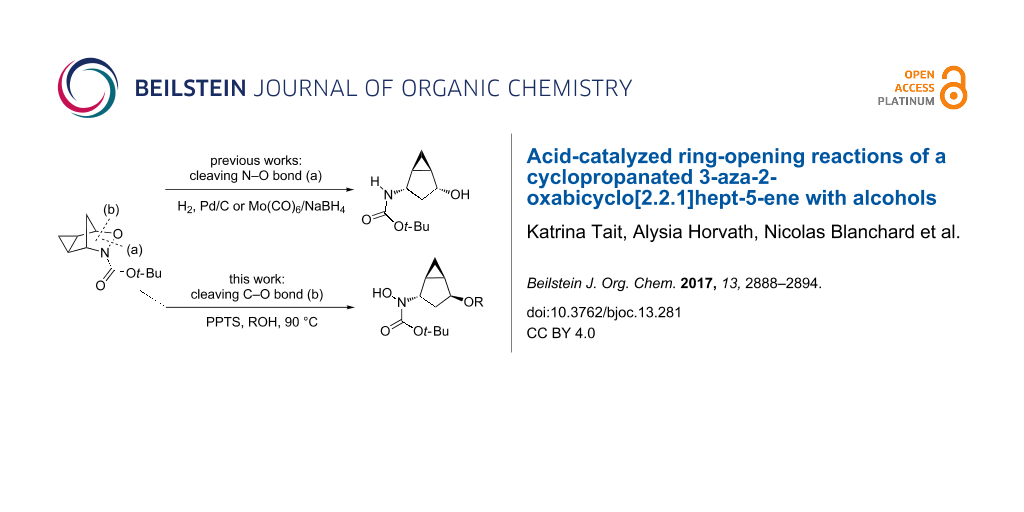
Abstract
The acid-catalyzed ring-opening reactions of a cyclopropanated 3-aza-2-oxabicylic alkene using alcohol nucleophiles were investigated. Although this acid-catalyzed ring-opening reaction did not cleave the cyclopropane unit as planned, this represent the first examples of ring-openings of cyclopropanated 3-aza-2-oxabicyclo[2.2.1]alkenes that lead to the cleavage of the C–O bond instead of the N–O bond. Different acid catalysts were tested and it was found that pyridinium toluenesulfonate in methanol gave the best yields in the ring-opening reactions. The scope of the reaction was successfully expanded to include primary, secondary, and tertiary alcohol nucleophiles. Through X-ray crystallography, the stereochemistry of the product was determined which confirmed an SN2-like mechanism to form the ring-opened product.

Graphical Abstract
Introduction
Heterobicyclic alkenes are useful templates to generate complex cyclic and acyclic systems [1,2]. 3-Aza-2-oxabicyclic alkenes are particularly interesting due to their asymmetric nature and the ability to modify selected components of the molecule to create vastly different products. 3-Aza-2-oxabicyclic alkenes are generally modified in one of four ways (Scheme 1). One of the most interesting manipulations of 3-aza-2-oxabicyclic alkenes is the modification of the alkene component. The manipulation of the olefin can lead to a wide variety of products often in a single step, which is synthetically useful to create many highly substituted products with different stereochemical outcomes (Scheme 2). There are many reported examples in the literature of the modification of the alkene component which includes the reduction to form alkane 8 [3], oxidative cleavage of the C=C bond to form 9 [4], ring-opening metathesis to form functionalized alkenes 10 and 11 [4], dihydroxylation to form diol 12 [5], ruthenium-catalyzed [2 + 2] cycloaddition with unsymmetrical alkynes to form regioisomers 13 and 14 [6], and cycloadditions using nitrile oxides to provide 15 and 16 [7].
![[1860-5397-13-281-i1]](/bjoc/content/inline/1860-5397-13-281-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: General reaction pathways for 3-aza-2-oxabicyclic alkenes.
Scheme 1: General reaction pathways for 3-aza-2-oxabicyclic alkenes.
![[1860-5397-13-281-i2]](/bjoc/content/inline/1860-5397-13-281-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Various reactions involving modification of the alkene component of 3-aza-2-oxabicyclic alkenes.
Scheme 2: Various reactions involving modification of the alkene component of 3-aza-2-oxabicyclic alkenes.
In the literature, there are also many examples of the cleavage of the C–O bond of 3-aza-2-oxabicyclic alkenes 1 (Scheme 3). This includes the use of protic acid [8], using metal catalysts such as Pd [9], Fe or Cu [10], In [11], organozinc or Grignard reagents [12], Rh [13], and Ru [14] catalysts.
![[1860-5397-13-281-i3]](/bjoc/content/inline/1860-5397-13-281-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Various reactions involving cleavage of the C–O bond of 3-aza-2-oxabicyclic alkenes.
Scheme 3: Various reactions involving cleavage of the C–O bond of 3-aza-2-oxabicyclic alkenes.
Another interesting modification of the alkene component is cyclopropanation. To date, there are a few reported examples in the literature of the cyclopropanation of 3-aza-2-oxabicyclic alkenes [15-17]. The addition of a cyclopropane unit adds ring-strain to the molecule that could lead to different pathways for ring-opening and further reactivity.
While the reactivity of 3-aza-2-oxabicyclo[2.2.1]hept-5-enes has been extensively studied (Scheme 1 and Scheme 2), there are only two examples in the literature investigating the reactivity of the cyclopropanated derivative (Scheme 4) and in both studies, cleavage of the N–O bond (b) was observed. While the Buono group demonstrated the reductive N–O bond cleavage to produce compound 25 as proof-of-principle [16], the Miller group reported the use of the cyclopropanated compound for the synthesis of 2’,3’-methano carbocyclic nucleosides via compound 24 (Scheme 5) [17]. Carbocyclic nucleosides are important synthetic targets because of their use as antiviral and antitumor agents [17]. Replacing the oxygen unit in the parent furanose ring with a methylene unit helps to stabilize the structure against cleavage by nucleoside phosphorylases or hydrolases [18,19]. The addition of a cyclopropane unit could provide further rigidity that could better stabilize the compound, thereby enhancing its biological activity. Both of these reported ring-openings of cyclopropanated 3-aza-2-oxabicyclo[2.2.1]alkenes reductively cleave the N–O bond (a) (Scheme 4), therefore, no examples cleaving the C–O bond have been reported in the literature. In this paper, we aim to explore the use of an acid catalyst with an alcohol nucleophile on the ring-opening of cyclopropanated 3-aza-2-oxabicyclic compound 19 for the cleavage of the C–O bond (b) (Scheme 4). We initially anticipated that the SN2’ type ring-opening would occur which would lead to the formation of ring-opened product 27 (Scheme 5). However, in all cases tested, only the SN2 type ring-opened product 26 was formed.
![[1860-5397-13-281-i4]](/bjoc/content/inline/1860-5397-13-281-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Ring-opening reactions of cyclopropanated 3-aza-2-oxabicyclic alkenes.
Scheme 4: Ring-opening reactions of cyclopropanated 3-aza-2-oxabicyclic alkenes.
![[1860-5397-13-281-i5]](/bjoc/content/inline/1860-5397-13-281-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Different possible ring-opening pathways of cyclopropanated 3-aza-2-oxabicyclic alkenes.
Scheme 5: Different possible ring-opening pathways of cyclopropanated 3-aza-2-oxabicyclic alkenes.
Results and Discussion
A variety of different acid catalysts was screened and the results are summarized in Table 1. In the presence of a Lewis acid catalyst (Table 1, entries 1–3), the reaction did not proceed as seen with FeCl3 (Table 1, entry 1) or produced ring-opened product 26 in low yields (Table 1, entries 2 and 3). The effect of inorganic protic acids was then investigated (Table 1, entries 4–6), producing moderate yields of the ring-opened product. The use of fluoroboric acid (Table 1, entry 4) and sulfuric acid (Table 1, entry 5) produced ring-opened product 26 in 45% and 48% yield, respectively, while using nitric acid increased the yield to 56% with trace amount of starting material 23a recovered. Finally, the effect of organic protic acids was investigated (Table 1, entries 7–9) which produced ring-opened product 26 in low to moderate yields. The use of p-toluenesulfonic acid monohydrate produced the ring-opened product at a yield of 38% (Table 1, entry 7) while using camphorsulfonic acid (CSA) increased the yield to 50% but took 46 hours to go to completion (Table 1, entry 8). The organic acid pyridinium p-toluenesulfonate (PPTS) produced the highest yield of ring-opened product with a 61% yield (Table 1, entry 9) and was chosen to further optimize reaction conditions.
Table 1: Effects of acid catalysts on the ring-opening reaction of cyclopropanated 3-aza-2-oxabicyclic alkene 23a with alcohols.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-281-i7.svg?max-width=637&scale=1.0)
|
|||
| Entry | Catalyst | Time (h) | Yield (%)a |
| 1 | FeCl3 | 22 | 0 |
| 2 | ZrCl4 | 24 | 15 |
| 3 | AlCl3 | 24 | 32 |
| 4 | HBF4 | 24 | 45 |
| 5 | H2SO4 | 24 | 48 |
| 6 | HNO3 | 24 | 56b |
| 7 | p-TsOH·H2O | 24 | 38b |
| 8 | CSA | 46 | 50 |
| 9 | PPTS | 24 | 61b |
aIsolated yield after column chromatography. b1–4% of starting material was recovered.
A variety of solvents was screened, including polar protic, aromatic, and polar aprotic solvents (Table 2). When methanol was used as the nucleophile and polar protic solvent, the reaction yielded the ring-opened product in a 61% yield (Table 2, entry 1). The use of the aromatic solvent toluene gave a moderate yield of 47% but took 48 hours to go to completion (Table 2, entry 2). Polar aprotic solvents DCE, THF, and 1,4-dioxane were investigated, which produced the ring-opened product 26 in moderate yields (Table 2, entries 3–5). Using DMF decreased the yield significantly to only 6%, with 57% of starting material 23a recovered after eight days (Table 2, entry 6). The use of the polar aprotic solvent acetonitrile decreased the yield of the reaction to 27% and took almost five days to complete with 3% of starting material 23a recovered (Table 2, entry 7). Finally, using DMSO decreased the yield to 32% with 5% of starting material recovered after 49 hours (Table 2, entry 8). Since the best result was obtained without the use of a cosolvent, the polar protic nucleophile will be used as both the nucleophile and solvent.
Table 2: Effect of solvent on the ring-opening reactions of cyclopropanated 3-aza-2-oxabicyclic alkene 23a with alcohols.
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-281-i8.svg?max-width=637&scale=1.0)
|
||||
| Entry | Solvent | Time (h) |
Yield
26a (%)a |
Recovered
23a (%)a |
| 1 | MeOH | 24 | 61 | 4 |
| 2 | toluene | 48 | 47 | 0 |
| 3 | DCE | 24 | 41 | 0 |
| 4 | THF | 48 | 39 | 0 |
| 5 | 1,4-dioxane | 24 | 39 | 3 |
| 6 | DMF | 190 | 6 | 57 |
| 7 | CH3CN | 117 | 27 | 3 |
| 8 | DMSO | 49 | 32 | 5 |
aIsolated yield after column chromatography.
To study the scope of the reaction, the use of different alcohol nucleophiles was tested (Table 3). With a primary alcohol, a decrease in reactivity was seen with increasing chain length (Me < Et < n-Bu; Table 3, entries 1–3) while maintaining reasonable yields. When 2-methoxyethanol was used as the nucleophile, the yield was decreased to 42% (Table 3, entry 4) while using isobutyl alcohol produced a similar yield of 41% (Table 3, entry 5). Using 2-methylbutanol produced a 34% yield of a 1:1 diastereomeric ratio of product (Table 3, entry 6) and using allyl alcohol produced ring-opened product 26g in a 38% yield (Table 3, entry 7). The use of secondary alcohols generally resulted in a decrease of yield of the ring-opened product (Table 3, entries 8–11). Isopropanol achieved a moderate yield of 51% (Table 3, entry 8) while using 2-butanol decreased the yield to 28% of a 1:1 diastereomeric ratio of product (Table 3, entry 9). The cyclic alcohols cyclohexanol and cyclopentanol (Table 3, entries 10 and 11) produced low amounts of the ring-opened alcohol in a 24% and 26% yield, respectively. The use of a tertiary alcohol surprisingly resulted in a moderate yield, with tert-butanol producing a 50% yield of product 26l (Table 3, entry 12, preparation of compound 26l from 23a and t-BuOH with PPTS was already published in reference [20]). When the aromatic alcohol phenol was investigated as a nucleophile, no reaction occurred though no starting material was recovered (Table 3, entry 13). Although in most cases, the starting material was completely consumed, the yields of these ring-opening reactions were only moderate (26–61%). This may be due to the decomposition or polymerization of the cyclopropanated 3-aza-2-oxabicyclic alkene under the reaction conditions. Through X-ray crystallography [20] and 1D NOESY 1H NMR the stereochemistry of the products was confirmed, with the nucleophile added syn to the cyclopropane ring and anti to the amino alcohol group.
Table 3: Scope of the reaction with different alcohol nucleophiles.
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-281-i9.svg?max-width=637&scale=1.0)
|
|||
| Entry | ROH | Products | Yield (%)a |
| 1 | MeOH | 26a | 61 |
| 2 | EtOH | 26b | 51 |
| 3 | n-BuOH | 26c | 36 |
| 4 |
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-281-i10.svg?max-width=637&scale=1.0)
|
26d | 42 |
| 5 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-13-281-i11.svg?max-width=637&scale=1.0)
|
26e | 41 |
| 6 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-13-281-i12.svg?max-width=637&scale=1.0)
|
26f | 34b |
| 7 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-13-281-i13.svg?max-width=637&scale=1.0)
|
26g | 38 |
| 8 | iPrOH | 26h | 51 |
| 9 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-13-281-i14.svg?max-width=637&scale=1.0)
|
26i | 28b |
| 10 | CyOH | 26j | 24 |
| 11 | cyclopentanol | 26k | 26 |
| 12 | t-BuOH | 26l | 50 |
| 13 | PhOH | 26m | 0 |
aIsolated yield after column chromatography. bProduced inseparable 1:1 diastereomeric products.
When forming the ring-opening product, there are two possible mechanisms (Scheme 6).
![[1860-5397-13-281-i6]](/bjoc/content/inline/1860-5397-13-281-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Possible mechanisms for the nucleophilic ring-opening of cyclopropanated 3-aza-2-oxabicyclic alkene 19.
Scheme 6: Possible mechanisms for the nucleophilic ring-opening of cyclopropanated 3-aza-2-oxabicyclic alkene ...
If the oxygen atom is first protonated followed by cleavage of the C–O bond as seen in path A, the free carbocation 28 would form in an SN1-like manner. The nucleophile could therefore attack from either the top or the bottom, forming products 26a and 29. Alternatively, in path B the oxygen atom could be protonated and undergo an SN2-like mechanism with the nucleophile attacking from the open top face seen in 30, forming sole product 26a. If a free carbocation was formed as shown in path A, both stereoisomers 26a and 29 should have been observed, which was not evident. Also, if a free carbocation was formed the product likely would have undergone rearrangement of the cyclopropyl cation to ring-open the cyclopropane ring and form either a five or six-membered product, however, the cyclopropane is proved to be intact. Therefore, since only one single product 26a was observed, it is confirmed the product is formed through an SN2-like pathway seen in path B.
Conclusion
In conclusion, we have demonstrated the first examples of acid-catalyzed nucleophilic ring-opening reactions of a cyclopropanated 3-aza-2-oxabicyclic alkene with alcohols. Although this acid-catalyzed ring-opening reaction did not cleave the cyclopropane unit as planned, this represent the first examples of ring-openings of cyclopropanated 3-aza-2-oxabicyclo[2.2.1]alkenes that lead to the cleavage of the C–O bond instead of the N–O bond. Different acid catalysts were tested and it was found that pyridinium toluenesulfonate in methanol gave the best yields in the ring-opening reactions. The scope of the reaction was successfully expanded to include primary, secondary, and tertiary alcohol nucleophiles. Through X-ray crystallography, the stereochemistry of the product was determined which confirmed an SN2-like mechanism to form the ring-opened product. Further investigation of the ring-opening reactions of cyclopropanated 3-aza-2-oxabicyclo[2.2.1]alkenes using other metal catalysts, such as those listed in Scheme 3, is ongoing in our laboratory.
Supporting Information
Experimental procedures and copies of 1H and 13C NMR spectra for compounds are provided in the Supporting Information.
| Supporting Information File 1: Experimental. | ||
| Format: PDF | Size: 466.3 KB | Download |
| Supporting Information File 2: NMR spectra. | ||
| Format: PDF | Size: 1.2 MB | Download |
References
-
Bournaud, C.; Chung, F.; Pérez Luna, A.; Pasco, M.; Errasti, G.; Lecourt, T.; Micouin, L. Synthesis 2009, 869–887. doi:10.1055/s-0028-1087980
Return to citation in text: [1] -
Tam, W.; Cockburn, N. Synlett 2010, 1170–1189. doi:10.1055/s-0029-1219780
Return to citation in text: [1] -
Just, G.; Cutrone, L. Can. J. Chem. 1976, 54, 867–870. doi:10.1139/v76-125
Return to citation in text: [1] -
Ellis, J. M.; King, S. B. Tetrahedron Lett. 2002, 43, 5833–5835.
Return to citation in text: [1] [2] -
Ranganathan, S.; George, K. S. Tetrahedron 1997, 53, 3347–3362. doi:10.1016/S0040-4020(97)00057-4
Return to citation in text: [1] -
Durham, R.; Mandel, J.; Blanchard, N.; Tam, W. Can. J. Chem. 2011, 89, 1494–1505. doi:10.1139/V11-135
Return to citation in text: [1] -
Bodnar, B. S.; Miller, M. J. J. Org. Chem. 2007, 72, 3929–3932.
Return to citation in text: [1] -
Muxworthy, J. P.; Wilkinson, J. A.; Procter, G. Tetrahedron Lett. 1995, 36, 7535–7538. doi:10.1016/0040-4039(95)01525-6
Return to citation in text: [1] -
Miller, A.; Procter, G. Tetrahedron Lett. 1990, 31, 1043–1046.
Return to citation in text: [1] -
Surman, M. D.; Miller, M. J. J. Org. Chem. 2001, 66, 2466–2469. doi:10.1021/jo010094a
Return to citation in text: [1] -
Yang, B.; Miller, M. J. J. Tetrahedron Lett. 2009, 74, 7990–7993. doi:10.1016/j.tetlet.2009.12.006
Return to citation in text: [1] -
Surman, M. D.; Mulvihill, M. J.; Miller, M. J. J. Org. Chem. 2002, 67, 4115–4121. doi:10.1351/pac200678020463
Return to citation in text: [1] -
Machin, B. P.; Ballantine, M.; Mandel, J.; Blanchard, N.; Tam, W. J. Org. Chem. 2009, 74, 7261–7266. doi:10.1021/jo901248w
Return to citation in text: [1] -
Machin, B. P.; Howell, J. H.; Mandel, J.; Blanchard, N.; Tam, W. Org. Lett. 2009, 11, 2077–2080. doi:10.1021/ol900454q
Return to citation in text: [1] -
Carlson, E.; Duret, G.; Blanchard, N.; Tam, W. Synth. Commun. 2015, 46, 55–62.
Return to citation in text: [1] -
Bigeault, J.; Giordano, L.; De Riggi, I.; Gimbert, Y.; Buono, G. Org. Lett. 2007, 9, 3567–3570. doi:10.1021/ol071386m
Return to citation in text: [1] [2] -
Ji, C.; Miller, M. J. Tetrahedron Lett. 2010, 51, 3789–3791. doi:10.1016/j.tetlet.2010.05.062
Return to citation in text: [1] [2] [3] -
Crimmins, M. T. Tetrahedron 1998, 54, 9229–9272. doi:10.1016/S0040-4020(98)00320-2
Return to citation in text: [1] -
Altona, C.; Sundaralingam, M. J. Am. Chem. Soc. 1972, 94, 8205–8212. doi:10.1021/ja00778a043
Return to citation in text: [1] -
Lough, A. J.; Tait, K.; Horvath, A.; Tam, W. IUCrData 2017, 2, x171419. doi:10.1107/S2414314617014195
Return to citation in text: [1] [2]
| 18. | Crimmins, M. T. Tetrahedron 1998, 54, 9229–9272. doi:10.1016/S0040-4020(98)00320-2 |
| 19. | Altona, C.; Sundaralingam, M. J. Am. Chem. Soc. 1972, 94, 8205–8212. doi:10.1021/ja00778a043 |
| 17. | Ji, C.; Miller, M. J. Tetrahedron Lett. 2010, 51, 3789–3791. doi:10.1016/j.tetlet.2010.05.062 |
| 17. | Ji, C.; Miller, M. J. Tetrahedron Lett. 2010, 51, 3789–3791. doi:10.1016/j.tetlet.2010.05.062 |
| 1. | Bournaud, C.; Chung, F.; Pérez Luna, A.; Pasco, M.; Errasti, G.; Lecourt, T.; Micouin, L. Synthesis 2009, 869–887. doi:10.1055/s-0028-1087980 |
| 2. | Tam, W.; Cockburn, N. Synlett 2010, 1170–1189. doi:10.1055/s-0029-1219780 |
| 5. | Ranganathan, S.; George, K. S. Tetrahedron 1997, 53, 3347–3362. doi:10.1016/S0040-4020(97)00057-4 |
| 15. | Carlson, E.; Duret, G.; Blanchard, N.; Tam, W. Synth. Commun. 2015, 46, 55–62. |
| 16. | Bigeault, J.; Giordano, L.; De Riggi, I.; Gimbert, Y.; Buono, G. Org. Lett. 2007, 9, 3567–3570. doi:10.1021/ol071386m |
| 17. | Ji, C.; Miller, M. J. Tetrahedron Lett. 2010, 51, 3789–3791. doi:10.1016/j.tetlet.2010.05.062 |
| 16. | Bigeault, J.; Giordano, L.; De Riggi, I.; Gimbert, Y.; Buono, G. Org. Lett. 2007, 9, 3567–3570. doi:10.1021/ol071386m |
| 13. | Machin, B. P.; Ballantine, M.; Mandel, J.; Blanchard, N.; Tam, W. J. Org. Chem. 2009, 74, 7261–7266. doi:10.1021/jo901248w |
| 14. | Machin, B. P.; Howell, J. H.; Mandel, J.; Blanchard, N.; Tam, W. Org. Lett. 2009, 11, 2077–2080. doi:10.1021/ol900454q |
| 11. | Yang, B.; Miller, M. J. J. Tetrahedron Lett. 2009, 74, 7990–7993. doi:10.1016/j.tetlet.2009.12.006 |
| 8. | Muxworthy, J. P.; Wilkinson, J. A.; Procter, G. Tetrahedron Lett. 1995, 36, 7535–7538. doi:10.1016/0040-4039(95)01525-6 |
| 12. | Surman, M. D.; Mulvihill, M. J.; Miller, M. J. J. Org. Chem. 2002, 67, 4115–4121. doi:10.1351/pac200678020463 |
| 20. | Lough, A. J.; Tait, K.; Horvath, A.; Tam, W. IUCrData 2017, 2, x171419. doi:10.1107/S2414314617014195 |
| 6. | Durham, R.; Mandel, J.; Blanchard, N.; Tam, W. Can. J. Chem. 2011, 89, 1494–1505. doi:10.1139/V11-135 |
| 10. | Surman, M. D.; Miller, M. J. J. Org. Chem. 2001, 66, 2466–2469. doi:10.1021/jo010094a |
| 20. | Lough, A. J.; Tait, K.; Horvath, A.; Tam, W. IUCrData 2017, 2, x171419. doi:10.1107/S2414314617014195 |
© 2017 Tait et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)