.; Magull, J.; de Meijere, A. Eur. J. Org. Chem. 2009, 2635–2641. doi:10.1002/ejoc.200900113 |
| 19. | Rassadin, V. A.; Tomashevskii, A. A.; Sokolov, V. V.; Potekhin, A. A. Chem. Heterocycl. Compd. 2008, 44, 474–485. doi:10.1007/s10593-008-0066-9 |
| 20. | Rassadin, V. A.; Grosheva, D. S.; Tomashevskiy, A. A.; Sokolov, V. V.; Yufit, D. S.; Kozhushkov, S. I.; de Meijere, A. Eur. J. Org. Chem. 2010, 3481–3486. doi:10.1002/ejoc.201000345 |
| 14. | Rassadin, V. A.; Grosheva, D. S.; Tomashevskii, A. A.; Sokolov, V. V. Chem. Heterocycl. Compd. 2013, 49, 39–65. doi:10.1007/s10593-013-1231-3 |
| 15. | Majumdar, K. C.; Mondal, S. Chem. Rev. 2011, 111, 7749–7773. doi:10.1021/cr1003776 |
| 16. | Szostak, M.; Aubé, J. Chem. Rev. 2013, 113, 5701–5765. doi:10.1021/cr4000144 |
| 38. | Yamaura, M.; Suzuki, T.; Hashimoto, H.; Yoshimura, J.; Okamoto, T.; Shin, C.-g. Bull. Chem. Soc. Jpn. 1985, 58, 1413–1420. doi:10.1246/bcsj.58.1413 |
| 39. | Yoshimura, J.; Yamaura, M.; Suzuki, T.; Hashimoto, H. Chem. Lett. 1983, 1001–1002. doi:10.1246/cl.1983.1001 |
| 8. | Wales, J. K.; Krees, S. V.; Grant, A. M.; Viktora, J. K.; Wolff, F. W. J. Pharmacol. Exp. Ther. 1968, 164, 421–432. |
| 9. | Wells, G. J.; Tao, M.; Josef, K. A.; Bihovsky, R. J. Med. Chem. 2001, 44, 3488–3503. doi:10.1021/jm010178b |
| 10. | Wang, Y.; Busch-Petersen, J.; Wang, F.; Ma, L.; Fu, W.; Kerns, J. K.; Jin, J.; Palovich, M. R.; Shen, J.-K.; Burman, M.; Foley, J. J.; Schmidt, D. B.; Hunsberger, G. E.; Sarau, H. M.; Widdowson, K. L. Bioorg. Med. Chem. Lett. 2007, 17, 3864–3867. doi:10.1016/j.bmcl.2007.05.011 |
| 11. | Combrink, K. D.; Gulgeze, H. B.; Thuring, J. W.; Yu, K.-L.; Civiello, R. L.; Zhang, Y.; Pearce, B. C.; Yin, Z.; Langley, D. R.; Kadow, K. F.; Cianci, C. W.; Li, Z.; Clarke, J.; Genovesi, E. V.; Medina, I.; Lamb, L.; Yang, Z.; Zadjura, L.; Krystal, M.; Meanwell, N. A. Bioorg. Med. Chem. Lett. 2007, 17, 4784–4790. doi:10.1016/j.bmcl.2007.06.065 |
| 12. | Cherney, R. J.; Duan, J. J.-W.; Voss, M. E.; Chen, L.; Wang, L.; Meyer, D. T.; Wasserman, Z. R.; Hardman, K. D.; Liu, R.-Q.; Covington, M. B.; Qian, M.; Mandlekar, S.; Christ, D. D.; Trzaskos, J. M.; Newton, R. C.; Magolda, R. L.; Wexler, R. R.; Decicco, C. P. J. Med. Chem. 2003, 46, 1811–1823. doi:10.1021/jm020475w |
| 13. | Giannotti, D.; Viti, G.; Sbraci, P.; Pestellini, V.; Volterra, G.; Borsini, F.; Lecci, A.; Meli, A.; Dapporto, P.; Paoli, P. J. Med. Chem. 1991, 34, 1356–1362. doi:10.1021/jm00108a018 |
| 40. | Roman, D. S.; Takahashi, Y.; Charette, A. B. Org. Lett. 2011, 13, 3242–3245. doi:10.1021/ol201160s |
| 41. | Zhang, H.; Hay, E. B.; Geib, S. J.; Curran, D. P. Beilstein J. Org. Chem. 2015, 11, 1649–1655. doi:10.3762/bjoc.11.181 |
| 42. | Grosheva, D. S.; Rassadin, V. A.; Sokolov, V. V. Eur. J. Org. Chem. 2015, 1355–1363. doi:10.1002/ejoc.201403416 |
| 27. | Moutrille, C.; Zard, S. Z. Tetrahedron Lett. 2004, 45, 4631–4634. doi:10.1016/j.tetlet.2004.04.111 |
| 28. | Grimm, J. B.; Katcher, M. H.; Witter, D. J.; Northrup, A. B. J. Org. Chem. 2007, 72, 8135–8138. doi:10.1021/jo701431j |
| 29. | Zhou, G.; Ting, P. C.; Aslanian, R. G. Tetrahedron Lett. 2010, 51, 939–941. doi:10.1016/j.tetlet.2009.12.035 |
| 30. | Kashin, A. N.; Mitin, A. V.; Beletskaya, I. P.; Wife, R. Tetrahedron Lett. 2002, 43, 2539–2542. doi:10.1016/S0040-4039(02)00321-0 |
| 31. | Uno, M.; Seto, K.; Takahashi, S. J. Chem. Soc., Chem. Commun. 1984, 932–933. doi:10.1039/c39840000932 |
| 32. | Uno, M.; Seto, K.; Ueda, W.; Masuda, M.; Takahashi, S. Synthesis 1985, 506–508. doi:10.1055/s-1985-31250 |
| 33. | Sakamoto, T.; Kayoh, E.; Kondo, Y.; Yamanaka, H. Chem. Pharm. Bull. 1988, 36, 1664–1668. doi:10.1248/cpb.36.1664 |
| 34. | Sakamoto, T.; Kayoh, E.; Kondo, Y.; Yamanaka, H. Chem. Pharm. Bull. 1990, 38, 1513–1517. doi:10.1248/cpb.38.1513 |
| 35. | Jiao, Z.; Chee, K. W.; Zhou, J. J. Am. Chem. Soc. 2016, 138, 16240–16243. doi:10.1021/jacs.6b11610 |
| 49. | Mitsunobu, O.; Yamada, M. Bull. Chem. Soc. Jpn. 1967, 40, 2380–2382. doi:10.1246/bcsj.40.2380 |
| 25. | Skorcz, J. A.; Suh, J. T.; Germershausen, R. L. J. Heterocycl. Chem. 1974, 11, 73–75. doi:10.1002/jhet.5570110116 |
| 26. | Kosinski, S.; Wojciechowski, K. Eur. J. Org. Chem. 2000, 1263–1270. doi:10.1002/1099-0690(200004)2000:7<1263::AID-EJOC1263>3.0.CO;2-E |
| 36. | Rogachev, V. O.; Metz, P. ARKIVOC 2007, No. 5, 167–190. |
| 37. | Semple, J. E.; Wang, P. C.; Lysenko, Z.; Joullie, M. M. J. Am. Chem. Soc. 1980, 102, 7505–7510. doi:10.1021/ja00545a018 |
| 18. | Rassadin, V. A.; Tomashevskiy, A. A.; Sokolov, V. V.; Ringe, A.; Magull, J.; de Meijere, A. Eur. J. Org. Chem. 2009, 2635–2641. doi:10.1002/ejoc.200900113 |
| 20. | Rassadin, V. A.; Grosheva, D. S.; Tomashevskiy, A. A.; Sokolov, V. V.; Yufit, D. S.; Kozhushkov, S. I.; de Meijere, A. Eur. J. Org. Chem. 2010, 3481–3486. doi:10.1002/ejoc.201000345 |
| 23. | Makosza, M.; Wojciechowski, K. Tetrahedron Lett. 1984, 25, 4791–4792. doi:10.1016/S0040-4039(01)81520-3 |
| 24. | Wojciechowski, K.; Makosza, M. Synthesis 1992, 571–576. doi:10.1055/s-1992-34113 |
| 45. | Wojciechowski, K. Tetrahedron 1993, 49, 10017–10026. doi:10.1016/S0040-4020(01)80198-8 |
| 21. | Yang, Z.; Xu, J. Chem. Commun. 2014, 50, 3616–3618. doi:10.1039/C4CC00250D |
| 22. | Huang, P.; Yang, Z.; Xu, J. Tetrahedron 2017, 73, 3255–3265. doi:10.1016/j.tet.2017.04.056 |
| 18. | Rassadin, V. A.; Tomashevskiy, A. A.; Sokolov, V. V.; Ringe, A.; Magull, J.; de Meijere, A. Eur. J. Org. Chem. 2009, 2635–2641. doi:10.1002/ejoc.200900113 |
| 19. | Rassadin, V. A.; Tomashevskii, A. A.; Sokolov, V. V.; Potekhin, A. A. Chem. Heterocycl. Compd. 2008, 44, 474–485. doi:10.1007/s10593-008-0066-9 |
| 46. | Padwa, A.; Kappe, C. O.; Cochran, J. E.; Snyder, J. P. J. Org. Chem. 1997, 62, 2786–2797. doi:10.1021/jo962358c |
| 47. | Samet, A. V.; Zakharov, E. P.; Semenov, V. V.; Buchanan, A. C.; Gakh, A. A. Synth. Commun. 2001, 31, 1441–1445. doi:10.1081/SCC-100104054 |
| 48. | Kappe, C. O.; Cochran, J. E.; Padwa, A. Tetrahedron Lett. 1995, 36, 9285–9288. doi:10.1016/0040-4039(95)02013-F |
© 2017 Rassadin et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)