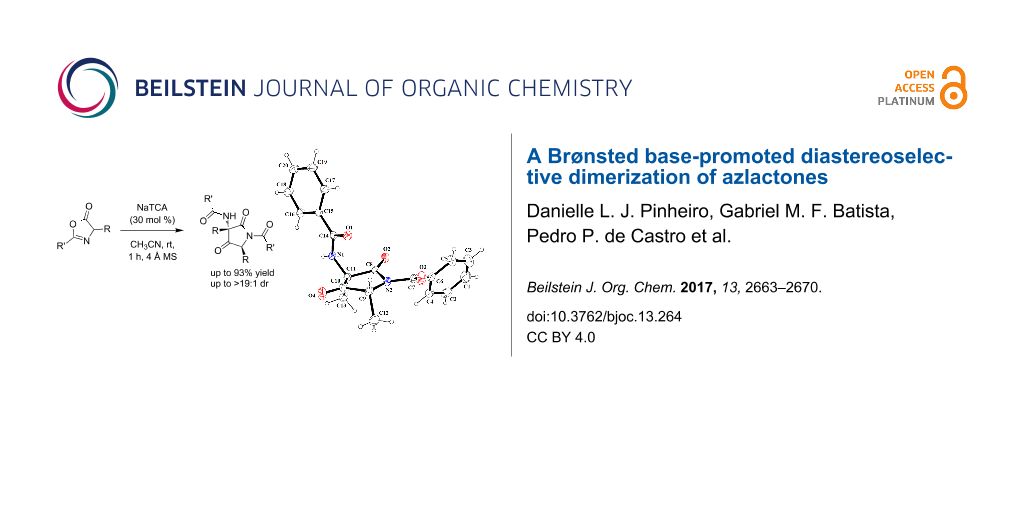
Abstract
A novel Brønsted base system for the diastereoselective dimerization of azlactones using trichloroacetate salts and acetonitrile has been developed. Desired products were obtained in good yields (60–93%) and with up to >19:1 dr after one hour of reaction. Additionally, the relative stereochemistry of the major dimer was assigned as being trans, by X-ray crystallographic analysis. The kinetic reaction profile was determined by using 1H NMR reaction monitoring and revealed a second order overall kinetic profile. Furthermore, by employing this methodology, a diastereoselective total synthesis of a functionalized analogue of streptopyrrolidine was accomplished in 65% overall yield.

Graphical Abstract
Findings
Azlactones have been acknowledged as a common nucleophilic reagent to introduce a quaternary carbon stereocenter in the α-position of α-amino acid derivatives. To this end, different catalytic transformations involving azlactones have been established [1-19]. For example, our research group has explored the potential of these heterocycles in the presence of Michael and Mannich acceptors [20-23].
Enolate anions derived from azlactone are effective intermediates for the functionalization of α-amino acids. However, the acylation of the enolate ion by another azlactone has been less reported in the literature [24-26]. This transformation affords an azlactone dimer, which is a versatile heterocycle containing two stereogenic centers (Figure 1).
![[1860-5397-13-264-1]](/bjoc/content/figures/1860-5397-13-264-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of an azlactone dimer.
Figure 1: Structure of an azlactone dimer.
Kobayashi and co-workers demonstrated the rich reactivity of azlactones conducting the dimerization reaction using strong bases as catalysts; however, higher temperatures and long reaction times were required [27]. Moreover, no evidence regarding either the diastereoselectivity and stereochemistry were reported. In 1998, Mazurkiewicz and co-workers developed a kinetic study of the base-catalyzed dimerization of azlactones. In this investigation, it was established that the dimerization reaction rates are strongly dependent on the base, as well as the substituent at the C2 position [24]. Once again, no comments concerning the stereochemistry were addressed.
Thus, we started this study envisioning the trichloromethylation of azlactones through the decarboxylative potassium trichloroacetate (KTCA) in DMSO. Although no trichloromethylation product was observed, we found out that by switching the solvent, the isolated major product comes from dimerization of azlactones. At this point, we turned our attention towards this product, envisioning the development of an atom-economic reaction for stereoselective C–C bond formation, such as a dimerization process.
We herein report a diastereoselective dimerization of azlactones using potassium or sodium trichloroacetate salts and acetonitrile as solvent. Besides, a reaction mechanism is proposed based on NMR studies and the relative stereochemistry of the major diastereomer was assigned by X-ray analysis. Finally, one of the products was selectively reduced to provide a functionalized analogue of streptopyrrolidine, a marine natural product isolated from Streptomyces sp.
As shown in Table 1, our studies started with the synthesis of dimer 2a using azlactone 1a in the presence of potassium trichlororacetate salt (2 equiv), DMSO as solvent, at room temperature. As preliminary results, we observed the formation of several undesired products, detected by 1H NMR analyses of the crude reaction mixtures. Several salts were evaluated, with great influence in reaction yields and dr; it is also worth mentioning that when employing KTFA, NaTFA and NaHCO3 no product was observed (Table 1, entries 16, 17 and 18). Besides, different solvents were also evaluated and the most promising result was found using acetonitrile (Table 1, entry 6). This probably was due to the formation of an intermediate between acetonitrile and the trichloroacetate salt (see mechanism discussion below). Under these reaction conditions the desired product was isolated in excellent conversion and with moderate dr 2:1. Encouraged by this result, we have then evaluated the effect of the salt concentration on the reaction outcome. Decreasing the amount of KTCA to 0.3 equiv, the diastereoselectivity slightly increased to 4:1 (Table 1, entry 9). Finally, NaTCA as salt gave 98% conversion and a good dr (6:1, Table 1, entry 13). Different basic conditions were evaluated without any improvements in terms of yields and selectivities.
Table 1: Optimization reaction conditions of azlactones dimerization.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-264-i6.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Salt/base | Equiv | Solvent | Temp. (°C) | Conv.a ( %) | dra |
| 1 | KTCAb | 2 | DMSO | rt | mix of products | – |
| 2 | KTCA | 1 | DMSO | rt | mix of products | – |
| 3 | KTCA | 2 | DMF | rt | mix of products | – |
| 4 | KTCA | 2 | DMF | 0 | – | – |
| 5 | KTCA | 2 | CH2Cl2 | rt | – | – |
| 6 | KTCA | 2 | CH3CN | rt | 95 | 2:1 |
| 7 | KTCA | 1 | CH3CN | rt | 95 | 2:1 |
| 8 | KTCA | 0.7 | CH3CN | rt | 95 | 3:1 |
| 9 | KTCA | 0.3 | CH3CN | rt | 94 | 4:1 |
| 10 | KTCA | 0.3 | CH3CN | 0 | – | – |
| 11 | KTCA | 0.2 | CH3CN | rt | 82 | 4:1 |
| 12 | NaTCAc | 0.2 | CH3CN | rt | 94 | 6:1 |
| 13 | NaTCA | 0.3 | CH3CN | rt | 98 | 6:1 |
| 14 | NaTCA | 0.3 | CH3CN | 0 | – | – |
| 15 | LiTCAd | 0.3 | CH3CN | rt | 88 | 3:1 |
| 16 | KTFAe | 0.3 | CH3CN | rt | – | – |
| 17 | NaTFAf | 0.3 | CH3CN | rt | – | – |
| 18 | NaHCO3 | 0.3 | CH3CN | rt | – | – |
| 19 | Et3N | 0.3 | CH3CN | rt | 34 | 1:1 |
| 20 | NaOH | 0.3 | CH3CN | rt | 10 | 1:1 |
| 21 | NaOH | 0.5 | CH3CN | rt | 98 | 2:1 |
aMeasured by ¹H NMR analysis of the crude reaction mixture. b Potassium trichloroacetate. cSodium trichloroacetate. dLithium trichloroacetate. ePotassium trifluoroacetate. fSodium trifluoroacetate.
With the optimal reaction conditions established, the scope of the reaction was then investigated (Scheme 1). The results revealed that the dimerization reaction shows tolerance to changes of functional groups at both the substituents R and R1. For example, in the presence of an electron-withdrawing aryl ring substituent R, the reaction worked well and afforded the product 2e in good yield and diastereoselectivity. When R1 was a benzyl group, the reaction tolerated the use of both electron-donating substituents and a non-substituted phenyl ring, affording the desired products in excellent yields and with up to dr >19:1 (products 2c, 2d and 2h). Substrates bearing an alkyl chain as an R1 group were well tolerated (2a and 2b). Importantly, azlactones containing a thioether or allyl substituents could be also accessed in good isolated yields (2f and 2g).
![[1860-5397-13-264-i1]](/bjoc/content/inline/1860-5397-13-264-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Diastereoselective dimerization of azlactones. Reactions were carried out using 0.45 mmol of 1 and 0.14 mmol of salt. Diastereomer ratio measured by 1H NMR analysis of the crude reaction mixture.
Scheme 1: Diastereoselective dimerization of azlactones. Reactions were carried out using 0.45 mmol of 1 and ...
The major diastereomer was isolated after purification through column chromatography or simple recrystallization in good to excellent yields. The relative configuration to C11 and C9 of the major diastereomer was assigned by X-ray crystallographic analysis of compound 2a (Figure 2) [28]. The other products were assigned by analogy.
![[1860-5397-13-264-2]](/bjoc/content/figures/1860-5397-13-264-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray crystallographic structure of 2a (30% ellipsoids probability).
Figure 2: X-ray crystallographic structure of 2a (30% ellipsoids probability).
An interesting observation was found when employing azlactones derived from more sterically bulky amino acids, such as valine or isoleucine. In this case, the dimerization process does not occur. We isolate the corresponding azlactone enol derivatives 2j and 2k, which are very stable and unreactive species (Scheme 2) [29].
![[1860-5397-13-264-i2]](/bjoc/content/inline/1860-5397-13-264-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Sterically bulky azlactone enol derivatives.
Scheme 2: Sterically bulky azlactone enol derivatives.
Although the mechanistic details of dimerization remain to be elucidated, a plausible mechanism was proposed based on 1H NMR evidence (Figure 3). The NMR experiments revealed that KTCA reacts with CH3CN to form a basic species 3 [30]. The enolate azlactone 1a’ attacks the carbonyl carbon of azlactone 1a forming intermediate 4 [27]. The diastereoselectivity is installed at this stage and kept the same until to the end of the reaction. Steric effects might be involved in this process and the biggest substituents positioned far away from each other, as shown in Figure 3. Furthermore, a six-membered ring involving the salt cation, the azlactone ring and the enolate might be involved in the addition step. Irreversible intramolecular cyclization of 4 gave 5 (for a reaction reversibility study of product 2h, see Supporting Information File 1), eventually affording 2a after protonation and thus regenerating the catalyst intermediate 3. 1H NMR analysis using CD3CN showed high levels of HCCl3, which could be form after decarboxylation of the basic species.
![[1860-5397-13-264-3]](/bjoc/content/figures/1860-5397-13-264-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Plausible mechanism for the dimerization of azlactone.
Figure 3: Plausible mechanism for the dimerization of azlactone.
In an attempt to obtain a better understanding at this dimerization reaction and to give new insights into this kind of transformation, we carried out some kinetics experiments. Taking into account the dimerization kinetic studies suggested by Mazurkiewicz et al. and experiments by NMR spectroscopy, we report here kinetic studies revealing mechanistic features on the dimerization reaction catalyzed by base species from trichloroacetate sodium salt [24].
The integrated rate law, suggested by Mazurkiewicz et al. was proposed for a 3/2 reaction order for the azlactone that results from the above mechanism, which is:
![[1860-5397-13-264-i4]](/bjoc/content/inline/1860-5397-13-264-i4.svg?max-width=590&scale=1.18182)
Where
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-264-i7.svg?max-width=637&scale=1.18182)
and
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-264-i8.svg?max-width=637&scale=1.18182)
so that [A], [A]0 and [B]0 are, respectively, azlactone 1a concentration, initial azlactone 1a concentration, and initial base species 3 concentration; k1 and k−1 are the direct and reverse reaction rate constants for the formation of an azlactone enolate derivative 1’ and the azlactone 1a, respectively.
In situ reaction monitoring by NMR spectroscopy allowed us to follow the disappearance of the characteristic signal of the azlactone 1a (q, 4.45 ppm) as well as the appearance of the characteristic signal of the dimer 2a (q, 5.18 ppm). The values found were plotted in Figure 4 and a linear relationship between
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-264-i9.svg?max-width=637&scale=1.18182)
and the reaction time was obtained with high correlation factor (R2 = 0.9905).
![[1860-5397-13-264-4]](/bjoc/content/figures/1860-5397-13-264-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4:
Plot of ![[Graphic 5]](/bjoc/content/inline/1860-5397-13-264-i10.svg?max-width=637&scale=1.18182) vs time for the dimerization of azlactone 1a.
vs time for the dimerization of azlactone 1a.
Figure 4:
Plot of vs time for the dimerization of azlactone 1a.
We then reasoned the second order kinetic profile to the global reaction, resulting from a 3/2 order for the azlactone 1a and 1/2 order for the base species 3 as could be shown in Equation 2 (please, refer to Supporting Information File 1 for a demonstration of the discussed equations):
![[1860-5397-13-264-i5]](/bjoc/content/inline/1860-5397-13-264-i5.svg?max-width=590&scale=1.18182)
The 3/2 reaction order for azlactone has also been confirmed using the technique of initial rates varying the initial concentrations of azlactone 1a (See Supporting Information File 1).
Finally, the synthetic utility of the dimerization products was demonstrated by stereocontrolled reduction of the dimer 2c (Scheme 3). Compound 2c was dissolved in a mixture of acetic acid and NaBH4, cooled to 0 °C to afford the highly functionalized (+/−)-streptopyrrolidine analogue 6 in 70% yield as a unique diastereomer. Streptopyrrolidine was isolated from the marine bacterium Streptomyces sp. and has exhibited a significant anti-angiogenesis activity [31]. It is worth mentioning that this product has three consecutive stereocenters and different sites of variation from the original natural product.
![[1860-5397-13-264-i3]](/bjoc/content/inline/1860-5397-13-264-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
The hydride comes from the less hindered side of the ketone moiety, leading to the formation of a hydroxy group in cis-position relative to both the benzyl substituents [32]. The relative stereochemistry of 6 was assigned by X-ray analysis (Figure 5).
![[1860-5397-13-264-5]](/bjoc/content/figures/1860-5397-13-264-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: X-ray crystallographic structure of 6 (30% ellipsoids probability).
Figure 5: X-ray crystallographic structure of 6 (30% ellipsoids probability).
Conclusion
In summary, we have demonstrated the first diastereoselective dimerization of azlactones mediated by a combination of acetonitrile and potassium or sodium trichloroacetate salts, which provides a straightforward and practical access to azlactone dimers with good to excellent yields and good diastereoselectivities. In addition, the relative stereochemistry of major azlactone dimer was assigned as being trans by X-ray analysis. A plausible mechanism was proposed by 1H NMR spectroscopic monitoring and revealed that the diastereoselectivity was determined during C–C bond formation step. A diastereoselective streptopyrrolidine analogue, containing three contiguous stereogenic centers, was smoothly obtained as a sole diastereomer in 70% yield.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, copies of 1H and 13C NMR spectra for the final products, NMR kinetic experiments as well as crystallographic data for 2a and 6. Demonstration of kinetic equations and results of the initial rate kinetic study. | ||
| Format: PDF | Size: 4.0 MB | Download |
Acknowledgements
The authors are grateful to the Fundação de Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG), the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), and the Rede Mineira de Química for financial support.
References
-
Wang, C.-M.; Xiao, J.-A.; Wang, J.; Wang, S.-S.; Deng, Z.-X.; Yang, H. J. Org. Chem. 2016, 81, 8001. doi:10.1021/acs.joc.6b01356
Return to citation in text: [1] -
Yu, X.-Y.; Chen, J.-R.; Wei, Q.; Cheng, H.-G.; Liu, Z.-C.; Xiao, W.-J. Chem. – Eur. J. 2016, 22, 6774. doi:10.1002/chem.201601227
Return to citation in text: [1] -
Liu, X.; Wang, Y.; Yang, D.; Zhang, J.; Liu, D.; Su, W. Angew. Chem., Int. Ed. 2016, 55, 8100. doi:10.1002/anie.201602880
Return to citation in text: [1] -
Choi, Y. S.; Park, S.; Park, Y. S. Eur. J. Org. Chem. 2016, 2539. doi:10.1002/ejoc.201600201
Return to citation in text: [1] -
Zhang, S.-Y.; Lv, M.; Yin, S.-J.; Li, N.-K.; Zhang, J.-Q.; Wang, X.-W. Adv. Synth. Catal. 2016, 358, 143. doi:10.1002/adsc.201500666
Return to citation in text: [1] -
De Castro, P. P.; Dos Santos, I. F.; Amarante, G. W. Curr. Org. Synth. 2016, 13, 440. doi:10.2174/1570179413999151211114542
Return to citation in text: [1] -
Wang, T.; Yu, Z.; Hoon, D. L.; Phee, C. Y.; Lan, Y.; Lu, Y. J. Am. Chem. Soc. 2016, 138, 265. doi:10.1021/jacs.5b10524
Return to citation in text: [1] -
Zhang, Z.; Sun, W.; Zhu, G.; Yang, J.; Zhang, M.; Hong, L.; Wang, R. Chem. Commun. 2016, 52, 1377. doi:10.1039/C5CC08989A
Return to citation in text: [1] -
Izumi, S.; Kobayashi, Y.; Takemoto, Y. Org. Lett. 2016, 18, 696. doi:10.1021/acs.orglett.5b03666
Return to citation in text: [1] -
de Castro, P. P.; Carpanez, A. G.; Amarante, G. W. Chem. – Eur. J. 2016, 22, 10294. doi:10.1002/chem.201600071
Return to citation in text: [1] -
Pinheiro, D. L. J.; Ávila, E. P.; Amarante, G. W. ChemistrySelect 2016, 1, 2960. doi:10.1002/slct.201600639
Return to citation in text: [1] -
Zheng, Y.; Deng, L. Chem. Sci. 2015, 6, 6510. doi:10.1039/C5SC02116B
Return to citation in text: [1] -
Zhu, C.-L.; Yang, L.-J.; Li, S.; Zheng, Y.; Ma, J.-A. Org. Lett. 2015, 17, 3442. doi:10.1021/acs.orglett.5b01450
Return to citation in text: [1] -
Qiao, B.; Liu, X.; Duan, S.; Yan, L.; Jiang, Z. Org. Lett. 2014, 16, 672. doi:10.1021/ol403303k
Return to citation in text: [1] -
Pereira, A. A.; de Castro, P. P.; de Mello, A. C.; Ferreira, B. R. V.; Eberlin, M. N.; Amarante, G. W. Tetrahedron 2014, 70, 3271. doi:10.1016/j.tet.2013.11.037
Return to citation in text: [1] -
Alba, A.-N. R.; Rios, R. Chem. – Asian J. 2011, 6, 720. doi:10.1002/asia.201000636
Return to citation in text: [1] -
Hewlett, N. M.; Hupp, C. D.; Tepe, J. J. Synthesis 2009, 2825. doi:10.1055/s-0029-1216924
Return to citation in text: [1] -
Mosey, R. A.; Fisk, J. S.; Tepe, J. J. Tetrahedron: Asymmetry 2008, 19, 2755. doi:10.1016/j.tetasy.2008.11.033
Return to citation in text: [1] -
Fisk, J. S.; Mosey, R. A.; Tepe, J. J. Chem. Soc. Rev. 2007, 36, 1432. doi:10.1039/b511113g
Return to citation in text: [1] -
Pinheiro, D. L. J.; Batista, G. M. F.; Gonçalves, J. R.; Duarte, T. N.; Amarante, G. W. Eur. J. Org. Chem. 2016, 459. doi:10.1002/ejoc.201501393
Return to citation in text: [1] -
Ávila, E. P.; Justo, R. M. S.; Gonçalves, V. P.; Pereira, A. A.; Diniz, R.; Amarante, G. W. J. Org. Chem. 2015, 80, 590. doi:10.1021/jo5024975
Return to citation in text: [1] -
Ávila, E. P.; de Mello, A. C.; Diniz, R.; Amarante, G. W. Eur. J. Org. Chem. 2013, 1881. doi:10.1002/ejoc.201300076
Return to citation in text: [1] -
Melhado, A. D.; Amarante, G. W.; Wang, Z. J.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 3517. doi:10.1021/ja1095045
Return to citation in text: [1] -
Mazurkiewicz, R.; Pierwocha, A. W.; Fryczkowska, B. Pol. J. Chem. 1998, 72, 113.
Return to citation in text: [1] [2] [3] -
Taylor, L. D.; Platt, T. E.; Mach, M. H. J. Polym. Sci., Part B: Polym. Lett. 1970, 8, 537. doi:10.1002/pol.1970.110080803
Return to citation in text: [1] -
Slavinskaya, V. A.; Sile, D. É.; Katkevich, M. Yu.; Korchagova, É. K.; Lukevits, É. Chem. Heterocycl. Compd. 1994, 30, 724. doi:10.1007/BF01166316
Return to citation in text: [1] -
Kobayashi, S.; Bryant, L. L., Jr.; Tsukamoto, Y.; Saegusa, T. Macromolecules 1986, 19, 1547. doi:10.1021/ma00160a012
Return to citation in text: [1] [2] -
Gakis, N.; Märky, M.; Hansen, H.-J.; Heimgartner, H.; Schmid, H.; Oberhänsli, W. E. Helv. Chim. Acta 1976, 59, 2149. doi:10.1002/hlca.19760590626
Return to citation in text: [1] -
Dong, S.; Liu, X.; Zhang, Y.; Lin, L.; Feng, X. Org. Lett. 2011, 13, 5060. doi:10.1021/ol2018888
Return to citation in text: [1] -
Galicia, M.; González, F. J. J. Electrochem. Soc. 2002, 149, D46. doi:10.1149/1.1450616
Return to citation in text: [1] -
Shin, H. J.; Kim, T. S.; Lee, H.-S.; Park, J. Y.; Choi, I.-K.; Kwon, H. J. Phytochemistry 2008, 69, 2363. doi:10.1016/j.phytochem.2008.05.020
Return to citation in text: [1] -
Shaameri, Z.; Ali, S. H. S.; Mohamat, M. F.; Yamin, B. M.; Hamzah, A. S. J. Heterocycl. Chem. 2013, 50, 320. doi:10.1002/jhet.1078
Return to citation in text: [1]
| 1. | Wang, C.-M.; Xiao, J.-A.; Wang, J.; Wang, S.-S.; Deng, Z.-X.; Yang, H. J. Org. Chem. 2016, 81, 8001. doi:10.1021/acs.joc.6b01356 |
| 2. | Yu, X.-Y.; Chen, J.-R.; Wei, Q.; Cheng, H.-G.; Liu, Z.-C.; Xiao, W.-J. Chem. – Eur. J. 2016, 22, 6774. doi:10.1002/chem.201601227 |
| 3. | Liu, X.; Wang, Y.; Yang, D.; Zhang, J.; Liu, D.; Su, W. Angew. Chem., Int. Ed. 2016, 55, 8100. doi:10.1002/anie.201602880 |
| 4. | Choi, Y. S.; Park, S.; Park, Y. S. Eur. J. Org. Chem. 2016, 2539. doi:10.1002/ejoc.201600201 |
| 5. | Zhang, S.-Y.; Lv, M.; Yin, S.-J.; Li, N.-K.; Zhang, J.-Q.; Wang, X.-W. Adv. Synth. Catal. 2016, 358, 143. doi:10.1002/adsc.201500666 |
| 6. | De Castro, P. P.; Dos Santos, I. F.; Amarante, G. W. Curr. Org. Synth. 2016, 13, 440. doi:10.2174/1570179413999151211114542 |
| 7. | Wang, T.; Yu, Z.; Hoon, D. L.; Phee, C. Y.; Lan, Y.; Lu, Y. J. Am. Chem. Soc. 2016, 138, 265. doi:10.1021/jacs.5b10524 |
| 8. | Zhang, Z.; Sun, W.; Zhu, G.; Yang, J.; Zhang, M.; Hong, L.; Wang, R. Chem. Commun. 2016, 52, 1377. doi:10.1039/C5CC08989A |
| 9. | Izumi, S.; Kobayashi, Y.; Takemoto, Y. Org. Lett. 2016, 18, 696. doi:10.1021/acs.orglett.5b03666 |
| 10. | de Castro, P. P.; Carpanez, A. G.; Amarante, G. W. Chem. – Eur. J. 2016, 22, 10294. doi:10.1002/chem.201600071 |
| 11. | Pinheiro, D. L. J.; Ávila, E. P.; Amarante, G. W. ChemistrySelect 2016, 1, 2960. doi:10.1002/slct.201600639 |
| 12. | Zheng, Y.; Deng, L. Chem. Sci. 2015, 6, 6510. doi:10.1039/C5SC02116B |
| 13. | Zhu, C.-L.; Yang, L.-J.; Li, S.; Zheng, Y.; Ma, J.-A. Org. Lett. 2015, 17, 3442. doi:10.1021/acs.orglett.5b01450 |
| 14. | Qiao, B.; Liu, X.; Duan, S.; Yan, L.; Jiang, Z. Org. Lett. 2014, 16, 672. doi:10.1021/ol403303k |
| 15. | Pereira, A. A.; de Castro, P. P.; de Mello, A. C.; Ferreira, B. R. V.; Eberlin, M. N.; Amarante, G. W. Tetrahedron 2014, 70, 3271. doi:10.1016/j.tet.2013.11.037 |
| 16. | Alba, A.-N. R.; Rios, R. Chem. – Asian J. 2011, 6, 720. doi:10.1002/asia.201000636 |
| 17. | Hewlett, N. M.; Hupp, C. D.; Tepe, J. J. Synthesis 2009, 2825. doi:10.1055/s-0029-1216924 |
| 18. | Mosey, R. A.; Fisk, J. S.; Tepe, J. J. Tetrahedron: Asymmetry 2008, 19, 2755. doi:10.1016/j.tetasy.2008.11.033 |
| 19. | Fisk, J. S.; Mosey, R. A.; Tepe, J. J. Chem. Soc. Rev. 2007, 36, 1432. doi:10.1039/b511113g |
| 24. | Mazurkiewicz, R.; Pierwocha, A. W.; Fryczkowska, B. Pol. J. Chem. 1998, 72, 113. |
| 27. | Kobayashi, S.; Bryant, L. L., Jr.; Tsukamoto, Y.; Saegusa, T. Macromolecules 1986, 19, 1547. doi:10.1021/ma00160a012 |
| 24. | Mazurkiewicz, R.; Pierwocha, A. W.; Fryczkowska, B. Pol. J. Chem. 1998, 72, 113. |
| 25. | Taylor, L. D.; Platt, T. E.; Mach, M. H. J. Polym. Sci., Part B: Polym. Lett. 1970, 8, 537. doi:10.1002/pol.1970.110080803 |
| 26. | Slavinskaya, V. A.; Sile, D. É.; Katkevich, M. Yu.; Korchagova, É. K.; Lukevits, É. Chem. Heterocycl. Compd. 1994, 30, 724. doi:10.1007/BF01166316 |
| 20. | Pinheiro, D. L. J.; Batista, G. M. F.; Gonçalves, J. R.; Duarte, T. N.; Amarante, G. W. Eur. J. Org. Chem. 2016, 459. doi:10.1002/ejoc.201501393 |
| 21. | Ávila, E. P.; Justo, R. M. S.; Gonçalves, V. P.; Pereira, A. A.; Diniz, R.; Amarante, G. W. J. Org. Chem. 2015, 80, 590. doi:10.1021/jo5024975 |
| 22. | Ávila, E. P.; de Mello, A. C.; Diniz, R.; Amarante, G. W. Eur. J. Org. Chem. 2013, 1881. doi:10.1002/ejoc.201300076 |
| 23. | Melhado, A. D.; Amarante, G. W.; Wang, Z. J.; Luparia, M.; Toste, F. D. J. Am. Chem. Soc. 2011, 133, 3517. doi:10.1021/ja1095045 |
| 27. | Kobayashi, S.; Bryant, L. L., Jr.; Tsukamoto, Y.; Saegusa, T. Macromolecules 1986, 19, 1547. doi:10.1021/ma00160a012 |
| 31. | Shin, H. J.; Kim, T. S.; Lee, H.-S.; Park, J. Y.; Choi, I.-K.; Kwon, H. J. Phytochemistry 2008, 69, 2363. doi:10.1016/j.phytochem.2008.05.020 |
| 30. | Galicia, M.; González, F. J. J. Electrochem. Soc. 2002, 149, D46. doi:10.1149/1.1450616 |
| 32. | Shaameri, Z.; Ali, S. H. S.; Mohamat, M. F.; Yamin, B. M.; Hamzah, A. S. J. Heterocycl. Chem. 2013, 50, 320. doi:10.1002/jhet.1078 |
| 29. | Dong, S.; Liu, X.; Zhang, Y.; Lin, L.; Feng, X. Org. Lett. 2011, 13, 5060. doi:10.1021/ol2018888 |
| 28. | Gakis, N.; Märky, M.; Hansen, H.-J.; Heimgartner, H.; Schmid, H.; Oberhänsli, W. E. Helv. Chim. Acta 1976, 59, 2149. doi:10.1002/hlca.19760590626 |
| 24. | Mazurkiewicz, R.; Pierwocha, A. W.; Fryczkowska, B. Pol. J. Chem. 1998, 72, 113. |
© 2017 Pinheiro et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)