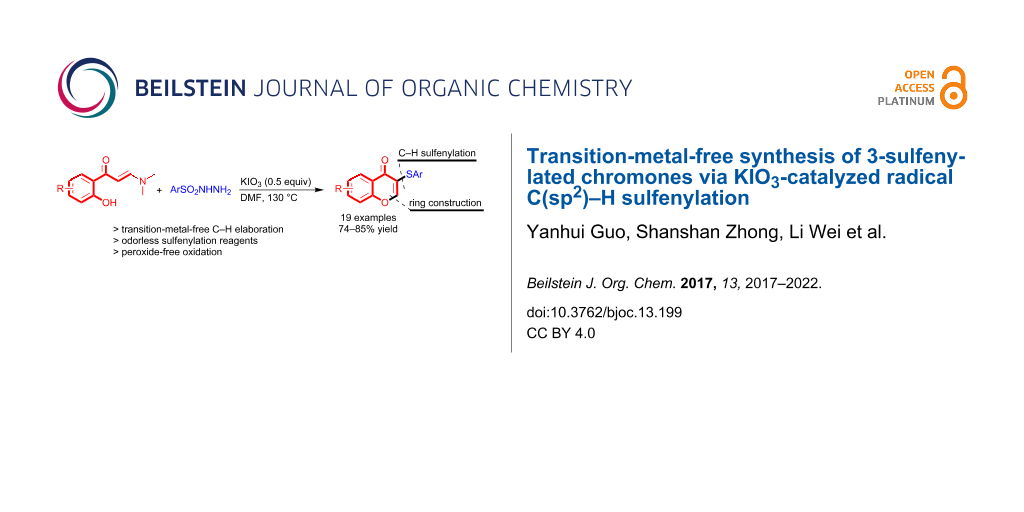
Abstract
The reactions between o-hydroxylphenyl-functionalized enaminones and sulfonyl hydrazines providing 3-sulfenylated chromones via domino chromone ring construction and C(sp2)–H bond sulfenylation have been achieved under transition-metal-free conditions by using KIO3 as the only catalyst.

Graphical Abstract
Introduction
The C–S bond-forming reactions occupy a significant position in organic synthesis as they are the major route to install sulfur fragments to organic compounds. As prevalent substructures in many natural products and biologically relevant organic molecules, the C(sp2)–S bonds such as the C–S(sulfenyl) bond are known to play crucial roles in determining the properties and biological functions of sulfur-containing compounds [1-3]. Therefore, research works in developing efficient and flexible methodologies to generate C(sp2)–S bonds have attracted sustainable interest throughout the advances of modern organic synthesis. Presently, the most popular approaches in constructing a C(sp2)–S bond are the transition-metal-catalyzed Ullmann C–S coupling reaction [4-8], Chan–Lam cross-coupling reaction [9-12] as well as the transition-metal-catalyzed C–H bond activation [13-15].
Recently, as a new trend, the transition-metal-free oxidative coupling has emerged as a powerful complementary tactic in C(sp2)–S bond forming reactions [16-27]. As a beneficial and sustainable approach, such transition-metal-free cross coupling transformations have been successfully employed in the synthesis of many useful organic compounds elaborated with sulfenyl groups. For example, the 3-sulfenylated chromones, a class of useful organic molecules with important biological profiles [28-31] and extensive application in synthetic chemistry [32-37], have been readily synthesized with several different transition-metal-free C–H cross-coupling approaches. Zhou and co-workers reported the NH4I-promoted synthesis of 3-sulfenylated chromones via the direct chromone C–H sulfenylation by using different sulfur sources (A, Scheme 1) [38,39]. Recently, our continuous efforts in exploring enaminone C(sp2)–H bond sulfenylation [40,41] reactions have led us to establish the synthesis of 3-sulfenylated chromones via KIO3-catalyzed tandem reactions of o-hydroxylphenylenaminone and thiophenols via tandem C–H sulfenylation and intramolecular C–N bond oxygenation (B, Scheme 1) [42]. Subsequently, Braga et al. reported a similar catalytic 3-sulfenylchromone synthesis by employing disfulfides as the sulfenyl sources (B, Scheme 1) [43]. Although the efficiency and application scope of the known routes are fine, the cost and limited variation on chromone substrates and/or the utility of the odorous thiophenols remain as restrictions. In this regard, devising alternative synthetic methods featuring simultaneously the advantages of easily variable, low-cost substrates and operationally practical sulfur sources is highly demanding. Herein, we report a new synthetic protocol toward these compounds through the tandem reactions between o-hydroxyphenylenaminones and sulfonyl hydrazines. In this method, the construction of the target products is furnished via the key C–H sulfenylation without using any transition metal catalyst or oxidative additive.
![[1860-5397-13-199-i1]](/bjoc/content/inline/1860-5397-13-199-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Methods on the synthesis of 3-sulfenylchromones.
Scheme 1: Methods on the synthesis of 3-sulfenylchromones.
Results and Discussion
Initially, the reaction of enaminone 1a and sulfonyl hydrazine 2a was tentatively subjected to different iodine reagents or catalyst-free conditions. The results indicated that no product was observed in the reaction without catalyst (entry 1, Table 1), and different types of iodine reagents such as KI, I2 or KIO3 could all catalyze the domino reaction to provide product 3a, wherein KIO3 displayed the highest catalytic activity (entries 2–4, Table 1). On the other hand, in the reactions performed in different media, including DMSO, ethyl lactate (EL), EtOH, MeCN, 1,4-dioxane and toluene, DMF was found as the most proper medium (entries 4–10, Table 1). Notably, increasing the reaction temperature to 130 °C promoted the production of 3a with evidently higher yields (entries 11–13, Table 1). While varying the loading of KIO3 catalyst did not led to better results (entries 14 and 15, Table 1), prolonging the reaction time to 24 h was found to be capable of further improving the yield of 3a (entries 16 and 17, Table 1).
Table 1: Optimization of the reaction conditions for the synthesis of sulfenylated chromonesa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-199-i5.svg?max-width=637&scale=1.0)
|
||||
| Entry | Catalyst | Solvent | T (°C) | Yield (%)b |
|---|---|---|---|---|
| 1 | no | DMF | 100 | nr |
| 2 | KI | DMF | 100 | 45 |
| 3 | I2 | DMF | 100 | 56 |
| 4 | KIO3 | DMF | 100 | 69 |
| 5 | KIO3 | DMSO | 100 | 61 |
| 6 | KIO3 | EL | 100 | 60 |
| 7 | KIO3 | EtOH | reflux | 56 |
| 8 | KIO3 | MeCN | reflux | 55 |
| 9 | KIO3 | 1,4-dioxane | 100 | 57 |
| 10 | KIO3 | toluene | 100 | 62 |
| 11 | KIO3 | DMF | 120 | 76 |
| 12 | KIO3 | DMF | 130 | 79 |
| 13 | KIO3 | DMF | 140 | 78 |
| 14c | KIO3 | DMF | 130 | 78 |
| 15d | KIO3 | DMF | 130 | 75 |
| 16e | KIO3 | DMF | 130 | 84 |
| 17f | KIO3 | DMF | 130 | 82 |
aGeneral conditions: 1a (0.3 mmol), 2a (0.36 mmol), catalyst (0.15 mmol) in 2 mL solvent, stirred for 12 h (nr = no reaction). bYield of isolated product based on 1a. cThe loading of KIO3 = 0.3 mmol. dThe loading of KIO3 = 0.09 mmol. eThe reaction time was 24 h. fThe reaction time was 30 h.
To examine the scope of the reaction, enaminones 1 containing different functional groups as well as various sulfonyl hydrazines 2 were subjected to the optimized standard reaction conditions. As shown in Scheme 2, functional groups with different properties, including electron-withdrawing and electron-donating ones at the phenyl ring of the enaminones and the sulfonyl hydrazines exhibited tolerance to the reaction conditions. The 3-sulfenylated products were obtained in generally good to excellent yields. A strong electron-withdrawing group such as a nitro group at the phenyl ring of the enaminone was found to have a negative influence on the product yields (3d and 3l, Scheme 2). On the other hand, the substituent on the aryl fragment of 2 also influenced the product yields. The main tendency was that a electron-withdrawing group on the aryl ring of the sulfonyl hydrazines and the group with ortho-steric hindrance gave the corresponding products with slightly lower yields than equivalent reactions using electron-donating-group-functionalized arylsulfonyl hydrazines (3p, 3q and 3s, Scheme 2).
![[1860-5397-13-199-i2]](/bjoc/content/inline/1860-5397-13-199-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of the 3-sulfenylated chromone synthesis. General conditions: 1 (0.3 mmol), 2 (0.36 mmol), KIO3 (0.15 mmol) in 2 mL DMF, stirred at 130 °C for 24 h; yield of isolated product based on 1 are reported.
Scheme 2: Scope of the 3-sulfenylated chromone synthesis. General conditions: 1 (0.3 mmol), 2 (0.36 mmol), KIO...
On the basis of the results in hand, some control experiments were also designed to probe the possible reaction mechanism. As outlined in Scheme 3, directly using enaminone 1b under standard conditions in the synthesis of products 3 was found to provide chromone 5 with high yield (reaction 1, Scheme 3). In addition, sulfonyl hydrazine 2b gave disulfide 6 under the same conditions (reaction 2, Scheme 3), suggesting that chromone 5 and disulfide 6 might be key intermediates in the domino reactions. Moreover, the same reaction in the presence of TEMPO gave no formation of 6, indicating that a free radical intermediate has also occurred in the generation of 6 (reaction 3, Scheme 3). The reactions of 5 with 6 and 2b were both found to yield the sulfenylated chromone 3i in excellent yield, respectively (reactions 4 and 5, Scheme 3). On the other hand, the reaction of 5 and 6 in the presence of TEMPO, however, provides only with trace amounts of 3i, supporting that products 3 are yielded via a free radical route (reaction 6, Scheme 3). In addition, the control reaction of 1b and 2b afforded also only trace amounts of product 3i in the presence of 1 equiv TEMPO (reaction 7, Scheme 3), further confirming that a radical intermediate was generated during the reaction process.
![[1860-5397-13-199-i3]](/bjoc/content/inline/1860-5397-13-199-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
According to the results obtained in the control experiments, the mechanism for this domino reaction is proposed (Scheme 4). As one of the key steps, the reductive coupling of two sulfonyl hydrazine molecules may first take place via the promotion of IO3− to afford intermediate 7, and the IO4− generated therein returns to IO3− in the presence of the simultaneously generated hydrogen in this step. According to a known route [44,45], 7 can be further reduced to yield disulfide 6. The disulfide is capable of either coupling chromone 5 to generate intermediate 10 by radical chain propagation (path a), or transforming into sulfur radical 8 [46,47] which can be captured by the in situ generated chromone 5 to yield radical intermediate 9 via reversible addition to the double bond. The repeated coupling of 9 with radical 8 may also provide dithiolated intermediate 10 (path b). And the elimination of the α-hydrogen and β-sulfur gives target product 3i and thiophenol 11. The thiophenol could be easily reoxidized into disulfide 6 even under air atmosphere [48,49], which enables the recycled production of sulfenylated chromone. The present method involving the free radical pathway is different from our previous work on synthesizing identical products via an ionic-based mechanism.
![[1860-5397-13-199-i4]](/bjoc/content/inline/1860-5397-13-199-i4.png?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: The proposed reaction mechanism.
Scheme 4: The proposed reaction mechanism.
Conclusion
In conclusion, starting from o-hydroxyphenyl-functionalized enaminones and sulfonyl hydrazines, we have successfully developed a flexible and facile method for the synthesis of 3-sulfenylated chromones via a transition-metal-free reaction involving a C(sp2)–H bond sulfenylation. Besides the notable feature of transition-metal-free operation, the present method also displays advantages in using odourless sulfur sources, free of any strong oxidants and neutral reaction conditions.
Supporting Information
| Supporting Information File 1: General experimental information, experimental details on the synthesis of products 3; full characterization data as well as 1H/13C NMR spectra of all products. | ||
| Format: PDF | Size: 2.3 MB | Download |
References
-
Damani, L. A., Ed. Sulphur-Containing Drugs and Related Organic Compounds; Wiley: New York, 1989.
Return to citation in text: [1] -
Lu, Q.; Zhang, J.; Wei, F.; Qi, Y.; Wang, H.; Liu, Z.; Lei, A. Angew. Chem., Int. Ed. 2013, 52, 7156. doi:10.1002/anie.201301634
Return to citation in text: [1] -
Xi, Y.; Dong, B.; McClain, E. J.; Wang, Q.; Gregg, T. L.; Akhmedov, N. G.; Petersen, J. L.; Shi, X. Angew. Chem., Int. Ed. 2014, 53, 4657. doi:10.1002/anie.201310142
Return to citation in text: [1] -
Kunz, K.; Scholz, U.; Ganzer, D. Synlett 2003, 2428. doi:10.1055/s-2003-42473
Return to citation in text: [1] -
Beletskaya, I. P.; Cheprakov, A. V. Coord. Chem. Rev. 2004, 248, 2337. doi:10.1016/j.ccr.2004.09.014
Return to citation in text: [1] -
Monnier, F.; Taillefer, M. Angew. Chem., Int. Ed. 2009, 48, 6954. doi:10.1002/anie.200804497
Return to citation in text: [1] -
Sambiagio, C.; Marsden, S. P.; Blacker, A. J.; McGowan, P. C. Chem. Soc. Rev. 2014, 43, 3525. doi:10.1039/C3CS60289C
Return to citation in text: [1] -
Lee, C.-F.; Liu, Y.-C.; Badsara, S. S. Chem. – Asian J. 2014, 9, 706. doi:10.1002/asia.201301500
Return to citation in text: [1] -
Rao, K. S.; Wu, T.-S. Tetrahedron 2012, 68, 7735. doi:10.1016/j.tet.2012.06.015
Return to citation in text: [1] -
Xu, H.-J.; Zhao, Y.-Q.; Feng, T.; Feng, Y.-S. J. Org. Chem. 2012, 77, 2878. doi:10.1021/jo300100x
Return to citation in text: [1] -
Yu, J.-T.; Guo, H.; Yi, Y.; Fei, H.; Jiang, Y. Adv. Synth. Catal. 2014, 356, 749. doi:10.1002/adsc.201300853
Return to citation in text: [1] -
Roy, S.; Sarma, M. J.; Kashyap, B.; Phukan, P. Chem. Commun. 2016, 52, 1170. doi:10.1039/C5CC04619J
Return to citation in text: [1] -
Matsumoto, K.; Sugiyama, H. Acc. Chem. Res. 2002, 35, 915. doi:10.1021/ar000103m
Return to citation in text: [1] -
Beletskaya, I. P.; Ananikov, V. P. Chem. Rev. 2011, 111, 1596. doi:10.1021/cr100347k
Return to citation in text: [1] -
Shen, C.; Zhang, P.; Sun, Q.; Bai, S.; Hor, T. S. A.; Liu, X. Chem. Soc. Rev. 2015, 44, 291. doi:10.1039/C4CS00239C
Return to citation in text: [1] -
Wang, P.; Tang, S.; Huang, P.; Lei, A. Angew. Chem., Int. Ed. 2017, 56, 3009. doi:10.1002/anie.201700012
Return to citation in text: [1] -
Huang, Z.; Zhang, D.; Qi, X.; Yan, Z.; Wang, M.; Yan, H.; Lei, A. Org. Lett. 2016, 18, 2351. doi:10.1021/acs.orglett.6b00764
Return to citation in text: [1] -
Yang, F.-L.; Tian, S.-K. Angew. Chem., Int. Ed. 2013, 52, 4929. doi:10.1002/anie.201301437
Return to citation in text: [1] -
Prasad, C. D.; Balkrishna, S. J.; Kumar, A.; Bhakuni, B. S.; Shrimali, K.; Biswas, S.; Kumar, S. J. Org. Chem. 2013, 78, 1434. doi:10.1021/jo302480j
Return to citation in text: [1] -
Hostier, T.; Ferey, V.; Ricci, G.; Pardo, D. G.; Cossy, J. Org. Lett. 2015, 17, 3898. doi:10.1021/acs.orglett.5b01889
Return to citation in text: [1] -
Varun, B. V.; Prabhu, K. R. J. Org. Chem. 2014, 79, 9655. doi:10.1021/jo501793q
Return to citation in text: [1] -
Ge, W.; Wei, Y. Green Chem. 2012, 14, 2066–2070. doi:10.1039/c2gc35337g
Return to citation in text: [1] -
Yang, Y.; Zhang, S.; Tang, L.; Hu, Y.; Zha, Z.; Wang, Z. Green Chem. 2016, 18, 2609. doi:10.1039/C6GC00313C
Return to citation in text: [1] -
Tudge, M.; Tamiya, M.; Savarin, C.; Humphrey, G. R. Org. Lett. 2006, 8, 565. doi:10.1021/ol052615c
Return to citation in text: [1] -
Siddaraju, Y.; Prabhu, K. R. J. Org. Chem. 2017, 82, 3084. doi:10.1021/acs.joc.7b00073
Return to citation in text: [1] -
Ravi, C.; Mohan, D. C.; Adimurthy, S. Org. Lett. 2014, 16, 2978. doi:10.1021/ol501117z
Return to citation in text: [1] -
Rafique, J.; Saba, S.; Rosário, A. R.; Braga, A. L. Chem. – Eur. J. 2016, 22, 11854. doi:10.1002/chem.201600800
Return to citation in text: [1] -
Sharma, S. K.; Kumar, S.; Chand, K.; Kathuria, A.; Gupta, A.; Jain, R. Curr. Med. Chem. 2011, 18, 3825. doi:10.2174/092986711803414359
Return to citation in text: [1] -
Choodej, S.; Sommit, D.; Pudhom, K. Bioorg. Med. Chem. Lett. 2013, 23, 3896. doi:10.1016/j.bmcl.2013.04.064
Return to citation in text: [1] -
Ibrahim, S. R. M.; Mohamed, G. A. Nat. Prod. Res. 2015, 29, 1489. doi:10.1080/14786419.2014.991323
Return to citation in text: [1] -
Han, S. H.; Kim, S.; De, U.; Mishra, N. K.; Park, J.; Sharma, S.; Kwak, J. H.; Han, S.; Kim, H. S.; Kim, I. S. J. Org. Chem. 2016, 81, 12416. doi:10.1021/acs.joc.6b02577
Return to citation in text: [1] -
Zhu, F.; Li, Y.; Wang, Z.; Wu, X.-F. Angew. Chem., Int. Ed. 2016, 55, 14151. doi:10.1002/anie.201608715
Return to citation in text: [1] -
Sun, P.; Gao, S.; Yang, C.; Guo, S.; Lin, A.; Yao, H. Org. Lett. 2016, 18, 6464. doi:10.1021/acs.orglett.6b03355
Return to citation in text: [1] -
Shen, C.; Li, W.; Yin, H.; Spannenberg, A.; Skrydstrup, T.; Wu, X.-F. Adv. Synth. Catal. 2016, 358, 466. doi:10.1002/adsc.201500858
Return to citation in text: [1] -
Wang, X.; Cheng, G.; Cui, X. Chem. Commun. 2014, 50, 652. doi:10.1039/C3CC48259F
Return to citation in text: [1] -
Zhao, J.; Zhao, Y.; Fu, H. Org. Lett. 2012, 14, 2710. doi:10.1021/ol300908g
Return to citation in text: [1] -
Vedachalam, S.; Wong, Q.-L.; Maji, B.; Zeng, J.; Ma, J.; Liu, X.-W. Adv. Synth. Catal. 2011, 353, 219. doi:10.1002/adsc.201000828
Return to citation in text: [1] -
Zhao, W.; Xie, P.; Bian, Z.; Zhou, A.; Ge, H.; Zhang, M.; Ding, Y.; Zheng, L. J. Org. Chem. 2015, 80, 9167. doi:10.1021/acs.joc.5b01602
Return to citation in text: [1] -
Zhao, W.; Zhou, A. ChemCatChem 2015, 7, 3464. doi:10.1002/cctc.201500673
Return to citation in text: [1] -
Wan, J.-P.; Zhong, S.; Xie, L.; Cao, X.; Liu, Y.; Wei, L. Org. Lett. 2016, 18, 584. doi:10.1021/acs.orglett.5b03608
Return to citation in text: [1] -
Gao, Y.; Wei, L.; Liu, Y.; Wan, J.-P. Org. Biomol. Chem. 2017, 15, 4631. doi:10.1039/C7OB00619E
Return to citation in text: [1] -
Zhong, S.; Liu, Y.; Cao, X.; Wan, J.-P. ChemCatChem 2017, 9, 465. doi:10.1002/cctc.201601273
Return to citation in text: [1] -
Rafique, J.; Saba, S.; Schneider, A. R.; Franco, M. S.; Silva, S. M.; Braga, A. L. ACS Omega 2017, 2, 2280. doi:10.1021/acsomega.7b00445
Return to citation in text: [1] -
Kang, X.; Yan, R.; Yu, G.; Pang, X.; Liu, X.; Li, X.; Xiang, L.; Huang, G. J. Org. Chem. 2014, 79, 10605. doi:10.1021/jo501778h
Return to citation in text: [1] -
Pang, X.; Xiang, L.; Yang, X.; Yan, R. Adv. Synth. Catal. 2016, 358, 321. doi:10.1002/adsc.201500943
Return to citation in text: [1] -
Deng, Y.; Wei, X.-J.; Wang, H.; Sun, Y.; Noël, T.; Wang, X. Angew. Chem., Int. Ed. 2017, 56, 832. doi:10.1002/anie.201607948
Return to citation in text: [1] -
Zhu, X.; Li, P.; Shi, Q.; Wang, L. Green Chem. 2016, 18, 6373. doi:10.1039/C6GC01487A
Return to citation in text: [1] -
Liu, Y.; Wang, H.; Wang, C.; Wan, J.-P.; Wen, C. RSC Adv. 2013, 3, 21369. doi:10.1039/c3ra42915f
Return to citation in text: [1] -
Wang, H.; Huang, G.; Sun, Y.; Liu, Y. J. Chem. Res. 2014, 38, 96. doi:10.3184/174751914X13892888669706
Return to citation in text: [1]
| 1. | Damani, L. A., Ed. Sulphur-Containing Drugs and Related Organic Compounds; Wiley: New York, 1989. |
| 2. | Lu, Q.; Zhang, J.; Wei, F.; Qi, Y.; Wang, H.; Liu, Z.; Lei, A. Angew. Chem., Int. Ed. 2013, 52, 7156. doi:10.1002/anie.201301634 |
| 3. | Xi, Y.; Dong, B.; McClain, E. J.; Wang, Q.; Gregg, T. L.; Akhmedov, N. G.; Petersen, J. L.; Shi, X. Angew. Chem., Int. Ed. 2014, 53, 4657. doi:10.1002/anie.201310142 |
| 16. | Wang, P.; Tang, S.; Huang, P.; Lei, A. Angew. Chem., Int. Ed. 2017, 56, 3009. doi:10.1002/anie.201700012 |
| 17. | Huang, Z.; Zhang, D.; Qi, X.; Yan, Z.; Wang, M.; Yan, H.; Lei, A. Org. Lett. 2016, 18, 2351. doi:10.1021/acs.orglett.6b00764 |
| 18. | Yang, F.-L.; Tian, S.-K. Angew. Chem., Int. Ed. 2013, 52, 4929. doi:10.1002/anie.201301437 |
| 19. | Prasad, C. D.; Balkrishna, S. J.; Kumar, A.; Bhakuni, B. S.; Shrimali, K.; Biswas, S.; Kumar, S. J. Org. Chem. 2013, 78, 1434. doi:10.1021/jo302480j |
| 20. | Hostier, T.; Ferey, V.; Ricci, G.; Pardo, D. G.; Cossy, J. Org. Lett. 2015, 17, 3898. doi:10.1021/acs.orglett.5b01889 |
| 21. | Varun, B. V.; Prabhu, K. R. J. Org. Chem. 2014, 79, 9655. doi:10.1021/jo501793q |
| 22. | Ge, W.; Wei, Y. Green Chem. 2012, 14, 2066–2070. doi:10.1039/c2gc35337g |
| 23. | Yang, Y.; Zhang, S.; Tang, L.; Hu, Y.; Zha, Z.; Wang, Z. Green Chem. 2016, 18, 2609. doi:10.1039/C6GC00313C |
| 24. | Tudge, M.; Tamiya, M.; Savarin, C.; Humphrey, G. R. Org. Lett. 2006, 8, 565. doi:10.1021/ol052615c |
| 25. | Siddaraju, Y.; Prabhu, K. R. J. Org. Chem. 2017, 82, 3084. doi:10.1021/acs.joc.7b00073 |
| 26. | Ravi, C.; Mohan, D. C.; Adimurthy, S. Org. Lett. 2014, 16, 2978. doi:10.1021/ol501117z |
| 27. | Rafique, J.; Saba, S.; Rosário, A. R.; Braga, A. L. Chem. – Eur. J. 2016, 22, 11854. doi:10.1002/chem.201600800 |
| 13. | Matsumoto, K.; Sugiyama, H. Acc. Chem. Res. 2002, 35, 915. doi:10.1021/ar000103m |
| 14. | Beletskaya, I. P.; Ananikov, V. P. Chem. Rev. 2011, 111, 1596. doi:10.1021/cr100347k |
| 15. | Shen, C.; Zhang, P.; Sun, Q.; Bai, S.; Hor, T. S. A.; Liu, X. Chem. Soc. Rev. 2015, 44, 291. doi:10.1039/C4CS00239C |
| 9. | Rao, K. S.; Wu, T.-S. Tetrahedron 2012, 68, 7735. doi:10.1016/j.tet.2012.06.015 |
| 10. | Xu, H.-J.; Zhao, Y.-Q.; Feng, T.; Feng, Y.-S. J. Org. Chem. 2012, 77, 2878. doi:10.1021/jo300100x |
| 11. | Yu, J.-T.; Guo, H.; Yi, Y.; Fei, H.; Jiang, Y. Adv. Synth. Catal. 2014, 356, 749. doi:10.1002/adsc.201300853 |
| 12. | Roy, S.; Sarma, M. J.; Kashyap, B.; Phukan, P. Chem. Commun. 2016, 52, 1170. doi:10.1039/C5CC04619J |
| 46. | Deng, Y.; Wei, X.-J.; Wang, H.; Sun, Y.; Noël, T.; Wang, X. Angew. Chem., Int. Ed. 2017, 56, 832. doi:10.1002/anie.201607948 |
| 47. | Zhu, X.; Li, P.; Shi, Q.; Wang, L. Green Chem. 2016, 18, 6373. doi:10.1039/C6GC01487A |
| 4. | Kunz, K.; Scholz, U.; Ganzer, D. Synlett 2003, 2428. doi:10.1055/s-2003-42473 |
| 5. | Beletskaya, I. P.; Cheprakov, A. V. Coord. Chem. Rev. 2004, 248, 2337. doi:10.1016/j.ccr.2004.09.014 |
| 6. | Monnier, F.; Taillefer, M. Angew. Chem., Int. Ed. 2009, 48, 6954. doi:10.1002/anie.200804497 |
| 7. | Sambiagio, C.; Marsden, S. P.; Blacker, A. J.; McGowan, P. C. Chem. Soc. Rev. 2014, 43, 3525. doi:10.1039/C3CS60289C |
| 8. | Lee, C.-F.; Liu, Y.-C.; Badsara, S. S. Chem. – Asian J. 2014, 9, 706. doi:10.1002/asia.201301500 |
| 48. | Liu, Y.; Wang, H.; Wang, C.; Wan, J.-P.; Wen, C. RSC Adv. 2013, 3, 21369. doi:10.1039/c3ra42915f |
| 49. | Wang, H.; Huang, G.; Sun, Y.; Liu, Y. J. Chem. Res. 2014, 38, 96. doi:10.3184/174751914X13892888669706 |
| 40. | Wan, J.-P.; Zhong, S.; Xie, L.; Cao, X.; Liu, Y.; Wei, L. Org. Lett. 2016, 18, 584. doi:10.1021/acs.orglett.5b03608 |
| 41. | Gao, Y.; Wei, L.; Liu, Y.; Wan, J.-P. Org. Biomol. Chem. 2017, 15, 4631. doi:10.1039/C7OB00619E |
| 43. | Rafique, J.; Saba, S.; Schneider, A. R.; Franco, M. S.; Silva, S. M.; Braga, A. L. ACS Omega 2017, 2, 2280. doi:10.1021/acsomega.7b00445 |
| 38. | Zhao, W.; Xie, P.; Bian, Z.; Zhou, A.; Ge, H.; Zhang, M.; Ding, Y.; Zheng, L. J. Org. Chem. 2015, 80, 9167. doi:10.1021/acs.joc.5b01602 |
| 39. | Zhao, W.; Zhou, A. ChemCatChem 2015, 7, 3464. doi:10.1002/cctc.201500673 |
| 44. | Kang, X.; Yan, R.; Yu, G.; Pang, X.; Liu, X.; Li, X.; Xiang, L.; Huang, G. J. Org. Chem. 2014, 79, 10605. doi:10.1021/jo501778h |
| 45. | Pang, X.; Xiang, L.; Yang, X.; Yan, R. Adv. Synth. Catal. 2016, 358, 321. doi:10.1002/adsc.201500943 |
| 32. | Zhu, F.; Li, Y.; Wang, Z.; Wu, X.-F. Angew. Chem., Int. Ed. 2016, 55, 14151. doi:10.1002/anie.201608715 |
| 33. | Sun, P.; Gao, S.; Yang, C.; Guo, S.; Lin, A.; Yao, H. Org. Lett. 2016, 18, 6464. doi:10.1021/acs.orglett.6b03355 |
| 34. | Shen, C.; Li, W.; Yin, H.; Spannenberg, A.; Skrydstrup, T.; Wu, X.-F. Adv. Synth. Catal. 2016, 358, 466. doi:10.1002/adsc.201500858 |
| 35. | Wang, X.; Cheng, G.; Cui, X. Chem. Commun. 2014, 50, 652. doi:10.1039/C3CC48259F |
| 36. | Zhao, J.; Zhao, Y.; Fu, H. Org. Lett. 2012, 14, 2710. doi:10.1021/ol300908g |
| 37. | Vedachalam, S.; Wong, Q.-L.; Maji, B.; Zeng, J.; Ma, J.; Liu, X.-W. Adv. Synth. Catal. 2011, 353, 219. doi:10.1002/adsc.201000828 |
| 28. | Sharma, S. K.; Kumar, S.; Chand, K.; Kathuria, A.; Gupta, A.; Jain, R. Curr. Med. Chem. 2011, 18, 3825. doi:10.2174/092986711803414359 |
| 29. | Choodej, S.; Sommit, D.; Pudhom, K. Bioorg. Med. Chem. Lett. 2013, 23, 3896. doi:10.1016/j.bmcl.2013.04.064 |
| 30. | Ibrahim, S. R. M.; Mohamed, G. A. Nat. Prod. Res. 2015, 29, 1489. doi:10.1080/14786419.2014.991323 |
| 31. | Han, S. H.; Kim, S.; De, U.; Mishra, N. K.; Park, J.; Sharma, S.; Kwak, J. H.; Han, S.; Kim, H. S.; Kim, I. S. J. Org. Chem. 2016, 81, 12416. doi:10.1021/acs.joc.6b02577 |
| 42. | Zhong, S.; Liu, Y.; Cao, X.; Wan, J.-P. ChemCatChem 2017, 9, 465. doi:10.1002/cctc.201601273 |
© 2017 Guo et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)