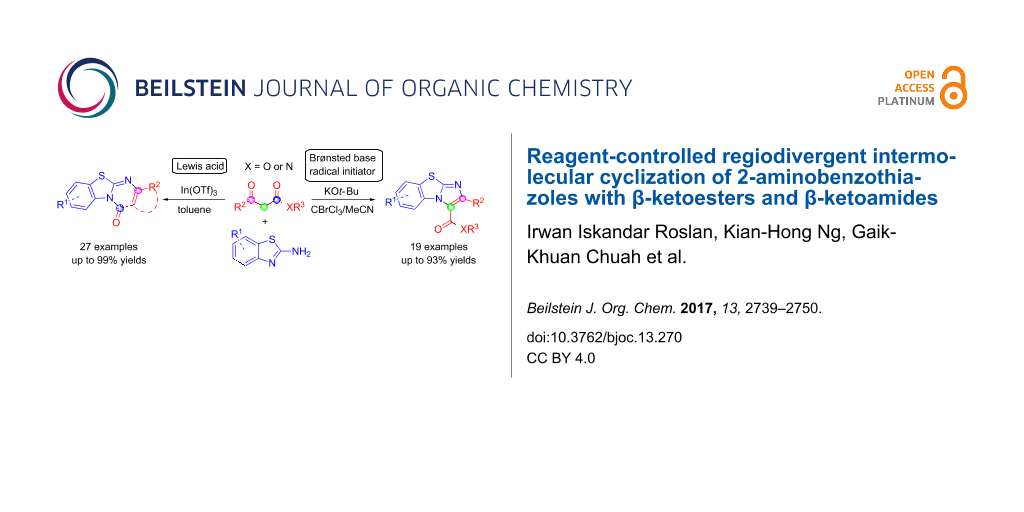
Abstract
Two regiodivergent approaches to intermolecular cyclization of 2-aminobenzothiazoles with β-ketoesters and amides have been developed, controlled by the reagents employed. With the Brønsted base KOt-Bu and CBrCl3 as radical initiator, benzo[d]imidazo[2,1-b]thiazoles are synthesized via attack at the α-carbon and keto carbon of the β-ketoester moiety. In contrast, switching to the Lewis acid catalyst, In(OTf)3, results in the regioselective nucleophilic attack at both carbonyl groups forming benzo[4,5]thiazolo[3,2-a]pyrimidin-4-ones instead.

Graphical Abstract
Introduction
β-Ketoesters are versatile substrates frequently used in heterocyclic synthesis, having both electrophilic keto and ester moieties as well as a nucleophilic α-carbon. They can act as dinucleophiles [1-4], dielectrophiles [5-7] or ambiphiles [8,9] in the presence of their complementary coupling partners. Furthermore, they can be prefunctionalized [10-13] with leaving groups, thus switching to an electrophile [14-16], or convert to an α-radical carbon with an oxidant [17-19]. β-Ketoesters are also inexpensive, abundant and commercially available, making them attractive substrates.
In our continuing effort to develop green and atom-efficient protocols, we have employed transition-metal-free approaches for the one-pot synthesis of imidazo[1,2-a]pyridines and thiazolamines by coupling β-ketoesters or their derivatives, phenylacetones and phenylacetophenones, with aminopyridines [20,21] and thioureas [22]. The strategy involves in situ bromination of the α-carbon using CBrCl3 as the Br source. This in situ halogenation strategy has been employed for the synthesis of quinoxalines [23], oxazoles [24,25], pyrido[1,2-a]benzimidazoles [26], imidazo[1,2-a]pyridines [27-30], thiazoles [31,32] and benzothiazoles [33,34]. With weak bicarbonate bases, direct bromination of the α-carbon does not occur. Instead, the Br is shuttled to the α-carbon by its coupling partner. With this tandem bromination and cyclization strategy, there is no need to presynthesize substrates, thus reducing the number of synthetic steps, time, chemicals and wastes. Here we describe the extension of this α-bromination shuttle system to 2-aminobenzothiazoles as substrates to synthesize benzo[d]imidazo[2,1-b]thiazoles.
The benzo[d]imidazo[2,1-b]thiazole backbone is found in many bioactive molecules and pharmaceutical compounds as evident by its use as antimicrobial [35,36], antitumor [37-39], antibacterial [40], and anti-allergic agents [41]. In addition, compounds with this backbone are employed as kinase inhibitors and receptors [42-44] and as a tracer for PET imaging of β-amyloid plaques [45,46]. The conventional approach for the construction of benzo[d]imidazo[2,1-b]thiazole is the condensation of 2-aminobenzothiazole with α-halo or tosyloxy ketone [47,48]. The requirement for prior functionalization of the ketone moiety is a drawback, and several more direct methods have been developed in recent years [49-52]. Zhang et al. coupled various 1,2-dihaloarenes with 2-mercaptobenzimidazole in a nucleophilic aromatic substitution reaction [53]. Zhu’s group used Cu salts as a promoter for the cycloaddition of isocyanides with benzothiazoles [54]. Benzo[d]imidazo[2,1-b]thiazoles have also been synthesized via coupling of 2-aminobenzothiazole with acetophenones [55] or aldehydes and nitroalkanes [56].
Because the bicyclic structure of 2-aminobenzothiazole is more stable than aminopyridines and thioureas, the nucleophilicity of the aromatic N atom is reduced. A stronger base, KOt-Bu, is therefore needed for in situ bromination to form the benzo[d]imidazo[2,1-b]thiazole derivatives via coupling of 2-aminobenzothiazole with the brominated β-ketoesters and amides. Over the past decade, there has been a lot of interest in KOt-Bu-mediated synthesis, especially after Itami’s group showed that KOt-Bu provides a metal-free approach to the traditional Pd-catalyzed aryl–heteroaryl coupling to biaryls [57]. Since then, KOt-Bu has been used as a mediator for various reactions including aryl–aryl coupling [58-63], inter- and intramolecular cyclizations [64-68], amidation [69], alkenylation [70], oxidation [71] and silylation [72]. In these reactions, the single electron transfer (SET) is initiated by KOt-Bu/DMF [63,67,69,71] or KOt-Bu in combination with additives such as bidentate diamine ligands [61-65], 18-crown-6 [70] or azobisisobutyronitrile (AIBN) [62,66].
Herein, we report the synthesis of benzo[d]imidazo[2,1-b]thiazoles via an in situ bromination strategy using a KOt-Bu/CBrCl3 system (Scheme 1a). By employing a base with radical susceptibility, the reaction mechanism is expected to differ substantially from that using bicarbonate bases which operates via the α-Br shuttle mechanism [20-22]. Interestingly, by replacing the radical initiator and Brønsted base system with a Lewis acid catalyst, benzo[4,5]thiazolo[3,2-a]pyrimidin-4-ones were formed instead (Scheme 1b). This highlights the versatility of β-ketoesters where the regioselectivity of the reaction is directed by the nature of the reagents.
![[1860-5397-13-270-i1]](/bjoc/content/inline/1860-5397-13-270-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Two different intermolecular cyclization pathways controlled by reagents used.
Scheme 1: Two different intermolecular cyclization pathways controlled by reagents used.
Results and Discussion
Optimization studies were carried out by reacting 1.2 mmol of 2-aminobenzothiazole (1a) with 1.0 mmol of methyl acetoacetate (2a) in the presence of 2 equivalents of base using a solvent mixture of CBrCl3/MeCN under reflux. After 16 h, the desired product 3a was not formed with KHCO3 (Table 1, entry 1). Instead, the N-acetylated side product 4 was obtained in moderate yields, together with trace amounts of the benzo[4,5]thiazolo[3,2-a]pyrimidin-4-one side product 5a. Even after switching to the stronger bases K2CO3 and K3PO4, only poor yields of 3a were obtained (Table 1, entries 2 and 3). These results are not surprising as the substrate 1a is a weaker dinucleophile than aminopyridine and thiourea used previously [20-22]. However, 56% yield of 3a was obtained with KOH (Table 1, entry 4). The use of KOH as a base in the α-bromination of 1,3-dicarbonyl compounds has been reported previously by Sasson’s group [73]. We propose that with a strong base like KOH, direct α-bromination of 2a occurs instead of relaying the Br through an α-bromination shuttle as observed in our previous studies [20-22]. Other strong bases like NaH and KOEt also proceeded with moderate yields of 3a (Table 1, entries 5 and 6). Gratifyingly, an even higher yield of 3a, 86%, was obtained with KOt-Bu (Table 1, entry 7).
Table 1: Optimization parameters for the synthesis of benzo[d]imidazo[2,1-b]thiazoles 3a.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-270-i5.svg?max-width=637&scale=1.0)
|
||||
| Entry | Base | Solvent | Yield of 3a (%)b,c | Yield of 4 (%)b,c |
| 1 | KHCO3 | MeCN | 0 | 40 (31) |
| 2 | K2CO3 | MeCN | trace | 45 (37) |
| 3 | K3PO4 | MeCN | 5 | 53 (46) |
| 4 | KOH | MeCN | 56 (48) | 31 |
| 5 | NaH | MeCN | 69 (63) | 20 |
| 6 | KOEt | MeCN | 61 (53) | 22 |
| 7 | KOt-Bu | MeCN | 86 (84) | 14 |
| 8d | KOt-Bu | DMF | 77 (75) | 5 |
| 9d | KOt-Bu | DMAc | 74 (70) | 7 |
| 10 | KOt-Bu | EtOAc | 33 (22) | 38 |
| 11 | KOt-Bu | DCE | 9 | 24 |
| 12d | KOt-Bu | toluene | trace | 44 (37) |
| 13e | KOt-Bu | MeCN | 89 (86) | 11 |
| 14f | KOt-Bu | MeCN | 65 (58) | 23 |
| 15g | KOt-Bu | MeCN | 78 (74) | 22 |
| 16h | KOt-Bu | MeCN | 46 (35) | 6 |
aReaction conditions: 1a (1.2 mmol), 2a (1.0 mmol), base (2.0 equiv), in 3 mL of 1:9 (v/v) CBrCl3/solvent (3 mmol CBrCl3) for 16 h. bYield (from GC) with respect to 2a, using biphenyl as an internal standard. cIsolated yields in parenthesis. dReaction conducted at 80 °C. e5.0 and f2.0 mmol of CBrCl3 was used. g2.0 mmol of 1a used. h1.0 mmol of 1a and 2.0 mmol of 2a used.
Next, various solvents besides MeCN were investigated. The polar aprotic solvents DMF and DMAc were suitable albeit with slightly lower yields of 3a (Table 1, entries 8 and 9). On the other hand, ethyl acetate and 1,2-dichloroethane fared poorly whereas toluene gave only the N-acetylated side product 4 (Table 1, entries 10–12). Unfortunately, further attempts to increase the yields of 3a, including increasing the equivalents of CBrCl3, 1a and 2a proved futile (Table 1, entries 13–16).
Hence, exploring the scope of the reaction was carried out using 1.0 mmol of methyl acetoacetate 2a with 1.2 mmol of CBrCl3/MeCN solvent mixture at refluxing conditions for 16 h. A wide array of 2-aminobenzothiazoles was screened (Scheme 2). Good to excellent yields were obtained with 2-aminobenzothiazoles bearing electron donating methyl, dimethyl or methoxy groups (Scheme 2, 3b–d). Halogen substituents F, Cl and Br were well tolerated under the optimized conditions. 2-Aminobenzothiazoles with an ester moiety also proceeded with good yields (Scheme 2, 3h). However, only trace amounts of 3i were obtained for 2-aminobenzothiazoles bearing the strongly electron-withdrawing CF3 group. It was encouraging to see both that the benzoxazole and thiazole derivatives reacted well with their respective β-ketoester coupling partners to form 3j and 3k with 84% and 92% yields, respectively.
![[1860-5397-13-270-i2]](/bjoc/content/inline/1860-5397-13-270-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of reaction. Reaction conditions: 1 (1.2 mmol), 2 (1.0 mmol), KOt-Bu (2 mmol), in 3 mL CBrCl3/MeCN 1:9 (v/v) under reflux for 16 h.
Scheme 2: Scope of reaction. Reaction conditions: 1 (1.2 mmol), 2 (1.0 mmol), KOt-Bu (2 mmol), in 3 mL CBrCl3...
A variety of β-ketoesters and β-ketoamides were tested using the optimized conditions. Alkyl acetoacetates, including pentyl, isopropyl, tert-butyl and methoxyethyl, reacted smoothly with excellent yields (Scheme 2, 3l–3o). β-Ketoamides were well tolerated (Scheme 2, 3h, 3p and 3q) while good yields were obtained with β-ketoesters containing n-propyl and n-butyl moieties (Scheme 2, 3r and 3s). However, significantly lower yields were obtained with the sterically more demanding iPr group (Scheme 2, 3t). Unfortunately, β-diketone (acetylacetone) was not suitable for the reaction as only trace amounts of 3u were formed, with the bulk of the substrate being converted to the N-acylated side product 4 instead.
As trace amounts of benzo[4,5]thiazolo[3,2-a]pyrimidin-4-ones, 5a, were observed in the synthesis of benzo[d]imidazo[2,1-b]-thiazoles, we searched for a suitable reagent to regioselectively form this instead of 3a and 4. This tricyclic backbone can be found in compounds that possess a wide range of medicinal properties including those with antimalarial [74], anticancer [75,76], antiallergic [77,78], antibacterial [79], and antimicrobial properties [80,81]. In addition, they have been found to be biologically active antagonists of adenosine receptors [82], inhibitors of cyclic-AMP-diphosphoesterase [83], and benzodiazepine receptor ligands [84,85]. Reported methods to access this structural motif include annulation between an aromatic amine and acid chloride [86] or via aza-Diels–Alder reaction [87]. Coupling between alkynoic acid and 2-aminobenzothiazole and the use of ionic liquids have also been developed [88]. Heterogeneous catalysts such as kaolin and hydrotalcites have been employed in the synthesis of benzo[4,5]thiazolo[3,2-a]pyrimidin-4-ones [89,90]. Polyphosphoric acid has also been used to access 5a by coupling 2-aminobenzothiazole with β-ketoesters [79,91,92].
We have previously shown that bismuth salts can catalyze the intermolecular cyclization of 2-aminopyridines and β-ketoesters to pyrido[1,2-a]pyrimidin-4-ones [6]. Using Bi(OTf)3 in 1.5 mmol 2-aminobenzolthiazole (1a) and 1.0 mmol methyl acetoacetate (2a) gave a relatively poor yield of 5a (Table 2, entry 1). This prompted us to switch the equivalents of 1a and 2a to 1.0 and 1.5 mmol, respectively, which improved the yield tremendously (Table 2, entry 2). Hence, the survey of Lewis acids was conducted with 10 mol % of the Lewis acid at 100 °C using toluene as solvent (Table 2). In(OTf)3 was a better catalyst for this reaction with 95% yield (Table 2, entry 3). Reactions with ZnII and YbIII triflates proceeded with moderate yields while AgI, CuII and CoII were unreactive (Table 2, entries 4–8). Other InIII salts screened were not as effective as its triflate salt (Table 2, entries 9 and 10). A wide range of solvents were also screened. Dioxane was fairly suitable for the reaction with moderate yields of 5a whereas nitromethane fared poorly (Table 2, entries 11 and 12). Propionitrile, DMF and DMSO were also unsuitable for the reaction (Table 2, entries 13–15). Hence, establishing the scope of reaction was carried out using In(OTf)3 as catalyst and toluene as solvent.
Table 2: Optimization parameters for the synthesis of benzo[4,5]thiazolo[3,2-a]pyrimidin-4-ones 5a.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-270-i6.svg?max-width=637&scale=1.0)
|
|||
| Entry | Catalyst | Solvent | Yield of 5ab,c (%)b,c |
| 1d | Bi(OTf)3 | toluene | 23 |
| 2 | Bi(OTf)3 | toluene | 73 (70) |
| 3 | In(OTf)3 | toluene | 96 (95) |
| 4 | Yb(OTf)3 | toluene | 59 (52) |
| 5 | Zn(OTf)2 | toluene | 67 (57) |
| 6 | Cu(OTf)2 | toluene | 0 |
| 7 | AgOTf | toluene | 0 |
| 8 | CoCl2 | toluene | 0 |
| 9 | InCl3 | toluene | 63 (58) |
| 10 | InBr3 | toluene | 50 (40) |
| 11 | In(OTf)3 | dioxane | 39 (26) |
| 12 | In(OTf)3 | NO2Me | 10 |
| 13 | In(OTf)3 | EtCN | 18 |
| 14 | In(OTf)3 | DMF | 0 |
| 15 | In(OTf)3 | DMSO | 0 |
aReaction conditions: 1a (1.0 mmol), 2a (1.5 mmol), catalyst (10 mol %), in 1.5 mL of solvent for 16 h at 100 °C. bYield (from GC) with respect to 1a, using biphenyl as an internal standard. cIsolated yields in parenthesis. dReaction using 1.5 mmol of 1a and 1.0 mmol of 2a instead.
Both ethyl acetoacetate and N,N-diethylacetamide reacted smoothly with 1a to give 5a in good yields (Scheme 3). A large variety of substituted 2-aminobenzothiazoles were then screened in this reaction. Electron-donating groups including methyl and methoxy were well tolerated under the reaction conditions (Scheme 3, 5b–d). 2-Aminobenzothiazoles with halogen substituents F, Cl and Br reacted smoothly with excellent yields (Scheme 3, 5e–g). Good to excellent yields were also obtained with electron-withdrawing ester and CF3 moieties (Scheme 3, 5h–j). Likewise, the reaction with 2-aminobenzoxazole proceeded smoothly giving 5k in 79% yield.
![[1860-5397-13-270-i3]](/bjoc/content/inline/1860-5397-13-270-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scope of the reaction. Reaction conditions: 1 (1.0 mmol), 2 (1.5 mmol), In(OTf)3 (0.1 mmol), in 1.5 mL toluene at 100 °C for 16 h. a2 mL of 2-butanol/toluene 1:3 (v/v) used as solvent instead. bIsopentanol/toluene.
Scheme 3: Scope of the reaction. Reaction conditions: 1 (1.0 mmol), 2 (1.5 mmol), In(OTf)3 (0.1 mmol), in 1.5...
Next, a variety of methyl or ethyl carboxylates of β-ketoesters were screened for this reaction. Good yields were obtained for β-ketoesters with n-propyl and n-butyl moieties at the C-4 carbon (Scheme 3, 5l and 5m). Bulkier isopropyl and tert-butyl substituents were less well tolerated, with significantly lower yields (Scheme 3, 5n and 5o). The best yield of 99% was obtained when ethyl 4,4,4-trifluoroacetoacetate was employed while the reaction with diethyl 3-oxopentanedioate proceeded with 92% yield (Scheme 3, 5p and 5q). β-Ketoesters with methyl or ethyl substituent at the α-carbon were well suited for this reaction (Scheme 3, 5r and 5s). Gratifyingly, the reaction also proceeded smoothly with various cyclic β-oxo esters ranging from cyclopentane to cyclooctane (Scheme 3, 5t–w). Interestingly, the ethers 5x and 5y were obtained when ethyl 4-chloroacetoacetate was reacted with 2-amino-6-methylbenzothiazole in a mixed solvent system of toluene with 2-butanol or isopentanol, respectively. The Cl is utilized as a leaving group during the nucleophilic attack by the corresponding alcohol.
A series of control experiments were conducted to gain insights into the mechanism of the two contrasting reactions. Firstly, two separate coupling reactions between 2-aminobenzothiazole (1a) and methyl acetoacetate (2a) were conducted using the optimized reaction conditions, one without KOt-Bu and the other without CBrCl3 (Scheme 4a and b). The absence of 3a in both cases shows that both KOt-Bu and CBrCl3 have to be present to form the desired product. Next, to confirm that the reaction proceeds via a radical pathway, 2,2,6,6-tetramethylpiperidin-1-yl oxyl (TEMPO, 2.0 equiv) was added to the reaction mixture as a radical scavenger (Scheme 4c). After 24 h, none of the desired product 3a had formed, indicating that the reaction pathway was a radical one in nature. Homolytic cleavage of the C–Br bond in CBrCl3 can occur under UVC irradiation or thermally [21]. The cleavage is facile and was also observed under blue LED light [93]. Finally, the α-brominated methyl acetoacetate 6 was detected in several reaction mixtures. To prove that 6 is indeed a reaction intermediate, the starting material 2a was replaced with 6 (Scheme 4d). After 16 h, 93% of 3a was formed, showing unambiguously that the reaction proceeds via 6 as an intermediate. We propose that KOt-Bu assists in α-bromination of 2a to form the intermediate 6 via single electron transfer (SET). Recent mechanistic work by Murphy and co-workers showed that in the presence of polyhalomethane CBr4, KOt-Bu undergoes a SET reaction, forming •CBr3 and t-BuO• radicals [95]. Furthermore, with other strong bases of similar strength including NaH and KOEt the reaction proceeded with significantly lower yields. This lends additional support to the hypothesis that KOt-Bu, besides being a base, has a secondary role of assisting in SET (Table 1, entries 5 and 6).
![[1860-5397-13-270-i4]](/bjoc/content/inline/1860-5397-13-270-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
The following control experiments were carried out for the synthesis of benzo[4,5]thiazolo[3,2-a]pyrimidin-4-ones. A blank reaction was done without the InIII catalyst in toluene under the optimized conditions (Scheme 4e). No desired product 5a was formed, implying the need for In(OTf)3 as catalyst in the reaction. A radical trapping experiment with 2 equivalents of TEMPO was also conducted using the optimized conditions (Scheme 4f). 89% of 5a was formed, implying that the reaction does not proceed via a radical pathway but an ionic one.
Based on these observations, the syntheses of benzo[d]imidazo[2,1-b]thiazoles and benzo[4,5]thiazolo[3,2-a]pyrimidin-4-ones are proposed to occur via the following mechanisms (Figure 1 and Figure 2). For the former, the reaction is initiated by SET from the tert-butoxide anion to CBrCl3, forming the tert-butoxy radical [94]. This radical attacks the α-hydrogen of 2a via hydrogen atom transfer (HAT), to form intermediate A with a radical at the α-carbon. A then undergoes α-bromination to form the intermediate 6 [95]. Attack at the α-carbon of 6 by 2-aminobenzothiazole (1a) via an Ortoleva–King type of reaction forms B [96,97]. This is followed by a nucleophilic addition and dehydration to form C. Upon deprotonation of the acidic proton at C by KOt-Bu, the desired product 3a is formed with release of KBr and tert-butanol. As proposed by Zeitler’s group, the •CCl3 radicals are quenched to CHCl3 via HAT [93].
![[1860-5397-13-270-1]](/bjoc/content/figures/1860-5397-13-270-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Proposed mechanism (benzo[d]imidazo[2,1-b]thiazoles).
Figure 1: Proposed mechanism (benzo[d]imidazo[2,1-b]thiazoles).
![[1860-5397-13-270-2]](/bjoc/content/figures/1860-5397-13-270-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Proposed mechanism (benzo[4,5]thiazolo[3,2-a]pyrimidin-4-ones).
Figure 2: Proposed mechanism (benzo[4,5]thiazolo[3,2-a]pyrimidin-4-ones).
The proposed catalytic cycle for the synthesis of benzo[4,5]thiazolo[3,2-a]pyrimidin-4-ones is as follows. The Lewis acidic InIII metal center coordinates to the more nucleophilic benzothiazole N atom, forming an adduct A [98]. This activates the N–H bonds and its subsequent cleavage by the triflate forms B [99]. The InIII metal center also coordinates to the keto carbon of methyl acetoacetate (2a) and thus brings both substrates in close proximity to one another [100,101]. An addition reaction ensues the formation of C which is followed by proton abstraction, releasing the catalyst as In(OTf)2OH [102] and forming D. An intramolecular condensation reaction occurs which forms the desired product 5a with extrusion of MeOH. The In(OTf)3 is regenerated by TfOH with release of a water molecule, and the catalytic cycle is repeated.
Conclusion
We have developed two regiodivergent protocols for the intermolecular cyclization of 2-aminobenzothiazoles with β-ketoesters and β-ketoamides that are determined by the reagents used. This is possible due to the versatility of β-ketoesters in switching polarities and reactivities in the presence of different reagents. With the Brønsted base and radical initiator system of KOt-BU/CBrCl3, in situ α-bromination occurs and nucleophilic attacks at the α-carbon and keto carbon lead to the formation of benzo[d]imidazo[2,1-b]thiazoles. On the other hand, the Lewis acidic catalyst In(OTf)3 allows for nucleophilic attacks at both carbonyl groups to form benzo[4,5]thiazolo[3,2-a]pyrimidin-4-ones. The scope of these regiodivergent protocols was demonstrated with 19 examples of tricyclic benzo[d]imidazo[2,1-b]thiazoles and 27 examples of tricyclic and tetracyclic benzo[4,5]thiazolo[3,2-a]pyrimidin-4-ones.
Experimental
Representative procedure for the synthesis of benzo[d]imidazo[2,1-b]thiazole: A 25 mL two-neck round-bottomed flask was charged with 2-aminobenzothiazole (1a, 180 mg, 1.2 mmol), methyl acetoacetate (2a, 108 μL, 1.0 mmol), in 3 mL of CBrCl3/MeCN 1:9 (v/v) solvent mixture. KOt-Bu (224 mg, 2.0 mmol) was added slowly at room temperature and the reaction mixture was stirred under reflux for 16 h. Upon completion, the reaction mixture was diluted with 30 mL of ethyl acetate, filtered through a short pad of silica gel and washed down with an additional 60 mL ethyl acetate. The filtrate was washed with distilled water (3 × 30 mL) and the organic phase was dried with anhydrous Na2SO4. After filtration, the solvent was removed by rotary evaporation and the residue was purified by column chromatography using hexane and ethyl acetate (v/v = 8:1) as eluent to afford 3a with 84% yield.
Methyl 2-methylbenzo[d]imidazo[2,1-b]thiazole-3-carboxylate (3a): Obtained as a yellow semi-solid (206 mg, 84%); 1H NMR (300 MHz, CDCl3) 8.95 (d, J = 8.1 Hz, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.45 (t, J = 8.0 Hz, 1H), 7.34 (t, J = 7.5 Hz, 1H), 3.97 (s, 3H), 2.63 (s, 3H); 13C NMR (75 MHz, CDCl3) 161.1, 154.4, 151.7, 134.0, 129.7,126.3, 125.0, 123.6, 118.3, 117.6, 51.6, 16.9; HRMS–ESI (m/z): [M + H]+: calcd for C12H11N2O2S, 247.0536; found, 247.0533.
Representative procedure for the synthesis of benzo[d]imidazo[2,1-b]thiazole: A 10 mL round-bottomed flask was charged with 2-aminobenzothiazole (1a, 150 mg, 1.0 mmol), methyl acetoacetate (2a, 162 μL, 1.5 mmol) and indium(III) trifluoromethanesulfonate (56 mg, 0.1 mmol) in 1.5 mL of toluene. After stirring at 100 °C for 16 h, the reaction was diluted with water and extracted with EtOAc (15 mL × 5). The combined organic layers were washed with brine and dried with anhydrous Na2SO4. After filtration, the solvent was removed by rotary evaporation, and the residue was cleaned up by column chromatography using hexane and ethyl acetate (v/v = 4:1) as eluent to afford 5a with 95% yield.
2-Methyl-4H-benzo[4,5]thiazolo[3,2-a]pyrimidin-4-one (5a): Obtained as a light yellow solid (206 mg, 95%); mp 202–204 °C; 1H NMR (300 MHz, CDCl3) δ 8.94 (d, J = 7.5 Hz, 1H), 7.57 (d, J = 7.2 Hz, 1H), 7.44–7.33 (m, 2H), 6.16 (s, 1H), 2.30 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 162.6, 161.1, 160.8, 135.8, 126.7, 126.6, 123.8, 121.5, 119.7, 106.9, 23.5; HRMS–ESI (m/z): [M + H]+ calcd for C11H9N2OS, 217.0430; found, 217.0432.
Supporting Information
| Supporting Information File 1: Experimental procedure, analytical data and NMR spectra. | ||
| Format: PDF | Size: 4.6 MB | Download |
References
-
He, C.; Guo, S.; Ke, J.; Hao, J.; Xu, H.; Chen, H.; Lei, A. J. Am. Chem. Soc. 2012, 134, 5766–5769. doi:10.1021/ja301153k
Return to citation in text: [1] -
Roslan, I. I.; Sun, J.; Chuah, G.-K.; Jaenicke, S. Adv. Synth. Catal. 2015, 357, 719–726. doi:10.1002/adsc.201400857
Return to citation in text: [1] -
Malakar, C. C.; Schmidt, D.; Conrad, J.; Beifuss, U. Org. Lett. 2011, 13, 1972–1975. doi:10.1021/ol200347g
Return to citation in text: [1] -
Duan, X.-h.; Lin, X.-y.; Guo, L.-n.; Liao, M.-c.; Liu, W.-M.; Liang, Y.-m. J. Org. Chem. 2005, 70, 6980–6983. doi:10.1021/jo050908d
Return to citation in text: [1] -
v. Pechmann, H. Ber. Dtsch. Chem. Ges. 1884, 17, 929–936. doi:10.1002/cber.188401701248
Return to citation in text: [1] -
Roslan, I. I.; Lim, Q.-X.; Han, A.; Chuah, G.-K.; Jaenicke, S. Eur. J. Org. Chem. 2015, 2351–2355. doi:10.1002/ejoc.201500227
Return to citation in text: [1] [2] -
Ritson, D. J.; Spiteri, C.; Moses, J. E. J. Org. Chem. 2011, 76, 3519–3522. doi:10.1021/jo1025332
Return to citation in text: [1] -
Wang, B.; Lu, B.; Jiang, Y.; Zhang, Y.; Ma, D. Org. Lett. 2008, 10, 2761–2763. doi:10.1021/ol800900a
Return to citation in text: [1] -
Yadav, J. S.; Reddy, B. V. S.; Premalatha, K. Synlett 2004, 963–966. doi:10.1055/s-2004-822898
Return to citation in text: [1] -
Huang, H.; Si, P.; Wang, L.; Xu, Y.; Xu, X.; Zhu, J.; Jiang, H.; Li, W.; Chen, L.; Li, J. ChemMedChem 2015, 10, 1184–1199. doi:10.1002/cmdc.201500136
Return to citation in text: [1] -
Zhang, Z.; Zhang, W.; Li, J.; Liu, Q.; Liu, T.; Zhang, G. J. Org. Chem. 2014, 79, 11226–11233. doi:10.1021/jo5018487
Return to citation in text: [1] -
Bagley, M. C.; Dale, J. W.; Bower, J. Chem. Commun. 2002, 1682–1683. doi:10.1039/B203900A
Return to citation in text: [1] -
Maiti, S.; Biswas, S.; Jana, U. J. Org. Chem. 2010, 75, 1674–1683. doi:10.1021/jo902661y
Return to citation in text: [1] -
Wang, C.; Zhang, J.; Wang, S.; Fan, J.; Wang, Z. Org. Lett. 2010, 12, 2338–2341. doi:10.1021/ol100688c
Return to citation in text: [1] -
Sadeghi, M.; Safari, J.; Zarnegar, Z. RSC Adv. 2016, 6, 64749–64755. doi:10.1039/C6RA11175K
Return to citation in text: [1] -
Wang, X.; Ma, L.; Yu, W. Synthesis 2011, 2445–2453. doi:10.1055/s-0030-1260106
Return to citation in text: [1] -
Wang, Y.-F.; Toh, K. K.; Chiba, S.; Narasaka, K. Org. Lett. 2008, 10, 5019–5022. doi:10.1021/ol802120u
Return to citation in text: [1] -
Guo, X.; Yu, R.; Li, H.; Li, Z. J. Am. Chem. Soc. 2009, 131, 17387–17393. doi:10.1021/ja907568j
Return to citation in text: [1] -
Liu, W.; Jiang, H.; Zhang, M.; Qi, C. J. Org. Chem. 2010, 75, 966–968. doi:10.1021/jo902375k
Return to citation in text: [1] -
Roslan, I. I.; Ng, K.-H.; Chuah, G.-K.; Jaenicke, S. Adv. Synth. Catal. 2016, 358, 364–369. doi:10.1002/adsc.201501012
Return to citation in text: [1] [2] [3] [4] -
Roslan, I. I.; Ng, K.-H.; Wu, J.-E.; Chuah, G.-K.; Jaenicke, S. J. Org. Chem. 2016, 81, 9167–9174. doi:10.1021/acs.joc.6b01714
Return to citation in text: [1] [2] [3] [4] [5] -
Roslan, I. I.; Ng, K.-H.; Chuah, G.-K.; Jaenicke, S. Eur. J. Org. Chem. 2017, 704–709. doi:10.1002/ejoc.201601410
Return to citation in text: [1] [2] [3] [4] -
Kumar, B. S. P. A.; Madhav, B.; Reddy, K. H. V.; Nageswar, Y. V. D. Tetrahedron Lett. 2011, 52, 2862–2865. doi:10.1016/j.tetlet.2011.03.110
Return to citation in text: [1] -
Xie, J.; Jiang, H.; Cheng, Y.; Zhu, C. Chem. Commun. 2012, 48, 979–981. doi:10.1039/C2CC15813B
Return to citation in text: [1] -
Gao, Q.-H.; Fei, Z.; Zhu, Y.-P.; Lian, M.; Jia, F.-C.; Liu, M.-C.; She, N.-F.; Wu, A.-X. Tetrahedron 2013, 69, 22–28. doi:10.1016/j.tet.2012.10.072
Return to citation in text: [1] -
Xie, Y.; Wu, J.; Che, X.; Chen, Y.; Huang, H.; Deng, G.-J. Green Chem. 2016, 18, 667–671. doi:10.1039/C5GC01978H
Return to citation in text: [1] -
Ma, L.; Wang, X.; Yu, W.; Han, B. Chem. Commun. 2011, 47, 11333–11335. doi:10.1039/c1cc13568f
Return to citation in text: [1] -
Lee, S. K.; Park, J. K. J. Org. Chem. 2015, 80, 3723–3729. doi:10.1021/acs.joc.5b00298
Return to citation in text: [1] -
Roslan, I. I.; Chuah, G.-K.; Jaenicke, S. Eur. J. Org. Chem. 2017, 671–675. doi:10.1002/ejoc.201601586
Return to citation in text: [1] -
Samanta, S.; Jana, S.; Mondal, S.; Monir, K.; Chandra, A. K.; Hajra, A. Org. Biomol. Chem. 2016, 14, 5073–5078. doi:10.1039/C6OB00656F
Return to citation in text: [1] -
Zhu, Y.-P.; Yuan, J.-J.; Zhao, Q.; Lian, M.; Gao, Q.-H.; Liu, M.-C.; Yang, Y.; Wu, A.-X. Tetrahedron 2012, 68, 173–178. doi:10.1016/j.tet.2011.10.074
Return to citation in text: [1] -
Shinde, M. H.; Kshirsagar, U. A. Green Chem. 2016, 18, 1455–1458. doi:10.1039/C5GC02771C
Return to citation in text: [1] -
Zhu, Y.-P.; Lian, M.; Jia, F.-C.; Liu, M.-C.; Yuan, J.-J.; Gao, Q.-H.; Wu, A.-X. Chem. Commun. 2012, 48, 9086–9088. doi:10.1039/c2cc34561g
Return to citation in text: [1] -
Zhao, J.; Huang, H.; Wu, W.; Chen, H.; Jiang, H. Org. Lett. 2013, 15, 2604–2607. doi:10.1021/ol400773k
Return to citation in text: [1] -
Farag, A. M.; Mayhoub, A. S.; Barakat, S. E.; Bayomi, A. H. Bioorg. Med. Chem. 2008, 16, 4569–4578. doi:10.1016/j.bmc.2008.02.043
Return to citation in text: [1] -
Al-Tel, T. H.; Al-Qawasmeh, R. A.; Zaarour, R. Eur. J. Med. Chem. 2011, 46, 1874–1881. doi:10.1016/j.ejmech.2011.02.051
Return to citation in text: [1] -
Andreani, A.; Burnelli, S.; Granaiola, M.; Leoni, A.; Locatelli, A.; Morigi, R.; Rambaldi, M.; Varoli, L.; Calonghi, N.; Cappadone, C.; Farruggia, G.; Zini, M.; Stefanelli, C.; Masotti, L.; Radin, N. S.; Shoemaker, R. H. J. Med. Chem. 2008, 51, 809–816. doi:10.1021/jm701246g
Return to citation in text: [1] -
Furlan, A.; Colombo, F.; Kover, A.; Issaly, N.; Tintori, C.; Angeli, L.; Leroux, V.; Letard, S.; Amat, M.; Asses, Y.; Maigret, B.; Dubreuil, P.; Botta, M.; Dono, R.; Bosch, J.; Piccolo, O.; Passarella, D.; Maina, F. Eur. J. Med. Chem. 2012, 47, 239–254. doi:10.1016/j.ejmech.2011.10.051
Return to citation in text: [1] -
Andreani, A.; Granaiola, M.; Locatelli, A.; Morigi, R.; Rambaldi, M.; Varoli, L.; Calonghi, N.; Cappadone, C.; Farruggia, G.; Stefanelli, C.; Masotti, L.; Nguyen, T. L.; Hamel, E.; Shoemaker, R. H. J. Med. Chem. 2012, 55, 2078–2088. doi:10.1021/jm2012694
Return to citation in text: [1] -
Palkar, M.; Noolvi, M.; Sankangoud, R.; Maddi, V.; Gadad, A.; Nargund, L. V. G. Arch. Pharm. 2010, 343, 353–359. doi:10.1002/ardp.200900260
Return to citation in text: [1] -
Ager, I. R.; Barnes, A. C.; Danswan, G. W.; Hairsine, P. W.; Kay, D. P.; Kennewell, P. D.; Matharu, S. S.; Miller, P.; Robson, P.; Rowlands, D. A.; Tully, W. R.; Westwood, R. J. Med. Chem. 1988, 31, 1098–1115. doi:10.1021/jm00401a009
Return to citation in text: [1] -
Andreani, A.; Granaiola, M.; Leoni, A.; Locatelli, A.; Morigi, R.; Rambaldi, M.; Varoli, L.; Lannigan, D.; Smith, J.; Scudiero, D.; Kondapaka, S.; Shoemaker, R. H. Eur. J. Med. Chem. 2011, 46, 4311–4323. doi:10.1016/j.ejmech.2011.07.001
Return to citation in text: [1] -
Chao, Q.; Sprankle, K. G.; Grotzfeld, R. M.; Lai, A. G.; Carter, T. A.; Velasco, A. M.; Gunawardane, R. N.; Cramer, M. D.; Gardner, M. F.; James, J.; Zarrinkar, P. P.; Patel, H. K.; Bhagwat, S. S. J. Med. Chem. 2009, 52, 7808–7816. doi:10.1021/jm9007533
Return to citation in text: [1] -
Shen, H. C.; Ding, F.-X.; Deng, Q.; Wilsie, L. C.; Krsmanovic, M. L.; Taggart, A. K.; Carballo-Jane, E.; Ren, N.; Cai, T. Q.; Wu, T.-J.; Wu, K. K.; Cheng, K.; Chen, Q.; Wolff, M. S.; Tong, X.; Holt, T. G.; Waters, M. G.; Hammond, M. L.; Tata, J. R.; Colletti, S. L. J. Med. Chem. 2009, 52, 2587–2602. doi:10.1021/jm900151e
Return to citation in text: [1] -
Yousefi, B. H.; Manook, A.; Drzezga, A.; von Reutern, B.; Schwaiger, M.; Wester, H.-J.; Henriksen, G. J. Med. Chem. 2011, 54, 949–956. doi:10.1021/jm101129a
Return to citation in text: [1] -
Yousefi, B. H.; Drzezga, A.; von Reutern, B.; Manook, A.; Schwaiger, M.; Wester, H.-J.; Henriksen, G. ACS Med. Chem. Lett. 2011, 2, 673–677. doi:10.1021/ml200123w
Return to citation in text: [1] -
Clements-Jewery, S.; Danswan, G.; Gardner, R. C.; Matharu, S. S.; Murdoch, R.; Tully, W. R.; Westwood, R. J. Med. Chem. 1988, 31, 1220–1226. doi:10.1021/jm00401a025
Return to citation in text: [1] -
Christodoulou, M. S.; Colombo, F.; Passarella, D.; Ieronimo, G.; Zuco, V.; De Cesare, M.; Zunino, F. Bioorg. Med. Chem. 2011, 19, 1649–1657. doi:10.1016/j.bmc.2011.01.039
Return to citation in text: [1] -
Guchhait, S. K.; Chaudhary, V. Org. Biomol. Chem. 2014, 12, 6694–6705. doi:10.1039/C4OB00882K
Return to citation in text: [1] -
Wu, Z.; Huang, Q.; Zhou, X.; Yu, L.; Li, Z.; Wu, D. Eur. J. Org. Chem. 2011, 5242–5245. doi:10.1002/ejoc.201100834
Return to citation in text: [1] -
Gao, J.; Zhu, J.; Chen, L.; Shao, Y.; Zhu, J.; Huang, Y.; Wang, X.; Lv, X. Tetrahedron Lett. 2014, 55, 3367–3373. doi:10.1016/j.tetlet.2014.04.070
Return to citation in text: [1] -
Shi, B.; Zhu, Z.; Zhu, Y.-S.; Zhou, D.; Wang, J.; Zhou, P.; Jing, H. Org. Biomol. Chem. 2016, 14, 2978–2984. doi:10.1039/C6OB00102E
Return to citation in text: [1] -
Zhang, X.; Jia, J.; Ma, C. Org. Biomol. Chem. 2012, 10, 7944–7948. doi:10.1039/c2ob26211h
Return to citation in text: [1] -
Wang, J.; Li, J.; Zhu, Q. Org. Lett. 2015, 17, 5336–5339. doi:10.1021/acs.orglett.5b02694
Return to citation in text: [1] -
Mishra, S.; Monir, K.; Mitra, S.; Hajra, A. Org. Lett. 2014, 16, 6084–6087. doi:10.1021/ol5028893
Return to citation in text: [1] -
Balwe, S. G.; Jeong, Y. T. RSC Adv. 2016, 6, 107225–107232. doi:10.1039/C6RA24183B
Return to citation in text: [1] -
Yanagisawa, S.; Ueda, K.; Taniguchi, T.; Itami, K. Org. Lett. 2008, 10, 4673–4676. doi:10.1021/ol8019764
Return to citation in text: [1] -
Liu, W.; Cao, H.; Zhang, H.; Zhang, H.; Chung, K. H.; He, C.; Wang, H.; Kwong, F. Y.; Lei, A. J. Am. Chem. Soc. 2010, 132, 16737–16740. doi:10.1021/ja103050x
Return to citation in text: [1] -
Shirakawa, E.; Itoh, K.-i.; Higashino, T.; Hayashi, T. J. Am. Chem. Soc. 2010, 132, 15537–15539. doi:10.1021/ja1080822
Return to citation in text: [1] -
Sun, C.-L.; Li, H.; Yu, D.-G.; Yu, M.; Zhou, X.; Lu, X.-Y.; Huang, K.; Zheng, S.-F.; Li, B.-J.; Shi, Z.-J. Nat. Chem. 2010, 2, 1044–1049. doi:10.1038/nchem.862
Return to citation in text: [1] -
Wu, Y.; Choy, P. Y.; Kwong, F. Y. Asian J. Org. Chem. 2014, 3, 1262–1265. doi:10.1002/ajoc.201402181
Return to citation in text: [1] [2] -
Stephens, D. E.; Lakey-Beitia, J.; Burch, J. E.; Arman, H. D.; Larionov, O. V. Chem. Commun. 2016, 52, 9945–9948. doi:10.1039/C6CC04816A
Return to citation in text: [1] [2] [3] -
Drapeau, M. P.; Fabre, I.; Grimaud, L.; Ciofini, I.; Ollevier, T.; Taillefer, M. Angew. Chem., Int. Ed. 2015, 54, 10587–10591. doi:10.1002/anie.201502332
Return to citation in text: [1] [2] [3] -
De, S.; Mishra, S.; Kakde, B. N.; Dey, D.; Bisai, A. J. Org. Chem. 2013, 78, 7823–7844. doi:10.1021/jo400890k
Return to citation in text: [1] [2] -
De, S.; Ghosh, S.; Bhunia, S.; Sheikh, J. A.; Bisai, A. Org. Lett. 2012, 14, 4466–4469. doi:10.1021/ol3019677
Return to citation in text: [1] [2] -
Rueping, M.; Leiendecker, M.; Das, A.; Poisson, T.; Bui, L. Chem. Commun. 2011, 47, 10629–10631. doi:10.1039/c1cc14297f
Return to citation in text: [1] [2] -
Bhakuni, B. S.; Kumar, A.; Balkrishna, S. J.; Sheikh, J. A.; Konar, S.; Kumar, S. Org. Lett. 2012, 14, 2838–2841. doi:10.1021/ol301077y
Return to citation in text: [1] [2] -
Wei, W.-T.; Cheng, Y.-J.; Hu, Y.; Chen, Y.-Y.; Zhang, X.-J.; Zou, Y.; Yan, M. Adv. Synth. Catal. 2015, 357, 3474–3478. doi:10.1002/adsc.201500647
Return to citation in text: [1] -
Zhang, M.-Z.; Guo, Q.-H.; Sheng, W.-B.; Guo, C.-C. Adv. Synth. Catal. 2015, 357, 2855–2861. doi:10.1002/adsc.201500551
Return to citation in text: [1] [2] -
Zhao, D.; Shen, Q.; Zhou, Y.-R.; Li, J.-X. Org. Biomol. Chem. 2013, 11, 5908–5912. doi:10.1039/c3ob41083h
Return to citation in text: [1] [2] -
Wang, H.; Wang, Z.; Huang, H.; Tan, J.; Xu, K. Org. Lett. 2016, 18, 5680–5683. doi:10.1021/acs.orglett.6b02914
Return to citation in text: [1] [2] -
Toutov, A. A.; Liu, W.-B.; Betz, K. N.; Fedorov, A.; Stoltz, B. M.; Grubbs, R. H. Nature 2015, 518, 80–84. doi:10.1038/nature14126
Return to citation in text: [1] -
Sasson, Y.; Webster, O. W. J. Chem. Soc., Chem. Commun. 1992, 1200–1201. doi:10.1039/c39920001200
Return to citation in text: [1] -
Bhattacharjee, A. K.; Hartell, M. G.; Nichols, D. A.; Hicks, R. P.; Stanton, B.; van Hamont, J. E.; Milhous, W. K. Eur. J. Med. Chem. 2004, 39, 59–67. doi:10.1016/j.ejmech.2003.10.004
Return to citation in text: [1] -
Shukla, G.; Tiwari, A. K.; Singh, V. K.; Bajpai, A.; Chandra, H.; Mishra, A. K. Chem. Biol. Drug Des. 2008, 72, 533–539. doi:10.1111/j.1747-0285.2008.00724.x
Return to citation in text: [1] -
El-Sherbeny, M. A. Arzneimittelforschung 2000, 50, 848–853. doi:10.1055/s-0031-1300300
Return to citation in text: [1] -
Wade, J. J.; Toso, C. B.; Matson, C. J.; Stelzer, V. L. J. Med. Chem. 1983, 26, 608–611. doi:10.1021/jm00358a031
Return to citation in text: [1] -
Yevich, J. P.; Temple, D. L., Jr.; Covington, R. R.; Owens, D. A.; Seidehamel, R. J.; Dungan, K. W. J. Med. Chem. 1982, 25, 864–868. doi:10.1021/jm00349a020
Return to citation in text: [1] -
Hilal, H. S.; Ali-Shtayeh, M. S.; Arafat, R.; Al-Tel, T.; Voelter, W.; Barakat, A. Eur. J. Med. Chem. 2006, 41, 1017–1024. doi:10.1016/j.ejmech.2006.03.025
Return to citation in text: [1] [2] -
Bhosale, V. N.; Vartale, S. P.; Deshmukh, V. K.; Kuberkar, S. V. J. Chem. Pharm. Res. 2010, 2, 51–58.
Return to citation in text: [1] -
Sharma, P. K.; Kumar, M.; Mohan, V. Res. Chem. Intermed. 2010, 36, 985–993. doi:10.1007/s11164-010-0211-9
Return to citation in text: [1] -
Glennon, R. A.; Tejani-Butt, S. M.; Padgett, W.; Daly, J. W. J. Med. Chem. 1984, 27, 1364–1367. doi:10.1021/jm00376a027
Return to citation in text: [1] -
Glennon, R. A.; Gaines, J. J.; Rogers, M. E. J. Med. Chem. 1981, 24, 766–769. doi:10.1021/jm00138a027
Return to citation in text: [1] -
Trapani, G.; Carotti, A.; Franco, M.; Latrofa, A.; Genchi, G.; Liso, G. Eur. J. Med. Chem. 1993, 28, 13–21. doi:10.1016/0223-5234(93)90074-O
Return to citation in text: [1] -
Trapani, G.; Franco, M.; Latrofa, A.; Genchi, G.; Iacobazzi, V.; Ghiani, C. A.; Maciocco, E.; Liso, G. Eur. J. Med. Chem. 1997, 32, 83–89. doi:10.1016/S0223-5234(97)84364-4
Return to citation in text: [1] -
Landreau, C.; Deniaud, D.; Evain, M.; Reliquet, A.; Meslin, J.-C. J. Chem. Soc., Perkin Trans. 1 2002, 741–745. doi:10.1039/b111639h
Return to citation in text: [1] -
Mellor, J. M.; Rataj, H. Tetrahedron Lett. 1996, 37, 2619–2622. doi:10.1016/0040-4039(96)00342-5
Return to citation in text: [1] -
Yadav, A. K.; Sharma, G. R.; Dhakad, P.; Yadav, T. Tetrahedron Lett. 2012, 53, 859–862. doi:10.1016/j.tetlet.2011.12.024
Return to citation in text: [1] -
Sahu, P. K.; Sahu, P. K.; Agarwal, D. D. RSC Adv. 2013, 3, 9854–9864. doi:10.1039/c3ra40993g
Return to citation in text: [1] -
Sahu, P. K.; Sahu, P. K.; Jain, R.; Yadav, R.; Agarwal, D. D. Catal. Sci. Technol. 2012, 2, 2465–2475. doi:10.1039/c2cy20067h
Return to citation in text: [1] -
Kumar, G.; Sharma, P. K.; Sharma, S.; Singh, S. J. Chem. Pharm. Res. 2015, 7, 710–714.
Return to citation in text: [1] -
Fogla, A. K.; Ankodia, V.; Sharma, P. K.; Kumar, M. Res. Chem. Intermed. 2009, 35, 35–41. doi:10.1007/s11164-008-0006-4
Return to citation in text: [1] -
Franz, J. F.; Kraus, W. B.; Zeitler, K. Chem. Commun. 2015, 51, 8280–8283. doi:10.1039/C4CC10270C
Return to citation in text: [1] [2] -
Barham, J. P.; Coulthard, G.; Emery, K. J.; Doni, E.; Cumine, F.; Nocera, G.; John, M. P.; Berlouis, L. E. A.; McGuire, T.; Tuttle, T.; Murphy, J. A. J. Am. Chem. Soc. 2016, 138, 7402–7410. doi:10.1021/jacs.6b03282
Return to citation in text: [1] -
Meyers, C. Y.; Chan-Yu-King, R.; Hua, D. H.; Kolb, V. M.; Matthews, W. S.; Parady, T. E.; Horii, T.; Sandrock, P. B.; Hou, Y.; Xie, S. J. Org. Chem. 2003, 68, 500–511. doi:10.1021/jo025781w
Return to citation in text: [1] [2] -
Stasyuk, A. J.; Banasiewicz, M.; Cyrański, M. K.; Gryko, D. T. J. Org. Chem. 2012, 77, 5552–5558. doi:10.1021/jo300643w
Return to citation in text: [1] -
Zhang, Y.; Chen, Z.; Wu, W.; Zhang, Y.; Su, W. J. Org. Chem. 2013, 78, 12494–12504. doi:10.1021/jo402134x
Return to citation in text: [1] -
Jamal, Z.; Teo, Y.-C. RSC Adv. 2015, 5, 26949–26953. doi:10.1039/C4RA17182A
Return to citation in text: [1] -
Denmark, S. E.; Beutner, G. L. Angew. Chem., Int. Ed. 2008, 47, 1560–1638. doi:10.1002/anie.200604943
Return to citation in text: [1] -
Tsuji, H.; Tanaka, I.; Endo, K.; Yamagata, K.-i.; Nakamura, M.; Nakamura, E. Org. Lett. 2009, 11, 1845–1847. doi:10.1021/ol9003542
Return to citation in text: [1] -
Thirupathaiah, B.; Seo, S. Chem. Commun. 2015, 51, 4216–4219. doi:10.1039/C4CC10016F
Return to citation in text: [1] -
Posevins, D.; Suta, K.; Turks, M. Eur. J. Org. Chem. 2016, 1414–1419. doi:10.1002/ejoc.201600013
Return to citation in text: [1]
| 58. | Liu, W.; Cao, H.; Zhang, H.; Zhang, H.; Chung, K. H.; He, C.; Wang, H.; Kwong, F. Y.; Lei, A. J. Am. Chem. Soc. 2010, 132, 16737–16740. doi:10.1021/ja103050x |
| 59. | Shirakawa, E.; Itoh, K.-i.; Higashino, T.; Hayashi, T. J. Am. Chem. Soc. 2010, 132, 15537–15539. doi:10.1021/ja1080822 |
| 60. | Sun, C.-L.; Li, H.; Yu, D.-G.; Yu, M.; Zhou, X.; Lu, X.-Y.; Huang, K.; Zheng, S.-F.; Li, B.-J.; Shi, Z.-J. Nat. Chem. 2010, 2, 1044–1049. doi:10.1038/nchem.862 |
| 61. | Wu, Y.; Choy, P. Y.; Kwong, F. Y. Asian J. Org. Chem. 2014, 3, 1262–1265. doi:10.1002/ajoc.201402181 |
| 62. | Stephens, D. E.; Lakey-Beitia, J.; Burch, J. E.; Arman, H. D.; Larionov, O. V. Chem. Commun. 2016, 52, 9945–9948. doi:10.1039/C6CC04816A |
| 63. | Drapeau, M. P.; Fabre, I.; Grimaud, L.; Ciofini, I.; Ollevier, T.; Taillefer, M. Angew. Chem., Int. Ed. 2015, 54, 10587–10591. doi:10.1002/anie.201502332 |
| 64. | De, S.; Mishra, S.; Kakde, B. N.; Dey, D.; Bisai, A. J. Org. Chem. 2013, 78, 7823–7844. doi:10.1021/jo400890k |
| 65. | De, S.; Ghosh, S.; Bhunia, S.; Sheikh, J. A.; Bisai, A. Org. Lett. 2012, 14, 4466–4469. doi:10.1021/ol3019677 |
| 66. | Rueping, M.; Leiendecker, M.; Das, A.; Poisson, T.; Bui, L. Chem. Commun. 2011, 47, 10629–10631. doi:10.1039/c1cc14297f |
| 67. | Bhakuni, B. S.; Kumar, A.; Balkrishna, S. J.; Sheikh, J. A.; Konar, S.; Kumar, S. Org. Lett. 2012, 14, 2838–2841. doi:10.1021/ol301077y |
| 68. | Wei, W.-T.; Cheng, Y.-J.; Hu, Y.; Chen, Y.-Y.; Zhang, X.-J.; Zou, Y.; Yan, M. Adv. Synth. Catal. 2015, 357, 3474–3478. doi:10.1002/adsc.201500647 |
| 69. | Zhang, M.-Z.; Guo, Q.-H.; Sheng, W.-B.; Guo, C.-C. Adv. Synth. Catal. 2015, 357, 2855–2861. doi:10.1002/adsc.201500551 |
| 62. | Stephens, D. E.; Lakey-Beitia, J.; Burch, J. E.; Arman, H. D.; Larionov, O. V. Chem. Commun. 2016, 52, 9945–9948. doi:10.1039/C6CC04816A |
| 66. | Rueping, M.; Leiendecker, M.; Das, A.; Poisson, T.; Bui, L. Chem. Commun. 2011, 47, 10629–10631. doi:10.1039/c1cc14297f |
| 20. | Roslan, I. I.; Ng, K.-H.; Chuah, G.-K.; Jaenicke, S. Adv. Synth. Catal. 2016, 358, 364–369. doi:10.1002/adsc.201501012 |
| 21. | Roslan, I. I.; Ng, K.-H.; Wu, J.-E.; Chuah, G.-K.; Jaenicke, S. J. Org. Chem. 2016, 81, 9167–9174. doi:10.1021/acs.joc.6b01714 |
| 22. | Roslan, I. I.; Ng, K.-H.; Chuah, G.-K.; Jaenicke, S. Eur. J. Org. Chem. 2017, 704–709. doi:10.1002/ejoc.201601410 |
| 61. | Wu, Y.; Choy, P. Y.; Kwong, F. Y. Asian J. Org. Chem. 2014, 3, 1262–1265. doi:10.1002/ajoc.201402181 |
| 62. | Stephens, D. E.; Lakey-Beitia, J.; Burch, J. E.; Arman, H. D.; Larionov, O. V. Chem. Commun. 2016, 52, 9945–9948. doi:10.1039/C6CC04816A |
| 63. | Drapeau, M. P.; Fabre, I.; Grimaud, L.; Ciofini, I.; Ollevier, T.; Taillefer, M. Angew. Chem., Int. Ed. 2015, 54, 10587–10591. doi:10.1002/anie.201502332 |
| 64. | De, S.; Mishra, S.; Kakde, B. N.; Dey, D.; Bisai, A. J. Org. Chem. 2013, 78, 7823–7844. doi:10.1021/jo400890k |
| 65. | De, S.; Ghosh, S.; Bhunia, S.; Sheikh, J. A.; Bisai, A. Org. Lett. 2012, 14, 4466–4469. doi:10.1021/ol3019677 |
| 70. | Zhao, D.; Shen, Q.; Zhou, Y.-R.; Li, J.-X. Org. Biomol. Chem. 2013, 11, 5908–5912. doi:10.1039/c3ob41083h |
| 72. | Toutov, A. A.; Liu, W.-B.; Betz, K. N.; Fedorov, A.; Stoltz, B. M.; Grubbs, R. H. Nature 2015, 518, 80–84. doi:10.1038/nature14126 |
| 63. | Drapeau, M. P.; Fabre, I.; Grimaud, L.; Ciofini, I.; Ollevier, T.; Taillefer, M. Angew. Chem., Int. Ed. 2015, 54, 10587–10591. doi:10.1002/anie.201502332 |
| 67. | Bhakuni, B. S.; Kumar, A.; Balkrishna, S. J.; Sheikh, J. A.; Konar, S.; Kumar, S. Org. Lett. 2012, 14, 2838–2841. doi:10.1021/ol301077y |
| 69. | Zhang, M.-Z.; Guo, Q.-H.; Sheng, W.-B.; Guo, C.-C. Adv. Synth. Catal. 2015, 357, 2855–2861. doi:10.1002/adsc.201500551 |
| 71. | Wang, H.; Wang, Z.; Huang, H.; Tan, J.; Xu, K. Org. Lett. 2016, 18, 5680–5683. doi:10.1021/acs.orglett.6b02914 |
| 70. | Zhao, D.; Shen, Q.; Zhou, Y.-R.; Li, J.-X. Org. Biomol. Chem. 2013, 11, 5908–5912. doi:10.1039/c3ob41083h |
| 71. | Wang, H.; Wang, Z.; Huang, H.; Tan, J.; Xu, K. Org. Lett. 2016, 18, 5680–5683. doi:10.1021/acs.orglett.6b02914 |
| 20. | Roslan, I. I.; Ng, K.-H.; Chuah, G.-K.; Jaenicke, S. Adv. Synth. Catal. 2016, 358, 364–369. doi:10.1002/adsc.201501012 |
| 21. | Roslan, I. I.; Ng, K.-H.; Wu, J.-E.; Chuah, G.-K.; Jaenicke, S. J. Org. Chem. 2016, 81, 9167–9174. doi:10.1021/acs.joc.6b01714 |
| 22. | Roslan, I. I.; Ng, K.-H.; Chuah, G.-K.; Jaenicke, S. Eur. J. Org. Chem. 2017, 704–709. doi:10.1002/ejoc.201601410 |
| 73. | Sasson, Y.; Webster, O. W. J. Chem. Soc., Chem. Commun. 1992, 1200–1201. doi:10.1039/c39920001200 |
| 20. | Roslan, I. I.; Ng, K.-H.; Chuah, G.-K.; Jaenicke, S. Adv. Synth. Catal. 2016, 358, 364–369. doi:10.1002/adsc.201501012 |
| 21. | Roslan, I. I.; Ng, K.-H.; Wu, J.-E.; Chuah, G.-K.; Jaenicke, S. J. Org. Chem. 2016, 81, 9167–9174. doi:10.1021/acs.joc.6b01714 |
| 22. | Roslan, I. I.; Ng, K.-H.; Chuah, G.-K.; Jaenicke, S. Eur. J. Org. Chem. 2017, 704–709. doi:10.1002/ejoc.201601410 |
| 83. | Glennon, R. A.; Gaines, J. J.; Rogers, M. E. J. Med. Chem. 1981, 24, 766–769. doi:10.1021/jm00138a027 |
| 84. | Trapani, G.; Carotti, A.; Franco, M.; Latrofa, A.; Genchi, G.; Liso, G. Eur. J. Med. Chem. 1993, 28, 13–21. doi:10.1016/0223-5234(93)90074-O |
| 85. | Trapani, G.; Franco, M.; Latrofa, A.; Genchi, G.; Iacobazzi, V.; Ghiani, C. A.; Maciocco, E.; Liso, G. Eur. J. Med. Chem. 1997, 32, 83–89. doi:10.1016/S0223-5234(97)84364-4 |
| 80. | Bhosale, V. N.; Vartale, S. P.; Deshmukh, V. K.; Kuberkar, S. V. J. Chem. Pharm. Res. 2010, 2, 51–58. |
| 81. | Sharma, P. K.; Kumar, M.; Mohan, V. Res. Chem. Intermed. 2010, 36, 985–993. doi:10.1007/s11164-010-0211-9 |
| 82. | Glennon, R. A.; Tejani-Butt, S. M.; Padgett, W.; Daly, J. W. J. Med. Chem. 1984, 27, 1364–1367. doi:10.1021/jm00376a027 |
| 77. | Wade, J. J.; Toso, C. B.; Matson, C. J.; Stelzer, V. L. J. Med. Chem. 1983, 26, 608–611. doi:10.1021/jm00358a031 |
| 78. | Yevich, J. P.; Temple, D. L., Jr.; Covington, R. R.; Owens, D. A.; Seidehamel, R. J.; Dungan, K. W. J. Med. Chem. 1982, 25, 864–868. doi:10.1021/jm00349a020 |
| 79. | Hilal, H. S.; Ali-Shtayeh, M. S.; Arafat, R.; Al-Tel, T.; Voelter, W.; Barakat, A. Eur. J. Med. Chem. 2006, 41, 1017–1024. doi:10.1016/j.ejmech.2006.03.025 |
| 74. | Bhattacharjee, A. K.; Hartell, M. G.; Nichols, D. A.; Hicks, R. P.; Stanton, B.; van Hamont, J. E.; Milhous, W. K. Eur. J. Med. Chem. 2004, 39, 59–67. doi:10.1016/j.ejmech.2003.10.004 |
| 75. | Shukla, G.; Tiwari, A. K.; Singh, V. K.; Bajpai, A.; Chandra, H.; Mishra, A. K. Chem. Biol. Drug Des. 2008, 72, 533–539. doi:10.1111/j.1747-0285.2008.00724.x |
| 76. | El-Sherbeny, M. A. Arzneimittelforschung 2000, 50, 848–853. doi:10.1055/s-0031-1300300 |
| 87. | Mellor, J. M.; Rataj, H. Tetrahedron Lett. 1996, 37, 2619–2622. doi:10.1016/0040-4039(96)00342-5 |
| 88. | Yadav, A. K.; Sharma, G. R.; Dhakad, P.; Yadav, T. Tetrahedron Lett. 2012, 53, 859–862. doi:10.1016/j.tetlet.2011.12.024 |
| 86. | Landreau, C.; Deniaud, D.; Evain, M.; Reliquet, A.; Meslin, J.-C. J. Chem. Soc., Perkin Trans. 1 2002, 741–745. doi:10.1039/b111639h |
| 1. | He, C.; Guo, S.; Ke, J.; Hao, J.; Xu, H.; Chen, H.; Lei, A. J. Am. Chem. Soc. 2012, 134, 5766–5769. doi:10.1021/ja301153k |
| 2. | Roslan, I. I.; Sun, J.; Chuah, G.-K.; Jaenicke, S. Adv. Synth. Catal. 2015, 357, 719–726. doi:10.1002/adsc.201400857 |
| 3. | Malakar, C. C.; Schmidt, D.; Conrad, J.; Beifuss, U. Org. Lett. 2011, 13, 1972–1975. doi:10.1021/ol200347g |
| 4. | Duan, X.-h.; Lin, X.-y.; Guo, L.-n.; Liao, M.-c.; Liu, W.-M.; Liang, Y.-m. J. Org. Chem. 2005, 70, 6980–6983. doi:10.1021/jo050908d |
| 14. | Wang, C.; Zhang, J.; Wang, S.; Fan, J.; Wang, Z. Org. Lett. 2010, 12, 2338–2341. doi:10.1021/ol100688c |
| 15. | Sadeghi, M.; Safari, J.; Zarnegar, Z. RSC Adv. 2016, 6, 64749–64755. doi:10.1039/C6RA11175K |
| 16. | Wang, X.; Ma, L.; Yu, W. Synthesis 2011, 2445–2453. doi:10.1055/s-0030-1260106 |
| 35. | Farag, A. M.; Mayhoub, A. S.; Barakat, S. E.; Bayomi, A. H. Bioorg. Med. Chem. 2008, 16, 4569–4578. doi:10.1016/j.bmc.2008.02.043 |
| 36. | Al-Tel, T. H.; Al-Qawasmeh, R. A.; Zaarour, R. Eur. J. Med. Chem. 2011, 46, 1874–1881. doi:10.1016/j.ejmech.2011.02.051 |
| 94. | Barham, J. P.; Coulthard, G.; Emery, K. J.; Doni, E.; Cumine, F.; Nocera, G.; John, M. P.; Berlouis, L. E. A.; McGuire, T.; Tuttle, T.; Murphy, J. A. J. Am. Chem. Soc. 2016, 138, 7402–7410. doi:10.1021/jacs.6b03282 |
| 10. | Huang, H.; Si, P.; Wang, L.; Xu, Y.; Xu, X.; Zhu, J.; Jiang, H.; Li, W.; Chen, L.; Li, J. ChemMedChem 2015, 10, 1184–1199. doi:10.1002/cmdc.201500136 |
| 11. | Zhang, Z.; Zhang, W.; Li, J.; Liu, Q.; Liu, T.; Zhang, G. J. Org. Chem. 2014, 79, 11226–11233. doi:10.1021/jo5018487 |
| 12. | Bagley, M. C.; Dale, J. W.; Bower, J. Chem. Commun. 2002, 1682–1683. doi:10.1039/B203900A |
| 13. | Maiti, S.; Biswas, S.; Jana, U. J. Org. Chem. 2010, 75, 1674–1683. doi:10.1021/jo902661y |
| 37. | Andreani, A.; Burnelli, S.; Granaiola, M.; Leoni, A.; Locatelli, A.; Morigi, R.; Rambaldi, M.; Varoli, L.; Calonghi, N.; Cappadone, C.; Farruggia, G.; Zini, M.; Stefanelli, C.; Masotti, L.; Radin, N. S.; Shoemaker, R. H. J. Med. Chem. 2008, 51, 809–816. doi:10.1021/jm701246g |
| 38. | Furlan, A.; Colombo, F.; Kover, A.; Issaly, N.; Tintori, C.; Angeli, L.; Leroux, V.; Letard, S.; Amat, M.; Asses, Y.; Maigret, B.; Dubreuil, P.; Botta, M.; Dono, R.; Bosch, J.; Piccolo, O.; Passarella, D.; Maina, F. Eur. J. Med. Chem. 2012, 47, 239–254. doi:10.1016/j.ejmech.2011.10.051 |
| 39. | Andreani, A.; Granaiola, M.; Locatelli, A.; Morigi, R.; Rambaldi, M.; Varoli, L.; Calonghi, N.; Cappadone, C.; Farruggia, G.; Stefanelli, C.; Masotti, L.; Nguyen, T. L.; Hamel, E.; Shoemaker, R. H. J. Med. Chem. 2012, 55, 2078–2088. doi:10.1021/jm2012694 |
| 8. | Wang, B.; Lu, B.; Jiang, Y.; Zhang, Y.; Ma, D. Org. Lett. 2008, 10, 2761–2763. doi:10.1021/ol800900a |
| 9. | Yadav, J. S.; Reddy, B. V. S.; Premalatha, K. Synlett 2004, 963–966. doi:10.1055/s-2004-822898 |
| 31. | Zhu, Y.-P.; Yuan, J.-J.; Zhao, Q.; Lian, M.; Gao, Q.-H.; Liu, M.-C.; Yang, Y.; Wu, A.-X. Tetrahedron 2012, 68, 173–178. doi:10.1016/j.tet.2011.10.074 |
| 32. | Shinde, M. H.; Kshirsagar, U. A. Green Chem. 2016, 18, 1455–1458. doi:10.1039/C5GC02771C |
| 93. | Franz, J. F.; Kraus, W. B.; Zeitler, K. Chem. Commun. 2015, 51, 8280–8283. doi:10.1039/C4CC10270C |
| 5. | v. Pechmann, H. Ber. Dtsch. Chem. Ges. 1884, 17, 929–936. doi:10.1002/cber.188401701248 |
| 6. | Roslan, I. I.; Lim, Q.-X.; Han, A.; Chuah, G.-K.; Jaenicke, S. Eur. J. Org. Chem. 2015, 2351–2355. doi:10.1002/ejoc.201500227 |
| 7. | Ritson, D. J.; Spiteri, C.; Moses, J. E. J. Org. Chem. 2011, 76, 3519–3522. doi:10.1021/jo1025332 |
| 33. | Zhu, Y.-P.; Lian, M.; Jia, F.-C.; Liu, M.-C.; Yuan, J.-J.; Gao, Q.-H.; Wu, A.-X. Chem. Commun. 2012, 48, 9086–9088. doi:10.1039/c2cc34561g |
| 34. | Zhao, J.; Huang, H.; Wu, W.; Chen, H.; Jiang, H. Org. Lett. 2013, 15, 2604–2607. doi:10.1021/ol400773k |
| 95. | Meyers, C. Y.; Chan-Yu-King, R.; Hua, D. H.; Kolb, V. M.; Matthews, W. S.; Parady, T. E.; Horii, T.; Sandrock, P. B.; Hou, Y.; Xie, S. J. Org. Chem. 2003, 68, 500–511. doi:10.1021/jo025781w |
| 23. | Kumar, B. S. P. A.; Madhav, B.; Reddy, K. H. V.; Nageswar, Y. V. D. Tetrahedron Lett. 2011, 52, 2862–2865. doi:10.1016/j.tetlet.2011.03.110 |
| 26. | Xie, Y.; Wu, J.; Che, X.; Chen, Y.; Huang, H.; Deng, G.-J. Green Chem. 2016, 18, 667–671. doi:10.1039/C5GC01978H |
| 6. | Roslan, I. I.; Lim, Q.-X.; Han, A.; Chuah, G.-K.; Jaenicke, S. Eur. J. Org. Chem. 2015, 2351–2355. doi:10.1002/ejoc.201500227 |
| 22. | Roslan, I. I.; Ng, K.-H.; Chuah, G.-K.; Jaenicke, S. Eur. J. Org. Chem. 2017, 704–709. doi:10.1002/ejoc.201601410 |
| 27. | Ma, L.; Wang, X.; Yu, W.; Han, B. Chem. Commun. 2011, 47, 11333–11335. doi:10.1039/c1cc13568f |
| 28. | Lee, S. K.; Park, J. K. J. Org. Chem. 2015, 80, 3723–3729. doi:10.1021/acs.joc.5b00298 |
| 29. | Roslan, I. I.; Chuah, G.-K.; Jaenicke, S. Eur. J. Org. Chem. 2017, 671–675. doi:10.1002/ejoc.201601586 |
| 30. | Samanta, S.; Jana, S.; Mondal, S.; Monir, K.; Chandra, A. K.; Hajra, A. Org. Biomol. Chem. 2016, 14, 5073–5078. doi:10.1039/C6OB00656F |
| 21. | Roslan, I. I.; Ng, K.-H.; Wu, J.-E.; Chuah, G.-K.; Jaenicke, S. J. Org. Chem. 2016, 81, 9167–9174. doi:10.1021/acs.joc.6b01714 |
| 20. | Roslan, I. I.; Ng, K.-H.; Chuah, G.-K.; Jaenicke, S. Adv. Synth. Catal. 2016, 358, 364–369. doi:10.1002/adsc.201501012 |
| 21. | Roslan, I. I.; Ng, K.-H.; Wu, J.-E.; Chuah, G.-K.; Jaenicke, S. J. Org. Chem. 2016, 81, 9167–9174. doi:10.1021/acs.joc.6b01714 |
| 89. | Sahu, P. K.; Sahu, P. K.; Agarwal, D. D. RSC Adv. 2013, 3, 9854–9864. doi:10.1039/c3ra40993g |
| 90. | Sahu, P. K.; Sahu, P. K.; Jain, R.; Yadav, R.; Agarwal, D. D. Catal. Sci. Technol. 2012, 2, 2465–2475. doi:10.1039/c2cy20067h |
| 17. | Wang, Y.-F.; Toh, K. K.; Chiba, S.; Narasaka, K. Org. Lett. 2008, 10, 5019–5022. doi:10.1021/ol802120u |
| 18. | Guo, X.; Yu, R.; Li, H.; Li, Z. J. Am. Chem. Soc. 2009, 131, 17387–17393. doi:10.1021/ja907568j |
| 19. | Liu, W.; Jiang, H.; Zhang, M.; Qi, C. J. Org. Chem. 2010, 75, 966–968. doi:10.1021/jo902375k |
| 24. | Xie, J.; Jiang, H.; Cheng, Y.; Zhu, C. Chem. Commun. 2012, 48, 979–981. doi:10.1039/C2CC15813B |
| 25. | Gao, Q.-H.; Fei, Z.; Zhu, Y.-P.; Lian, M.; Jia, F.-C.; Liu, M.-C.; She, N.-F.; Wu, A.-X. Tetrahedron 2013, 69, 22–28. doi:10.1016/j.tet.2012.10.072 |
| 79. | Hilal, H. S.; Ali-Shtayeh, M. S.; Arafat, R.; Al-Tel, T.; Voelter, W.; Barakat, A. Eur. J. Med. Chem. 2006, 41, 1017–1024. doi:10.1016/j.ejmech.2006.03.025 |
| 91. | Kumar, G.; Sharma, P. K.; Sharma, S.; Singh, S. J. Chem. Pharm. Res. 2015, 7, 710–714. |
| 92. | Fogla, A. K.; Ankodia, V.; Sharma, P. K.; Kumar, M. Res. Chem. Intermed. 2009, 35, 35–41. doi:10.1007/s11164-008-0006-4 |
| 42. | Andreani, A.; Granaiola, M.; Leoni, A.; Locatelli, A.; Morigi, R.; Rambaldi, M.; Varoli, L.; Lannigan, D.; Smith, J.; Scudiero, D.; Kondapaka, S.; Shoemaker, R. H. Eur. J. Med. Chem. 2011, 46, 4311–4323. doi:10.1016/j.ejmech.2011.07.001 |
| 43. | Chao, Q.; Sprankle, K. G.; Grotzfeld, R. M.; Lai, A. G.; Carter, T. A.; Velasco, A. M.; Gunawardane, R. N.; Cramer, M. D.; Gardner, M. F.; James, J.; Zarrinkar, P. P.; Patel, H. K.; Bhagwat, S. S. J. Med. Chem. 2009, 52, 7808–7816. doi:10.1021/jm9007533 |
| 44. | Shen, H. C.; Ding, F.-X.; Deng, Q.; Wilsie, L. C.; Krsmanovic, M. L.; Taggart, A. K.; Carballo-Jane, E.; Ren, N.; Cai, T. Q.; Wu, T.-J.; Wu, K. K.; Cheng, K.; Chen, Q.; Wolff, M. S.; Tong, X.; Holt, T. G.; Waters, M. G.; Hammond, M. L.; Tata, J. R.; Colletti, S. L. J. Med. Chem. 2009, 52, 2587–2602. doi:10.1021/jm900151e |
| 40. | Palkar, M.; Noolvi, M.; Sankangoud, R.; Maddi, V.; Gadad, A.; Nargund, L. V. G. Arch. Pharm. 2010, 343, 353–359. doi:10.1002/ardp.200900260 |
| 41. | Ager, I. R.; Barnes, A. C.; Danswan, G. W.; Hairsine, P. W.; Kay, D. P.; Kennewell, P. D.; Matharu, S. S.; Miller, P.; Robson, P.; Rowlands, D. A.; Tully, W. R.; Westwood, R. J. Med. Chem. 1988, 31, 1098–1115. doi:10.1021/jm00401a009 |
| 93. | Franz, J. F.; Kraus, W. B.; Zeitler, K. Chem. Commun. 2015, 51, 8280–8283. doi:10.1039/C4CC10270C |
| 95. | Meyers, C. Y.; Chan-Yu-King, R.; Hua, D. H.; Kolb, V. M.; Matthews, W. S.; Parady, T. E.; Horii, T.; Sandrock, P. B.; Hou, Y.; Xie, S. J. Org. Chem. 2003, 68, 500–511. doi:10.1021/jo025781w |
| 96. | Stasyuk, A. J.; Banasiewicz, M.; Cyrański, M. K.; Gryko, D. T. J. Org. Chem. 2012, 77, 5552–5558. doi:10.1021/jo300643w |
| 97. | Zhang, Y.; Chen, Z.; Wu, W.; Zhang, Y.; Su, W. J. Org. Chem. 2013, 78, 12494–12504. doi:10.1021/jo402134x |
| 56. | Balwe, S. G.; Jeong, Y. T. RSC Adv. 2016, 6, 107225–107232. doi:10.1039/C6RA24183B |
| 57. | Yanagisawa, S.; Ueda, K.; Taniguchi, T.; Itami, K. Org. Lett. 2008, 10, 4673–4676. doi:10.1021/ol8019764 |
| 54. | Wang, J.; Li, J.; Zhu, Q. Org. Lett. 2015, 17, 5336–5339. doi:10.1021/acs.orglett.5b02694 |
| 55. | Mishra, S.; Monir, K.; Mitra, S.; Hajra, A. Org. Lett. 2014, 16, 6084–6087. doi:10.1021/ol5028893 |
| 49. | Guchhait, S. K.; Chaudhary, V. Org. Biomol. Chem. 2014, 12, 6694–6705. doi:10.1039/C4OB00882K |
| 50. | Wu, Z.; Huang, Q.; Zhou, X.; Yu, L.; Li, Z.; Wu, D. Eur. J. Org. Chem. 2011, 5242–5245. doi:10.1002/ejoc.201100834 |
| 51. | Gao, J.; Zhu, J.; Chen, L.; Shao, Y.; Zhu, J.; Huang, Y.; Wang, X.; Lv, X. Tetrahedron Lett. 2014, 55, 3367–3373. doi:10.1016/j.tetlet.2014.04.070 |
| 52. | Shi, B.; Zhu, Z.; Zhu, Y.-S.; Zhou, D.; Wang, J.; Zhou, P.; Jing, H. Org. Biomol. Chem. 2016, 14, 2978–2984. doi:10.1039/C6OB00102E |
| 102. | Posevins, D.; Suta, K.; Turks, M. Eur. J. Org. Chem. 2016, 1414–1419. doi:10.1002/ejoc.201600013 |
| 53. | Zhang, X.; Jia, J.; Ma, C. Org. Biomol. Chem. 2012, 10, 7944–7948. doi:10.1039/c2ob26211h |
| 45. | Yousefi, B. H.; Manook, A.; Drzezga, A.; von Reutern, B.; Schwaiger, M.; Wester, H.-J.; Henriksen, G. J. Med. Chem. 2011, 54, 949–956. doi:10.1021/jm101129a |
| 46. | Yousefi, B. H.; Drzezga, A.; von Reutern, B.; Manook, A.; Schwaiger, M.; Wester, H.-J.; Henriksen, G. ACS Med. Chem. Lett. 2011, 2, 673–677. doi:10.1021/ml200123w |
| 99. | Denmark, S. E.; Beutner, G. L. Angew. Chem., Int. Ed. 2008, 47, 1560–1638. doi:10.1002/anie.200604943 |
| 47. | Clements-Jewery, S.; Danswan, G.; Gardner, R. C.; Matharu, S. S.; Murdoch, R.; Tully, W. R.; Westwood, R. J. Med. Chem. 1988, 31, 1220–1226. doi:10.1021/jm00401a025 |
| 48. | Christodoulou, M. S.; Colombo, F.; Passarella, D.; Ieronimo, G.; Zuco, V.; De Cesare, M.; Zunino, F. Bioorg. Med. Chem. 2011, 19, 1649–1657. doi:10.1016/j.bmc.2011.01.039 |
| 100. | Tsuji, H.; Tanaka, I.; Endo, K.; Yamagata, K.-i.; Nakamura, M.; Nakamura, E. Org. Lett. 2009, 11, 1845–1847. doi:10.1021/ol9003542 |
| 101. | Thirupathaiah, B.; Seo, S. Chem. Commun. 2015, 51, 4216–4219. doi:10.1039/C4CC10016F |
© 2017 Roslan et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)