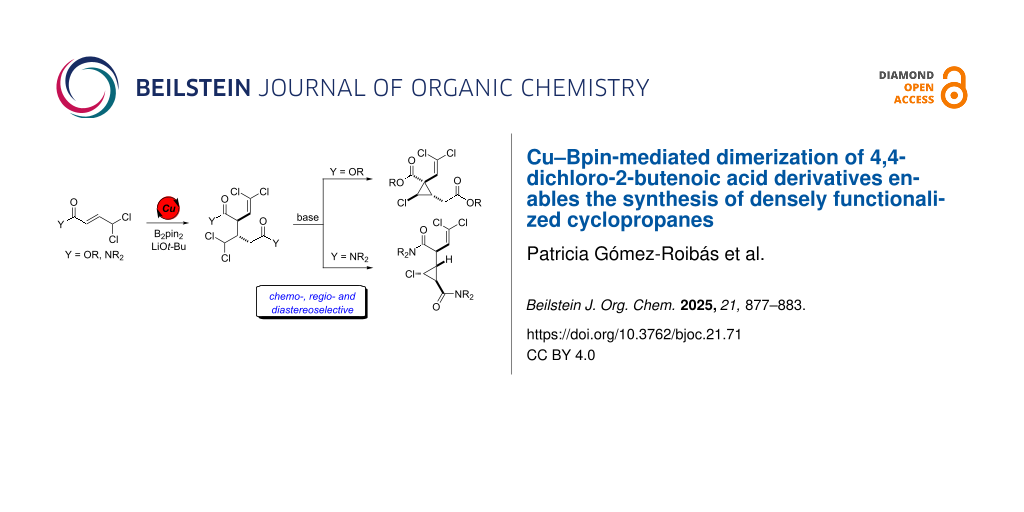
Abstract
4,4-Dichloro-2-butenoic acid derivatives are shown to undergo a rare dimerization process when reacted with bis(pinacolato)diboron under copper catalysis. The reaction provides densely functionalized products with excellent levels of chemo-, regio-, and diastereoselectivity. This high degree of functionalization makes these products versatile building blocks for the stereoselective synthesis of chlorocyclopropanes.

Graphical Abstract
Introduction
In the last years our group has been focused on the development of catalytic methodologies for the carboboration of unsaturated hydrocarbons [1-7]. In the course of our investigation of the copper-catalyzed borylative coupling of alkynes with allylic gem-dichlorides [3], we observed that alkyl 4,4-dichloro-2-butenoates deviated from the general reactivity trend. While allylic gem-dichlorides bearing aromatic and aliphatic substituents efficiently provided the allylboration product (Scheme 1a), the use of ester derivative 1 under same reaction conditions led to the formation of an unexpected product arising from the coupling of two dichloride molecules with no alkyne incorporation (Scheme 1b). We have studied this reaction and now report a catalytic methodology for the diastereoselective synthesis of these dimeric structures. The high degree of functionalization present in these molecules, which feature two ester groups, an aliphatic gem-dichloride and a dichloroalkene unit, offers ample opportunities for further functionalization. This is illustrated by their chemo- and diastereoselective conversion into densely functionalized cyclopropanes (Scheme 1c).
![[1860-5397-21-71-i1]](/bjoc/content/inline/1860-5397-21-71-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Chemodivergent reactivity observed in copper-catalyzed borylative couplings of allylic gem-dichlorides.
Scheme 1: Chemodivergent reactivity observed in copper-catalyzed borylative couplings of allylic gem-dichlori...
Results and Discussion
Based on our preliminary result, we started our study by exploring the reaction between ethyl 4,4-dichloro-2-butenoate (1) and B2pin2 (Table 1). By using NaOt-Bu as base and toluene as solvent, the Cu/SIMes catalyst provided compound 2 as single reaction product, albeit in low yield and with low diastereoselectivity (Table 1, entry 1). Lowering the amount of B2pin2 to 1 equivalent was found to be beneficial (Table 1, entry 2), although the use of sub-stoichiometric amounts led to a significant decrease in reaction yield (Table 1, entry 3). We also tried to reduce the amount of base, but this caused a drop in the reaction efficiency (Table 1, entry 4). Evaluation of different bases demonstrated the important role of the base metal cation. Gratifyingly, we observed that the use of LiOt-Bu led to the formation of product 2 as a single diastereomer (Table 1, entry 5). A slightly lower diastereoselectivity was observed when KOt-Bu was used, which also gave rise to 2 in diminished yield (Table 1, entry 6). The nature of the solvent also played a role in the reaction outcome. A decrease both in efficiency and diastereoselectivity was observed when THF was used (Table 1, entry 7). The use of dichloromethane eroded the diastereoselectivity and also the chemoselectivity as shown with the additional formation of 3 as a mixture of Z:E isomers (Table 1, entry 8). Having identified the proper combination of base and solvent, we then screened different copper catalysts. Different NHCs, bisphosphines and phosphines were tested (Table 1, entries 9–14) and excellent chemo- and diastereoselectivity was observed in all cases, with SIPr providing the best result (Table 1, entry 11). Under these optimized conditions, product 2 was isolated in 60% yield as a single diastereomer. The relative configuration of 2 was determined by two-dimensional NMR analysis (see Supporting Information File 1 for details).
Table 1: Optimization studies.
![[Graphic 1]](/bjoc/content/inline/1860-5397-21-71-i6.svg?max-width=637&scale=1.0)
|
|||||
| Entrya | Base | B2pin2 (equiv) | Ligand | Yield 2b | 2 drc |
| 1 | NaOt-Bu | 2.0 | SIMes | 34 | 73:27 |
| 2 | NaOt-Bu | 1.0 | SIMes | 56 | 70:30 |
| 3 | NaOt-Bu | 0.5 | SIMes | 8 | n.d. |
| 4 | NaOt-Bud | 1.0 | SIMes | 22 | 72:28 |
| 5 | LiOt-Bu | 1.0 | SIMes | 49 | >95:5 |
| 6 | KOt-Bu | 1.0 | SIMes | 27 | 92:8 |
| 7e | LiOt-Bu | 1.0 | SIMes | 20 | 93:7 |
| 8f | LiOt-Bu | 1.0 | SIMes | 41g | 92:8 |
| 9 | LiOt-Bu | 1.0 | IMes | 35 | >95:5 |
| 10 | LiOt-Bu | 1.0 | IPr | 50 | >95:5 |
| 11 | LiOt-Bu | 1.0 | SIPr | 60 | >95:5 |
| 12 | LiOt-Bu | 1.0 | DPEphos | 57 | >95:5 |
| 13 | LiOt-Bu | 1.0 | Xantphos | 39 | >95:5 |
| 14 | LiOt-Bu | 1.0 | PCy3 | 51 | >95:5 |
aReactions run on a 0.2 mmol scale. bYield of isolated product. cDiastereomeric ratio determined by GC analysis of reaction crude (structure of major diastereomer shown). d1.0 equiv of NaOt-Bu. eTHF used as solvent. fCH2Cl2 used as solvent. gProduct 3 was also obtained in 15% yield as a 1:1 mixture of Z:E isomers.
This transformation could be efficiently applied to the dimerization of other 4,4-dichloro-2-butenoates and 4,4-dichloro-2-butenamides. The corresponding products 8 and 9 were obtained in good yield and excellent diastereoselectivity (Scheme 2). In sharp contrast, the use of gem-dichlorides bearing a ketone group did not result in the formation of the dimerization product and complex mixtures of products were observed in these cases.
![[1860-5397-21-71-i2]](/bjoc/content/inline/1860-5397-21-71-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Cu-Bpin-mediated dimerization of 4,4-dichoro-2-butenoic acid derivatives.
Scheme 2: Cu-Bpin-mediated dimerization of 4,4-dichoro-2-butenoic acid derivatives.
To gather insight into the reaction mechanism, several control experiments were performed (Scheme 3).
![[1860-5397-21-71-i3]](/bjoc/content/inline/1860-5397-21-71-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
We first observed that the reaction does not take place in the absence of B2pin2 (Scheme 3a). Based on the well accepted metathesis reaction of Cu(I) alkoxides with B2pin2 and the reactivity of the resulting Cu–Bpin complex towards α,β-unsaturated esters and hydrocarbons [8-15], we hypothesized that the first step of the reaction may deal with the insertion of the copper–boron bond into 1. The dual functionality of this substrate imposed a question related to the regioselectivity of the Cu–Bpin insertion since it can potentially behave as an α,β-unsaturated ester or an allylic substrate [16-19]. To shed some light into this issue, we ran the reaction in the presence of MeOH in order to trap the potential copper intermediate by protonation. When 2 equiv of MeOH were used, we still obtained the dimerization product 2. Nevertheless, when a catalytic amount of base was used, we only observed the formation of β-borylation product 12 (Scheme 3b). This result suggests that Cu–Bpin insertion into 1 generates a copper enolate which may engage in further steps for the formation of the dimerization product. The presence of the two chlorine atoms was found to be key for the outcome of the reaction. No dimerization product was observed when the reaction was carried out under standard conditions with ethyl crotonate derivatives bearing a methyl group, hydrogen or bromine atoms at the γ position. Either β-borylation or decomposition products were obtained in those cases (Scheme 3c).
On the basis of our experimental results, we propose the following mechanism for the copper-catalyzed diastereoselective dimerization of 4,4-dichoro-2-butenoic acid derivatives (Scheme 4). Initially, the LCu–pin complex generated through reaction between LCu–Ot-Bu and B2pin2 undergoes coordination and regioselective insertion into 1 giving rise to β-borylated organocopper species A which is in equilibrium with the Cu–O enolate B [11]. In the presence of excess of LiOt-Bu, a salt metathesis reaction between this base and intermediate B generates lithium enolate C and LCuOt-Bu to close the copper catalytic cycle. The formation of a lithium enolate is consistent with the different diastereoselectivity observed when other bases featuring different metal cations were used (Table 1, entry 2 vs entry 5), and the absence of any significant stereochemical influence from the copper complex (Table 1, entries 9–14). Lithium enolate C would then undergo a diastereoselective conjugate addition to a second molecule of 1. Given the negative results observed for other crotonate derivatives (Scheme 3c), coordination between the Li cation and the two chlorine atoms via proposed transition state D may be crucial not only for diastereoselective control but also for the viability of this step. Finally, the new enolate E evolves through intramolecular proton abstraction and elimination of boryllithium [20,21]. The formation of side product 3 observed when dichloromethane was used as a solvent could be explained by protonation of intermediate A, followed by transmetalation of the resulting organoboron compound with CuOt-Bu and subsequent SN2’-selective allylic alkylation of 1.
![[1860-5397-21-71-i4]](/bjoc/content/inline/1860-5397-21-71-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed mechanism for the Cu-catalyzed dimerization of 4,4-dichoro-2-butenoic acid derivatives.
Scheme 4: Proposed mechanism for the Cu-catalyzed dimerization of 4,4-dichoro-2-butenoic acid derivatives.
The densely functionalized structure of these dimerization products offers a versatile synthetic handle for further chemoselective functionalization. Considering the presence of two enolizable esters together with the aliphatic gem-dichloride, we explored the feasibility of a base-mediated formation of chlorocyclopropanes (Table 2).
Table 2: Synthesis of densely functionalized (2,2-dichlorovinyl)cyclopropanes by base-promoted intramolecular cyclization.
![[Graphic 2]](/bjoc/content/inline/1860-5397-21-71-i7.svg?max-width=637&scale=1.0)
|
|||||
| Entrya | Y | Base | T (°C) | Product, yield (%)b | drc |
| 1 | OEt | CsF | 70 | 16, 80 | 50:50 |
| 2 | OEt | Cs2CO3 | 70 | 16, 55 | 61:39 |
| 3 | OEt | TBAF | 70 | 16, 32d | 80:20 |
| 4 | OEt | TBAF | 50 | 16, 70 | 83:17 |
| 5 | Ot-Bu | TBAF | 50 | 17, 61 | 81:19 |
| 6 | N(Me)(OMe) | TBAF | 50 | 18, – | – |
aReactions run on a 0.1 mmol scale. bYield of isolated product. cDiastereomeric ratio determined by 1H NMR analysis of reaction crude. dProduct 19 was also obtained in 23% yield.
Evaluation of bases such as metal tert-butoxides, phosphates, acetates, and organic amines resulted in either low conversion or decomposition of 2 (see Supporting Information File 1 for details). In contrast, the use of CsF in dioxane at 70 °C proved to be efficient and selectively provided cyclopropane 9 in good yield, albeit with no diastereoselectivity (Table 2, entry 1). Cs2CO3 was also selective for this cyclization and provided a slight increase in diastereoselectivity, although still far from satisfactory (Table 2, entry 2). A major improvement was observed when TBAF was used as base. At 70 °C, product 16 was obtained with good diastereoselectivity (80:20 dr), although in low yield mainly due to the formation of side product 19. However, 16 could be obtained as a single product in 70% yield with 83:17 dr by decreasing the reaction temperature to 50 °C (Table 2, entry 4). Under the same optimal conditions, compound 8 could be transformed into cyclopropane 17 in 61% yield with 81:19 dr (Table 2, entry 5). It is important to note that (2,2-dichlorovinyl)cyclopropanes represent an important class of compounds present in a range of bioactive compounds such as permethrin or alpha-cypermethrin, which are commonly used as insecticides [22,23]. The present transformation provides access to densely functionalized (2,2-dichlorovinyl)cyclopropanes, thus representing a potential platform for the synthetic diversification on these important scaffolds.
Surprisingly, when the TBAF-mediated cyclization was attempted on bisamide 9 the corresponding cyclopropane 18 was not formed (Table 2, entry 6). The use of other mild bases at different temperatures also resulted unproductive. However, when the reaction was carried out with KOt-Bu, we observed the selective conversion of 9 into product 20 featuring a different cyclopropane scaffold. Slight modification of the reaction conditions allowed us to obtain product 20 in 56% yield as a single diastereomer (Scheme 5a). Taking advantage of the twofold utility of KOt-Bu, we explored the formation of this new cyclopropane structure directly from gem-dichloride 5. By using this base under standard conditions, product 20 was selectively obtained in similar yield, albeit with diminished diastereoselectivity (Scheme 5b).
![[1860-5397-21-71-i5]](/bjoc/content/inline/1860-5397-21-71-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: a) KOt-Bu-mediated intramolecular cyclization of 9. b) Direct formation of cyclopropane 20 from gem-dichloride 5 using KOt-Bu as base.
Scheme 5: a) KOt-Bu-mediated intramolecular cyclization of 9. b) Direct formation of cyclopropane 20 from gem...
Conclusion
In summary, we have discovered an unanticipated Cu–Bpin-promoted diastereoselective dimerization of 4,4-dichloro-2-butenoic acid derivatives. The reaction occurs via initial Cu–Bpin insertion followed by keto–enol isomerization and salt metathesis to generate a lithium enolate which is then trapped by a second molecule of the 4,4-dichloro-2-butenoic acid derivative. We have observed that the use of lithium as base metal cation is key to achieve excellent levels of diastereoselectivity. Our study also highlights, how the dimerization products can be selectively converted into different densely functionalized cyclopropane scaffolds depending on the nature of the carboxylic acid derivative.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data and copies of NMR spectra. | ||
| Format: PDF | Size: 2.5 MB | Download |
Funding
Financial support from the AEI (PID2020-118237RB-I00), European Research Council (863914), Xunta de Galicia (ED431C 2022/27; Centro de investigación do Sistema universitario de Galicia accreditation 2023–2027, ED431G 2023/03) and the European Regional Development Fund (ERDF) is gratefully acknowledged.
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Rivera‐Chao, E.; Fañanás‐Mastral, M. Angew. Chem., Int. Ed. 2018, 57, 9945–9949. doi:10.1002/anie.201806334
Return to citation in text: [1] -
Rivera‐Chao, E.; Mitxelena, M.; Varela, J. A.; Fañanás‐Mastral, M. Angew. Chem., Int. Ed. 2019, 58, 18230–18234. doi:10.1002/anie.201910707
Return to citation in text: [1] -
Chaves-Pouso, A.; Álvarez-Constantino, A. M.; Fañanás-Mastral, M. Angew. Chem., Int. Ed. 2022, 61, e202117696. doi:10.1002/anie.202117696
Return to citation in text: [1] [2] -
Pérez-Saavedra, B.; Velasco-Rubio, Á.; Rivera-Chao, E.; Varela, J. A.; Saá, C.; Fañanás-Mastral, M. J. Am. Chem. Soc. 2022, 144, 16206–16216. doi:10.1021/jacs.2c07969
Return to citation in text: [1] -
Piñeiro-Suárez, M.; Álvarez-Constantino, A. M.; Fañanás-Mastral, M. ACS Catal. 2023, 13, 5578–5583. doi:10.1021/acscatal.3c00536
Return to citation in text: [1] -
Álvarez‐Constantino, A. M.; Chaves‐Pouso, A.; Fañanás‐Mastral, M. Angew. Chem., Int. Ed. 2024, 63, e202407813. doi:10.1002/anie.202407813
Return to citation in text: [1] -
Vázquez-Galiñanes, N.; Sciortino, G.; Piñeiro-Suárez, M.; Tóth, B. L.; Maseras, F.; Fañanás-Mastral, M. J. Am. Chem. Soc. 2024, 146, 21977–21988. doi:10.1021/jacs.4c07188
Return to citation in text: [1] -
Ito, H.; Yamanaka, H.; Tateiwa, J.-i.; Hosomi, A. Tetrahedron Lett. 2000, 41, 6821–6825. doi:10.1016/s0040-4039(00)01161-8
Return to citation in text: [1] -
Takahashi, K.; Ishiyama, T.; Miyaura, N. J. Organomet. Chem. 2001, 625, 47–53. doi:10.1016/s0022-328x(00)00826-3
Return to citation in text: [1] -
Mun, S.; Lee, J.-E.; Yun, J. Org. Lett. 2006, 8, 4887–4889. doi:10.1021/ol061955a
Return to citation in text: [1] -
Dang, L.; Lin, Z.; Marder, T. B. Organometallics 2008, 27, 4443–4454. doi:10.1021/om8006294
Return to citation in text: [1] [2] -
Lillo, V.; Bonet, A.; Fernández, E. Dalton Trans. 2009, 2899–2908. doi:10.1039/b819237e
Return to citation in text: [1] -
Neeve, E. C.; Geier, S. J.; Mkhalid, I. A. I.; Westcott, S. A.; Marder, T. B. Chem. Rev. 2016, 116, 9091–9161. doi:10.1021/acs.chemrev.6b00193
Return to citation in text: [1] -
Hemming, D.; Fritzemeier, R.; Westcott, S. A.; Santos, W. L.; Steel, P. G. Chem. Soc. Rev. 2018, 47, 7477–7494. doi:10.1039/c7cs00816c
Return to citation in text: [1] -
Das, K. K.; Mahato, S.; Hazra, S.; Panda, S. Chem. Rec. 2022, 22, e202100290. doi:10.1002/tcr.202100290
Return to citation in text: [1] -
Ito, H.; Kawakami, C.; Sawamura, M. J. Am. Chem. Soc. 2005, 127, 16034–16035. doi:10.1021/ja056099x
Return to citation in text: [1] -
Guzman-Martinez, A.; Hoveyda, A. H. J. Am. Chem. Soc. 2010, 132, 10634–10637. doi:10.1021/ja104254d
Return to citation in text: [1] -
Gao, P.; Yuan, C.; Zhao, Y.; Shi, Z. Chem 2018, 4, 2201–2211. doi:10.1016/j.chempr.2018.07.003
Return to citation in text: [1] -
Akiyama, S.; Kubota, K.; Mikus, M. S.; Paioti, P. H. S.; Romiti, F.; Liu, Q.; Zhou, Y.; Hoveyda, A. H.; Ito, H. Angew. Chem., Int. Ed. 2019, 58, 11998–12003. doi:10.1002/anie.201906283
Return to citation in text: [1] -
Segawa, Y.; Yamashita, M.; Nozaki, K. Science 2006, 314, 113–115. doi:10.1126/science.1131914
Return to citation in text: [1] -
Segawa, Y.; Suzuki, Y.; Yamashita, M.; Nozaki, K. J. Am. Chem. Soc. 2008, 130, 16069–16079. doi:10.1021/ja8057919
Return to citation in text: [1] -
Li, N.; Xu, H.-H.; Liu, Z.-Y.; Yang, Z.-H. J. Photochem. Photobiol., B 2009, 96, 170–177. doi:10.1016/j.jphotobiol.2009.06.002
Return to citation in text: [1] -
Walker, K. J.; Williams, C. T.; Oladepo, F. O.; Lucas, J.; Malone, D.; Paine, M. J. I.; Ismail, H. M. Sci. Rep. 2022, 12, 9715. doi:10.1038/s41598-022-13768-z
Return to citation in text: [1]
| 1. | Rivera‐Chao, E.; Fañanás‐Mastral, M. Angew. Chem., Int. Ed. 2018, 57, 9945–9949. doi:10.1002/anie.201806334 |
| 2. | Rivera‐Chao, E.; Mitxelena, M.; Varela, J. A.; Fañanás‐Mastral, M. Angew. Chem., Int. Ed. 2019, 58, 18230–18234. doi:10.1002/anie.201910707 |
| 3. | Chaves-Pouso, A.; Álvarez-Constantino, A. M.; Fañanás-Mastral, M. Angew. Chem., Int. Ed. 2022, 61, e202117696. doi:10.1002/anie.202117696 |
| 4. | Pérez-Saavedra, B.; Velasco-Rubio, Á.; Rivera-Chao, E.; Varela, J. A.; Saá, C.; Fañanás-Mastral, M. J. Am. Chem. Soc. 2022, 144, 16206–16216. doi:10.1021/jacs.2c07969 |
| 5. | Piñeiro-Suárez, M.; Álvarez-Constantino, A. M.; Fañanás-Mastral, M. ACS Catal. 2023, 13, 5578–5583. doi:10.1021/acscatal.3c00536 |
| 6. | Álvarez‐Constantino, A. M.; Chaves‐Pouso, A.; Fañanás‐Mastral, M. Angew. Chem., Int. Ed. 2024, 63, e202407813. doi:10.1002/anie.202407813 |
| 7. | Vázquez-Galiñanes, N.; Sciortino, G.; Piñeiro-Suárez, M.; Tóth, B. L.; Maseras, F.; Fañanás-Mastral, M. J. Am. Chem. Soc. 2024, 146, 21977–21988. doi:10.1021/jacs.4c07188 |
| 11. | Dang, L.; Lin, Z.; Marder, T. B. Organometallics 2008, 27, 4443–4454. doi:10.1021/om8006294 |
| 16. | Ito, H.; Kawakami, C.; Sawamura, M. J. Am. Chem. Soc. 2005, 127, 16034–16035. doi:10.1021/ja056099x |
| 17. | Guzman-Martinez, A.; Hoveyda, A. H. J. Am. Chem. Soc. 2010, 132, 10634–10637. doi:10.1021/ja104254d |
| 18. | Gao, P.; Yuan, C.; Zhao, Y.; Shi, Z. Chem 2018, 4, 2201–2211. doi:10.1016/j.chempr.2018.07.003 |
| 19. | Akiyama, S.; Kubota, K.; Mikus, M. S.; Paioti, P. H. S.; Romiti, F.; Liu, Q.; Zhou, Y.; Hoveyda, A. H.; Ito, H. Angew. Chem., Int. Ed. 2019, 58, 11998–12003. doi:10.1002/anie.201906283 |
| 8. | Ito, H.; Yamanaka, H.; Tateiwa, J.-i.; Hosomi, A. Tetrahedron Lett. 2000, 41, 6821–6825. doi:10.1016/s0040-4039(00)01161-8 |
| 9. | Takahashi, K.; Ishiyama, T.; Miyaura, N. J. Organomet. Chem. 2001, 625, 47–53. doi:10.1016/s0022-328x(00)00826-3 |
| 10. | Mun, S.; Lee, J.-E.; Yun, J. Org. Lett. 2006, 8, 4887–4889. doi:10.1021/ol061955a |
| 11. | Dang, L.; Lin, Z.; Marder, T. B. Organometallics 2008, 27, 4443–4454. doi:10.1021/om8006294 |
| 12. | Lillo, V.; Bonet, A.; Fernández, E. Dalton Trans. 2009, 2899–2908. doi:10.1039/b819237e |
| 13. | Neeve, E. C.; Geier, S. J.; Mkhalid, I. A. I.; Westcott, S. A.; Marder, T. B. Chem. Rev. 2016, 116, 9091–9161. doi:10.1021/acs.chemrev.6b00193 |
| 14. | Hemming, D.; Fritzemeier, R.; Westcott, S. A.; Santos, W. L.; Steel, P. G. Chem. Soc. Rev. 2018, 47, 7477–7494. doi:10.1039/c7cs00816c |
| 15. | Das, K. K.; Mahato, S.; Hazra, S.; Panda, S. Chem. Rec. 2022, 22, e202100290. doi:10.1002/tcr.202100290 |
| 3. | Chaves-Pouso, A.; Álvarez-Constantino, A. M.; Fañanás-Mastral, M. Angew. Chem., Int. Ed. 2022, 61, e202117696. doi:10.1002/anie.202117696 |
| 22. | Li, N.; Xu, H.-H.; Liu, Z.-Y.; Yang, Z.-H. J. Photochem. Photobiol., B 2009, 96, 170–177. doi:10.1016/j.jphotobiol.2009.06.002 |
| 23. | Walker, K. J.; Williams, C. T.; Oladepo, F. O.; Lucas, J.; Malone, D.; Paine, M. J. I.; Ismail, H. M. Sci. Rep. 2022, 12, 9715. doi:10.1038/s41598-022-13768-z |
| 20. | Segawa, Y.; Yamashita, M.; Nozaki, K. Science 2006, 314, 113–115. doi:10.1126/science.1131914 |
| 21. | Segawa, Y.; Suzuki, Y.; Yamashita, M.; Nozaki, K. J. Am. Chem. Soc. 2008, 130, 16069–16079. doi:10.1021/ja8057919 |
© 2025 Gómez-Roibás et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.