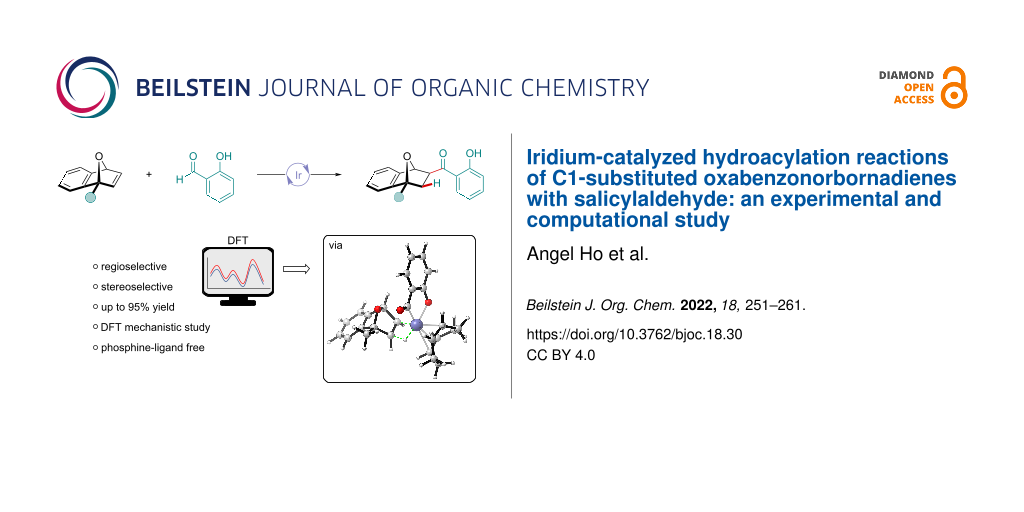
Abstract
An experimental and theoretical investigation on the iridium-catalyzed hydroacylation of C1-substituted oxabenzonorbornadienes with salicylaldehyde is reported. Utilizing commercially available [Ir(COD)Cl]2 in the presence of 5 M KOH in dioxane at 65 °C, provided a variety of hydroacylated bicyclic adducts in up to a 95% yield with complete stereo- and regioselectivity. The mechanism and origins of selectivity in the iridium-catalyzed hydroacylation reaction has been examined at the M06/Def2TZVP level of theory. The catalytic cycle consists of three key steps including oxidative addition into the aldehyde C–H bond, insertion of the olefin into the iridium hydride, and C–C bond-forming reductive elimination. Computational results indicate the origin of regioselectivity is involved in the reductive elimination step.

Graphical Abstract
Introduction
Organic synthesis is the art and science of selective molecular engineering [1]. To date, organic synthesis has largely been governed by the interconversion of pre-existing functional groups through the use of more traditional transition-metal-catalyzed cross-coupling reactions [2-5]. Although these reactions have revolutionized the modern chemist’s synthetic toolbox, prior installation of these functional groups requires a number of steps, leading to undesired side-products and reduced overall yield. An attractive alternative is the catalytic activation and subsequent functionalization of otherwise inert carbon–hydrogen bonds [6-13]. Hydroacylation reactions, the formal addition of an aldehyde C–H bond across a C–C π-system, has emerged as a powerful, and highly atom-economic approach to synthesize ketones. As such, C–H functionalizations are inherently both environmentally benign and economically attractive.
Transition-metal-catalyzed reactions of strained bicyclic derivatives have been an intense area of research in the last 20 years (Scheme 1) [14-17]. Of particular interest is oxabenzonorbornadiene (OBD, 1), as it bears multiple points of reactivity that allow for diverse functionalization. Over the years, several interesting transformations have been investigated such as cycloadditions 4 [18-23], dimerizations 3 [24-27], isomerizations [28-31], among other reactions that have been reported [32-38]. The nucleophilic ring-opening reactions of heterobicyclic alkenes are of particular interest [39-53], as they provide access to a broad family of synthetic building blocks bearing multiple stereocenters in a single step 2 [54]. Application of these functionalized intermediates have found use in the total synthesis of (+)-norchelidonine (an isoquinoline alkaloid) [55], sertraline (an antidepressant) [56], and arnottin I (an anti-inflammatory) [57].
Although OBD 1 has been shown to undergo many different modes of reactivity in both a stereo- and enantioselective manner, the regioselectivity of such reactions is still undefined (Scheme 1) [58]. While the chemistry of symmetric OBD derivatives is well established, their unsymmetrically substituted counterparts 5 have remained underexplored (Scheme 1). Upon C1-substitution, the reactivity of C1-substituted OBDs 5 can greatly differ, as described by Allen and co-workers in their 2007 report on rhodium-catalyzed cyclodimerization reactions [59]. Moreover, desymmetrization of OBD produces more unique sites of reactivity allowing for the production of regioisomeric products. In 2019, Deng et al. described syn-stereocontrolled ring-opening reactions of oxa- and azabicyclic olefins with dialkylzinc reagents catalyzed by a nickel compound (Scheme 1) [60]. The reaction was entirely stereoselective; however, unsymmetrical OBDs 5 produced mixtures of regioisomers 6 and 7. In the same year, Hill and co-workers published a study about the regioselective nucleophilic ring opening of C1-substituted OBDs 5 with water and alcohol and with an iridium compound as a catalyst (Scheme 1) [61]. Their study found the electronic nature of the C1-substituent controlled the regioselectivity of the reaction. Electron-donating groups (EDGs) led to naphthol compounds 9, while electron-withdrawing groups (EWGs) led to the anticipated ring-opened 1,1,2-trisubstituted naphthalene framework 10 [61]. On the other hand, Edmunds and co-workers described a ring-opening reaction of C1-substituted OBDs 5 with arylboronic acids that was catalyzed by rhodium/diene to afford the 1,2,4-trisubstituted naphthalene framework 8 with complete regio- and stereocontrol (Scheme 1) [62,63].
![[1860-5397-18-30-i1]](/bjoc/content/inline/1860-5397-18-30-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Previously reported metal-catalyzed reactions of heterobicyclic alkenes and applications towards the synthesis of biologically active compounds (top). Representative examples of regioselectivity in different metal-catalyzed ring-opening reactions of C1-substituted oxabenzonorbornadiene derivatives (bottom left). Nishimura’s seminal report on iridium-catalyzed hydroacylation reactions of bicyclic alkenes and the context of this work.
Scheme 1: Previously reported metal-catalyzed reactions of heterobicyclic alkenes and applications towards th...
In 2015, the Nishimura group reported the first iridium-catalyzed addition of salicylaldehydes 14 to bicyclic alkenes 11 (Scheme 1) [64]. Although a variety of carbo- and heterobicyclic alkenes was investigated, the study was limited by the number of unsymmetrical coupling partners. In their seminal report, the authors were able to produce hydroacylated adducts 15a and 15b in good yield. On the basis of the aforementioned literature, several different products can be formed based on a complex relationship between the reactants, C1-substituent, and reaction conditions [58]; therefore, it is paramount to understand of the effects that C1-substitution has on the reactions. Inspired by the initial work of Nishimura and co-workers [64], we pursued a study on the effects of C1-substitution on the iridium-catalyzed hydroacylation reactions of unsymmetrical OBDs with salicylaldehyde. To further understand the observed regioselectivity, an in-depth investigation into the reaction mechanism of the iridium-catalyzed hydroacylation reaction was carried out by preforming density functional theory calculations. We set out to confirm the catalytic cycle in detail, the geometries of the intermediates, the energy profiles of the reactions, and most importantly, the origin of regioselectivity.
Results and Discussion
Experimental
We began our investigation with C1-methyl-substituted OBD (MeOBD, 13b) (Table 1). The use of [Ir(COD)Cl]2 (5 mol %) and 5 M KOH in H2O (10 mol %) in 1,4-dioxane at 65 °C for 20 h were the optimal conditions for the hydroacylation reaction (Table 1, entry 1) exclusively affording the C3-regioisomer 15b in a 61% isolated yield. To the effect of the dummy ligand present on the active iridium species, tetrabutylammonium salts were added (Table 1, entries 4 and 5); however, these were not as efficient in the reaction. Other iridium sources (Table 1, entries 2 and 3) proved to be not as effective in promoting the reaction, with Vaska’s complex failing to react. Alternative bases (Table 1, entries 7–10) were tested; however, the reaction produced isomerized naphthol derivative 17 rather than the predicted addition product. These results indicate the formation of a phenoxoiridium(I) species assists in the oxidative addition of the C–H bond, as previously put forth by Nishimura and co-workers [64]. Isomerization of the oxabicyclic starting material to 17 may operate through a similar process described at Hill and co-workers (Scheme 1) [61]. Irreversible C–O insertion of the chloroiridium(I) species at the more electron-dense C1-position affords an enyliridium(III) alkoxide complex which eventually leads to the formation of isomerized 1-naphthol products [61]. Interestingly, the loading of the iridium precatalyst (Table 1, entries 11–13) also had a substantial effect on the isomerization of 13b, with increased loading producing more byproduct. Other solvents (Table 1, entries 16–20) were explored but were not as efficient in the reaction, producing mixtures of 15b and 17.
Table 1: Optimization of the hydroacylation reaction of MeOBD 13b with salicylaldehyde (14).
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-30-i4.svg?max-width=637&scale=1.0)
|
|||
| entry | deviation from standard conditions | yield 15b (%)a | yield 17 (%)a |
| 1 | none | 61 | 0 |
| 2 | Ir(CO)Cl(PPh3)2 instead of [Ir(COD)Cl]2 | 0 | 0 |
| 3 | [Ir(COD)2]BF4 instead of [Ir(COD)Cl]2 | 23 | 0 |
| 4 | 10% TBAIb as an additive | 37 | 0 |
| 5 | 10% TBABrc as an additive | 28 | 0 |
| 7 | Na2CO3 instead of KOH | 45 | 45 |
| 8 | NaOH instead of KOH | 34 | 22 |
| 9 | Cs2CO3 instead of KOH | 22 | 49 |
| 10 | no base | 13 | 64 |
| 11 | 1% [Ir(COD)Cl]2 loading | 0 | 0 |
| 12 | 10% [Ir(COD)Cl]2 loading | 25 | 23 |
| 13 | 20% [Ir(COD)Cl]2 loading | 19 | 57 |
| 14 | 90 °C instead of 65 °C | 32 | 25 |
| 15 | 110 °C instead of 65 °C | 36 | 34 |
| 16 | DME instead of 1,4-dioxane | 38 | 13 |
| 17 | DMF instead of 1,4-dioxane | 31 | 0 |
| 18 | MeCN instead of 1,4-dioxane | 23 | 0 |
| 19 | DCE instead of 1,4-dioxane | 0 | 61 |
| 20 | THF instead of 1,4-dioxane | 0 | 39 |
aIsolated yields. bTetrabutylammonium iodide. cTetrabutylammonium bromide.
The scope of the reaction was expanded to include different C1-substituted OBDs to investigate the electronic and steric effects of the C1 functionality on the hydroacylation reaction (Scheme 2). Satisfyingly, the reaction exclusively afforded the C3-hydroacylated regioisomer 15 in all cases. Moreover, the reaction was stereoselective for the formation of the exo-adduct rather than a mixture of endo/exo products as previously reported by Tanaka/Suemune [65] and Bolm [66], who independently studied the rhodium-catalyzed intermolecular hydroacylation reaction of salicylaldehydes with norbornadiene derivatives. It was found that electron-donating moieties at the C1-position were well tolerated in the hydroacylation reaction giving methyl- (15b), ethyl- (15c), and t-Bu- (15d) adducts in a 61%, 52%, and 76% yield, respectively. A significant decrease in the yield was observed for electron-withdrawing C1-substituted OBDs with ketone- (15i) and ester- (15j) substituted adducts only being produced in a 5% and 9% yield, respectively; however, unreacted starting material was recovered. Interestingly, C1-substitution with a trimethylsilyl (TMS) group resulted in the corresponding adduct 15k as well as the ring-opened 2,4-substituted naphthol product 16k. It was noted the insertion of a methylene unit at the C1-position allowed for electron-withdrawing substituents to be present in the reaction (15e, 15f, 15h). Sensitive functional groups like alkyl iodides were tolerated in the reaction, although product yields were slightly diminished. The relative stereo- and regiochemistry of the adducts was confirmed through NMR experiments and X-ray crystallography (15h) [67].
![[1860-5397-18-30-i2]](/bjoc/content/inline/1860-5397-18-30-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Iridium-catalyzed hydroacylation of C1-substituted OBDs 13a–k with salicylaldehyde 14.
Scheme 2: Iridium-catalyzed hydroacylation of C1-substituted OBDs 13a–k with salicylaldehyde 14.
We next sought to determine the effect of the electron-withdrawing group on the efficacy of the reaction. Interested if the C1-substituted ketone OBD 13i was merely unreactive, we subjected it to a competition reaction against C1-substituted methyl OBD 13b (Scheme 3). In the presence of 13i, the C1-substituted methyl OBD 13b failed to react, giving a total <5% product yield as an inseparable mixture of both 15b and 15i. Noteworthy, both C1-substituted OBDs were recovered, with very little side-product formation. Although we are unable to confirm the precise cause of the deleterious effect, we suspect C1-substitution with electron-withdrawing groups inactivates the iridium catalyst, perhaps by chelation with the carbonyl and the bridging oxygen atom.
![[1860-5397-18-30-i3]](/bjoc/content/inline/1860-5397-18-30-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Competition reaction of different C1-substituted OBDs.
Scheme 3: Competition reaction of different C1-substituted OBDs.
Computational
Computational details
All density functional theory (DFT) calculations in this study were carried out with the Gaussian 16, C.01 suite of programs [68]. Geometry optimizations of all the intermediates and transition states were carried out with the Minnesota functional M06 [69] with the double-ζ basis set def2SVP [70] and Grimme’s dispersion (GD3) [71]. Harmonic vibrational frequencies were computed to verify the nature of the stationary points. The normal modes of all local minima have only real frequencies, while transition-state structures were characterized by exactly one imaginary frequency. Solvent effects (solvent = 1,4-dioxane) were taken into account using the polarized continuum model (PCM) of Tomasi and co-workers [72] and were involved in all geometry optimization and frequency calculations. Frequency analyses and single-point energies were calculated with the M06 functional [69] with the triple-ζ basis set def2TZVP [70] with the PCM (1,4-dioxane) solvent model [72]. The Gibbs free energies of formation of the reactants, products, and transition states were calculated from the optimized structures by single-point calculations by adding thermochemical corrections to the electronic energy. Optimized structures are illustrated using CYLview [73].
In order to further understand the iridium-catalyzed hydroacylation reaction, we carried out DFT calculations on the mechanism. Although an in-depth investigation into the mechanism of iridium-catalyzed hydroacylation reactions has never been carried out, the mechanistic pathways should parallel those of other analogously reactive d7 metals. On the basis of pioneering mechanistic investigations into rhodium-catalyzed hydroacylation reactions [74-78], we propose a catalytic cycle utilizing iridium that proceeds with three key steps: (1) iridium(I) oxidative addition into the aldehyde C–H bond, (2) insertion of the olefin into the iridium hydride, and (3) C–C bond-forming reductive elimination.
The hydroacylation reaction with C1-substituted methyl OBD (MeOBD) with salicylaldehyde catalyzed by [Ir(COD)OH]2 was chosen as the model reaction. As the reaction is in the presence of 5 M KOH, the potassium salt of salicylaldehyde was used rather than the protonated species for all calculations. Likewise, [Ir(COD)OH]2, and its derivatives were used in all calculations rather than the experimentally used precatalyst [Ir(COD)Cl]2, as ligand exchange with the hydroxide ions present likely generates the [Ir(COD)OH]2 species in solution. Upon monomerization of [Ir(COD)OH]2, either through solvolysis or coordination of the substrate, the active catalyst Ir(COD)OH will undergo oxidative addition into the aldehyde C–H bond. Next, the iridium hydride species will undergo exo-η2-coordination with the olefin of MeOBD to generate intermediates IN1a and IN1b (Figure 1). It is typically assumed exo-η2-coordination is preferential over endo-η2-coordination, as the less congested convex face would impose reduced steric requirements. There are two potential isomeric intermediates following η2-coordination to the exo-face of MeOBD concerning the relative orientation of the COD ligand, acyl group, and C1-substituent on the oxabicyclic alkene. In IN1a, the chelated acyl group is positioned syn to the C1-methyl substituent while in IN1b, they are positioned anti to one another. IN1b is 1.8 kcal/mol higher in energy than its isomer which can be attributed to the increased steric interactions between the bulky COD ligand and the C1-methyl substituent. In both cases, the C–C’ distance of the olefin (1.40 Å) in the Ir–C–C’ coordination is marginally lengthened with respect to the separated olefin (1.33 Å) from the π back donation π*-antibonding orbital of the ligand. The next process concerns the insertion of the olefin into the iridium–hydride bond to form hydrometalated intermediates IN2a and IN2b (Figure 1). Two possible transition states, which exhibit a distorted Ir–H–C–C’ four-membered ring geometry, can be located. The concerning free energy barrier about IN1a to IN2a, via 1aTS2a is 8.6 kcal/mol whereas it is 4.9 kcal/mol to form IN2b, via 1bTS2b. Understandably, the steric interaction between the C1-substituent and the acyl group is responsible for this energy difference. The relative energy of the hydrometalated intermediates (IN2a and IN2b) are comparable to their preceding intermediates with IN2a being 0.5 kcal/mol less stable while IN2b is 2.0 kcal/mol more stable (Figure 1). As such, it is likely these two intermediates are strongly in equilibrium with no thermodynamic driving force favoring one over the other; moreover, the activation energies of the forward and reverse reactions are proportional.
![[1860-5397-18-30-1]](/bjoc/content/figures/1860-5397-18-30-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Potential energy profile of the PCM solvation model for the hydrometalation/reductive elimination pathway of the Ir/diene-catalyzed hydroacylation of MeOBD with salicylaldehyde (14) in 1,4-dioxane, as evaluated by DFT calculation (M06 [69]/def2SVP [70]/PCM [72]//M06 [69]/def2TZVP [70]/PCM [72]). Calculated Gibbs free energies (in kcal/mol; T = 338.15 K) are with respect to separated reactants Ir(COD)OH (cat) + SaliK + MeOBD. The dotted lines in the illustration of the transition states represent bonds being broken/formed. The bond lengths are given in Å.
Figure 1: Potential energy profile of the PCM solvation model for the hydrometalation/reductive elimination p...
The last key step in the catalytic cycle involves the C–C bond-forming reductive elimination to form the final ketone intermediate IN3a or IN3b (Figure 1). Two possible transition states, 2aTS3a and 2bTS3b, can be located. The concerning free energy barrier about IN2a to IN3a, via 2aTS3a is 10.8 kcal/mol whereas it is 5.9 kcal/mol to form IN3b, via 2bTS3b. The Ir(I) alkoxide intermediate IN3b (IN3a) is rather thermodynamically stable (Figure 1), 31.5 (30.6) kcal/mol lower in energy than the preceding intermediate IN2b (IN2a) indicating a strongly exergonic process. By comparing all the competing transition states in both reaction pathways, it is determined the reductive elimination step is the rate-determining step (RDS) for the active bond-forming hydroacylation catalytic cycle, as it possesses the greatest free energy barriers. This parallels that determined by Morehead and Sargent who hypothesized the reductive elimination step was the RDS for rhodium-catalyzed intramolecular hydroacylation reactions [74]. Based on the activation energy of the reverse step of reductive elimination 3TS2 (37.4–41.4 kcal/mol), we predict reductive elimination, and subsequent C–C formation, to be irreversible under the experimental reaction conditions. As such, we theorize the origin of regioselectivity for the title reaction is the reductive elimination step. Based on the relative kinetics, we predict the point of selectivity must occur before the irreversible C–C forming step. Comparing the two competing reductive elimination transition states, 2bTS3b is the more energetically accessible transition state with an energy barrier of 5.9 kcal/mol, that is 4.9 kcal/mol lower in energy compared to 2aTS3a. This large difference in activation energy (ΔΔG‡) between the two competing transition states offers explanation towards the sole production of the experimentally observed anti-acylated product 15.
Although Weller and co-workers have elegantly demonstrated hydride migration, rather than the alternative carbometalation, occurs during rhodium-catalyzed intermolecular alkyne hydroacylation [78], no such experiment has been carried out under iridium catalysis. As such, we set out to explore the possibility acyl migration is favored over hydride migration in iridium-catalyzed hydroacylation reactions (Figure 2). Beginning with the two exo-η2-coordinated intermediates IN1a and IN1b, two possible transition states for the acyl migration, which exhibit a distorted Ir–C–C’–C’’ four-membered ring geometry, can be located. Directly comparing the hydrometalation process (Figure 1) to the carbometalation process, we see that acyl migration is unfavored, exhibiting activation energies of 19.2 (1bTS2d) to 20.7 (1aTS2c) kcal/mol. The carbometalated intermediates IN2c and IN2d are 10.3 and 11.7 kcal/mol less stable than their preceding intermediates (IN1a and IN1b), indicating the carbometalation step is an endergonic process. Subsequent reductive elimination of the hydride ligand, via 2cTS3a and 2dTS3b, requires an activation energy of 4.8 to 5.6 kcal/mol, respectively, to produce the aforementioned thermodynamically stable Ir(I) alkoxide intermediates INa3 and INb3. Based on the extremely high energy barrier required for acyl migration over hydride migration, we hypothesize iridium-catalyzed hydroacylation reactions proceed via the hydride migration pathway, like that reported for the rhodium-catalyzed hydroacylation reactions.
![[1860-5397-18-30-2]](/bjoc/content/figures/1860-5397-18-30-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Potential energy profile of the PCM solvation model for the carbometalation/reductive elimination pathway of the Ir/diene-catalyzed hydroacylation of MeOBD with salicylaldehyde (14) in 1,4-dioxane, as evaluated by DFT calculation (M06 [69]/def2SVP [70]/PCM [72]//M06 [69]/def2TZVP [70]/PCM [72]). Calculated Gibbs free energies (in kcal/mol; T = 338.15 K) are with respect to separated reactants Ir(COD)OH (Cat) + SaliK + MeOBD.
Figure 2: Potential energy profile of the PCM solvation model for the carbometalation/reductive elimination p...
As mentioned above, it is typically assumed exo-η2-coordination is preferential over endo-η2-coordination; however, for greater completeness, we investigated the endo-hydroacylation of MeOBD as a potential competing reaction (Figure 3). There are two potential isomeric intermediates following endo-η2-coordination of MeOBD concerning the relative orientation of the COD ligand, acyl group, and C1-substituent on the oxabicyclic alkene. In IN1e, the chelated acyl group is positioned syn to the C1-methyl substituent while in IN1f, they are positioned anti to one another. Compared to the exo-η2-coordinated intermediates, the endo-isomers are 5.0 to 7.3 kcal/mol higher in energy. The concerning free energy barrier about IN1e to IN2e, via 1eTS2e is 10.0 kcal/mol whereas it is 9.5 kcal/mol to form IN1f via 1fTS2f. Although the calculated barriers are not prohibitively high, they are much greater than the exo-hydrometalation pathway. These results suggest the reaction’s stereoselectivity originates from the imposed steric constraints of the more-hindered endo-face.
![[1860-5397-18-30-3]](/bjoc/content/figures/1860-5397-18-30-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Potential energy profile of the PCM solvation model for the endo hydrometalation/reductive elimination pathway of the Ir/diene-catalyzed hydroacylation of MeOBD with salicylaldehyde (14) in 1,4-dioxane, as evaluated by DFT calculation (M06 [69]/def2SVP [70]/PCM [72]//M06 [69]/def2TZVP [70]/PCM [72]). Calculated Gibbs free energies (in kcal/mol; T = 338.15 K) are with respect to separated reactants Ir(COD)OH (Cat) + SaliK + MeOBD.
Figure 3: Potential energy profile of the PCM solvation model for the endo hydrometalation/reductive eliminat...
Based on the calculated energies of the optimized intermediates and transition states for the three investigated pathways, an overall reaction mechanism can be proposed. It is predicted pathway B is the most accessible pathway for the hydroacylation reaction which corresponds with the production of the experimentally observed C3-exo-product 15. First, the active iridium catalyst will undergo oxidative addition into the aldehyde C–H bond (Figure 4). Next, the iridium hydride species will undergo exo-η2-coordination with the olefin of MeOBD to generate intermediate IN1b. Insertion of the olefin into the iridium-hydride bond to form hydrometalated intermediate IN2b proceeds via 1bTS2b which exhibits a distorted Ir–H–C–C’ four-membered ring geometry, requiring an activation of 4.9 kcal/mol. Reductive elimination through 2bTS3b crosses an energy barrier of 5.9 kcal/mol to generate the extremely stable Ir(I) alkoxide intermediate IN3b. The final reductive elimination step possesses the greatest energy of activation in pathway B, acting as both the RDS and the origin of regioselectivity.
![[1860-5397-18-30-4]](/bjoc/content/figures/1860-5397-18-30-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Potential energy profile of the PCM solvation model for the Ir/diene-catalyzed hydroacylation of MeOBD with salicylaldehyde (14) in 1,4-dioxane, as evaluated by DFT calculation (M06 [69]/def2SVP [70]/PCM [72]//M06 [69]/def2TZVP [70]/PCM [72]). Calculated Gibbs free energies (in kcal/mol; T = 338.15 K) are with respect to separated reactants Ir(COD)OH (cat) + SaliK + MeOBD.
Figure 4: Potential energy profile of the PCM solvation model for the Ir/diene-catalyzed hydroacylation of Me...
Conclusion
In conclusion, we have successfully investigated the regioselectivity of iridium-catalyzed hydroacylation reactions of C1-substituted OBDs with salicylaldehyde. Utilizing commercially available [Ir(COD)Cl]2 in the presence of 5 M KOH in dioxane at 65 °C, a variety of hydroacylated bicyclic adducts were obtained in up to a 95% yield with complete stereo- and regioselectivity. It was observed the addition of the acyl group occurred entirely at the less hindered position, exclusively producing the C3-exo-product. Experimental and theoretical studies were undertaken in order to understand the mechanism. Although not fully discerned, electron-withdrawing C1-substituents seem to deactivate the catalyst, leading to severely diminished product yields. Using DFT calculations, we investigated the [Ir(COD)OH]2-catalyzed hydroacylation reactions of C1-substituted OBDs. From these results, we found the reductive elimination step is the rate-determining step and the origin of regioselectivity for the catalytic cycle. Moreover, we determined the stereoselectivity of the reaction arises from the unattractive interactions imposed from the sterically hindered endo-face of the bicyclic alkene. The mechanistic insights gained from this combined experimental and theoretical study will facilitate further future methodology development in this field.
Supporting Information
| Supporting Information File 1: Experimental procedures, compound characterization, and 1H and 13C NMR spectra of compounds. | ||
| Format: PDF | Size: 1.5 MB | Download |
| Supporting Information File 2: Cartesian coordinates and selected energy values for all calculated structures. | ||
| Format: PDF | Size: 360.8 KB | Download |
References
-
Nicolaou, K. C. Proc. R. Soc. A 2014, 470, 20130690. doi:10.1098/rspa.2013.0690
Return to citation in text: [1] -
Negishi, E.-i. Angew. Chem., Int. Ed. 2011, 50, 6738–6764. doi:10.1002/anie.201101380
Return to citation in text: [1] -
Ruiz-Castillo, P.; Buchwald, S. L. Chem. Rev. 2016, 116, 12564–12649. doi:10.1021/acs.chemrev.6b00512
Return to citation in text: [1] -
Suzuki, A. Angew. Chem., Int. Ed. 2011, 50, 6722–6737. doi:10.1002/anie.201101379
Return to citation in text: [1] -
Biffis, A.; Centomo, P.; Del Zotto, A.; Zecca, M. Chem. Rev. 2018, 118, 2249–2295. doi:10.1021/acs.chemrev.7b00443
Return to citation in text: [1] -
Kundu, K.; McCullagh, J. V.; Morehead, A. T. J. Am. Chem. Soc. 2005, 127, 16042–16043. doi:10.1021/ja0564416
Return to citation in text: [1] -
Colby, D. A.; Bergman, R. G.; Ellman, J. A. Chem. Rev. 2010, 110, 624–655. doi:10.1021/cr900005n
Return to citation in text: [1] -
Leung, J. C.; Krische, M. J. Chem. Sci. 2012, 3, 2202–2209. doi:10.1039/c2sc20350b
Return to citation in text: [1] -
Willis, M. C. Chem. Rev. 2010, 110, 725–748. doi:10.1021/cr900096x
Return to citation in text: [1] -
Newton, C. G.; Wang, S.-G.; Oliveira, C. C.; Cramer, N. Chem. Rev. 2017, 117, 8908–8976. doi:10.1021/acs.chemrev.6b00692
Return to citation in text: [1] -
Zeng, X. Chem. Rev. 2013, 113, 6864–6900. doi:10.1021/cr400082n
Return to citation in text: [1] -
Yang, L.; Huang, H. Chem. Rev. 2015, 115, 3468–3517. doi:10.1021/cr500610p
Return to citation in text: [1] -
Murphy, S. K.; Dong, V. M. Chem. Commun. 2014, 50, 13645–13649. doi:10.1039/c4cc02276a
Return to citation in text: [1] -
Rayabarapu, D. K.; Cheng, C.-H. Acc. Chem. Res. 2007, 40, 971–983. doi:10.1021/ar600021z
Return to citation in text: [1] -
Lautens, M.; Fagnou, K.; Hiebert, S. Acc. Chem. Res. 2003, 36, 48–58. doi:10.1021/ar010112a
Return to citation in text: [1] -
Boutin, R.; Koh, S.; Tam, W. Curr. Org. Synth. 2019, 16, 460–484. doi:10.2174/1570179416666181122094643
Return to citation in text: [1] -
Vivek Kumar, S.; Yen, A.; Lautens, M.; Guiry, P. J. Chem. Soc. Rev. 2021, 50, 3013–3093. doi:10.1039/d0cs00702a
Return to citation in text: [1] -
Villeneuve, K.; Riddell, N.; Jordan, R. W.; Tsui, G. C.; Tam, W. Org. Lett. 2004, 6, 4543–4546. doi:10.1021/ol048111g
Return to citation in text: [1] -
Riddell, N.; Tam, W. J. Org. Chem. 2006, 71, 1934–1937. doi:10.1021/jo052295a
Return to citation in text: [1] -
Villeneuve, K.; Riddell, N.; Tam, W. Tetrahedron 2006, 62, 3823–3836. doi:10.1016/j.tet.2005.11.081
Return to citation in text: [1] -
Qin, H.; Chen, J.; Li, K.; He, Z.; Zhou, Y.; Fan, B. Chem. – Asian J. 2018, 13, 2431–2434. doi:10.1002/asia.201800492
Return to citation in text: [1] -
Yang, Q.; Yu, L.; Xu, J.; Li, S.; Liu, S.; Chen, H.; Zhou, Y.; Wang, L.; Fan, B. Tetrahedron: Asymmetry 2014, 25, 957–961. doi:10.1016/j.tetasy.2014.06.007
Return to citation in text: [1] -
Chao, K. C.; Rayabarapu, D. K.; Wang, C.-C.; Cheng, C.-H. J. Org. Chem. 2001, 66, 8804–8810. doi:10.1021/jo010609y
Return to citation in text: [1] -
Shih, H.-T.; Shih, H.-H.; Cheng, C.-H. Org. Lett. 2001, 3, 811–814. doi:10.1021/ol0069204
Return to citation in text: [1] -
Jack, K.; Tam, W. J. Org. Chem. 2013, 78, 3416–3420. doi:10.1021/jo400104q
Return to citation in text: [1] -
Huang, D.-J.; Cheng, C.-H. J. Organomet. Chem. 1995, 490, C1–C7. doi:10.1016/0022-328x(94)05335-9
Return to citation in text: [1] -
Nishimura, T.; Kawamoto, T.; Sasaki, K.; Tsurumaki, E.; Hayashi, T. J. Am. Chem. Soc. 2007, 129, 1492–1493. doi:10.1021/ja068488c
Return to citation in text: [1] -
Ballantine, M.; Menard, M. L.; Tam, W. J. Org. Chem. 2009, 74, 7570–7573. doi:10.1021/jo901504n
Return to citation in text: [1] -
Yen, A.; Choo, K.-L.; Yazdi, S. K.; Franke, P. T.; Webster, R.; Franzoni, I.; Loh, C. C. J.; Poblador-Bahamonde, A. I.; Lautens, M. Angew. Chem. 2017, 129, 6404–6408. doi:10.1002/ange.201700632
Return to citation in text: [1] -
Villeneuve, K.; Tam, W. J. Am. Chem. Soc. 2006, 128, 3514–3515. doi:10.1021/ja058621l
Return to citation in text: [1] -
Peng, F.; Fan, B.; Shao, Z.; Pu, X.; Li, P.; Zhang, H. Synthesis 2008, 3043–3046. doi:10.1055/s-2008-1067268
Return to citation in text: [1] -
Yang, J.; Sekiguchi, Y.; Yoshikai, N. ACS Catal. 2019, 9, 5638–5644. doi:10.1021/acscatal.9b00655
Return to citation in text: [1] -
Sawano, T.; Ou, K.; Nishimura, T.; Hayashi, T. Chem. Commun. 2012, 48, 6106–6108. doi:10.1039/c2cc31880f
Return to citation in text: [1] -
Chen, W.; Yang, W.; Wu, R.; Yang, D. Green Chem. 2018, 20, 2512–2518. doi:10.1039/c7gc03772d
Return to citation in text: [1] -
Lin, Q.; Yang, W.; Yao, Y.; Chen, S.; Tan, Y.; Chen, D.; Yang, D. Org. Lett. 2019, 21, 7244–7247. doi:10.1021/acs.orglett.9b02452
Return to citation in text: [1] -
Sakae, R.; Hirano, K.; Satoh, T.; Miura, M. Angew. Chem., Int. Ed. 2015, 54, 613–617. doi:10.1002/anie.201409104
Return to citation in text: [1] -
Lee, H.; Lee, B. Y.; Yun, J. Org. Lett. 2015, 17, 764–766. doi:10.1021/ol503598w
Return to citation in text: [1] -
Prakash, S.; Chang, Y.-C.; Cheng, C.-H. Chem. – Asian J. 2018, 13, 1664–1668. doi:10.1002/asia.201800433
Return to citation in text: [1] -
Koh, S.; Pounder, A.; Brown, E.; Tam, W. Eur. J. Org. Chem. 2020, 4558–4562. doi:10.1002/ejoc.202000672
Return to citation in text: [1] -
Koh, S.; Pounder, A.; Brown, E.; Tam, W. Org. Lett. 2020, 22, 3433–3437. doi:10.1021/acs.orglett.0c00900
Return to citation in text: [1] -
Cheng, H.; Yang, D. J. Org. Chem. 2012, 77, 9756–9765. doi:10.1021/jo3018507
Return to citation in text: [1] -
Yang, X.; Yang, W.; Yao, Y.; Deng, Y.; Yang, D. Org. Chem. Front. 2019, 6, 1151–1156. doi:10.1039/c8qo01403e
Return to citation in text: [1] -
Long, Y.; Li, X.; Pan, X.; Ding, D.; Xu, X.; Zuo, X.; Yang, D.; Wang, S.; Li, C. Catal. Lett. 2014, 144, 419–433. doi:10.1007/s10562-013-1136-x
Return to citation in text: [1] -
Yang, D.; Long, Y.; Zhang, J.; Zeng, H.; Wang, S.; Li, C. Organometallics 2010, 29, 3477–3480. doi:10.1021/om100384q
Return to citation in text: [1] -
Chen, J.; Zou, L.; Zeng, C.; Zhou, Y.; Fan, B. Org. Lett. 2018, 20, 6859–6862. doi:10.1021/acs.orglett.8b02980
Return to citation in text: [1] -
Jack, K.; Fatila, E.; Hillis, C.; Tam, W. Synth. Commun. 2013, 43, 1181–1187. doi:10.1080/00397911.2011.626140
Return to citation in text: [1] -
Li, L.-P.; Rayabarapu, D. K.; Nandi, M.; Cheng, C.-H. Org. Lett. 2003, 5, 1621–1624. doi:10.1021/ol034251z
Return to citation in text: [1] -
Tan, Y.; Yao, Y.; Yang, W.; Lin, Q.; Huang, G.; Tan, M.; Chen, S.; Chen, D.; Yang, D. Adv. Synth. Catal. 2020, 362, 139–145. doi:10.1002/adsc.201901152
Return to citation in text: [1] -
Shen, G.; Khan, R.; Yang, F.; Yang, Y.; Pu, D.; Gao, Y.; Zhan, Y.; Luo, Y.; Fan, B. Asian J. Org. Chem. 2019, 8, 97–102. doi:10.1002/ajoc.201800569
Return to citation in text: [1] -
Li, Y.; Yang, W.; Cheng, G.; Yang, D. J. Org. Chem. 2016, 81, 4744–4750. doi:10.1021/acs.joc.6b00667
Return to citation in text: [1] -
Millet, R.; Gremaud, L.; Bernardez, T.; Palais, L.; Alexakis, A. Synthesis 2009, 2101–2112. doi:10.1055/s-0029-1216838
Return to citation in text: [1] -
Yang, D.; Liang, N. Org. Biomol. Chem. 2014, 12, 2080–2086. doi:10.1039/c3ob42199f
Return to citation in text: [1] -
Yang, D.; Hu, P.; Long, Y.; Wu, Y.; Zeng, H.; Wang, H.; Zuo, X. Beilstein J. Org. Chem. 2009, 5, No. 53. doi:10.3762/bjoc.5.53
Return to citation in text: [1] -
Lautens, M. Synlett 1993, 177–185. doi:10.1055/s-1993-22393
Return to citation in text: [1] -
Fleming, M. J.; McManus, H. A.; Rudolph, A.; Chan, W. H.; Ruiz, J.; Dockendorff, C.; Lautens, M. Chem. – Eur. J. 2008, 14, 2112–2124. doi:10.1002/chem.200701775
Return to citation in text: [1] -
Lautens, M.; Rovis, T. Tetrahedron 1999, 55, 8967–8976. doi:10.1016/s0040-4020(99)00456-1
Return to citation in text: [1] -
Madan, S.; Cheng, C.-H. J. Org. Chem. 2006, 71, 8312–8315. doi:10.1021/jo061477h
Return to citation in text: [1] -
Pounder, A.; Ho, A.; Macleod, M.; Tam, W. Curr. Org. Synth. 2021, 18, 446–474. doi:10.2174/1570179417666210105121115
Return to citation in text: [1] [2] -
Allen, A.; Le Marquand, P.; Burton, R.; Villeneuve, K.; Tam, W. J. Org. Chem. 2007, 72, 7849–7857. doi:10.1021/jo7012884
Return to citation in text: [1] -
Deng, Y.; Yang, W.; Yao, Y.; Yang, X.; Zuo, X.; Yang, D. Org. Biomol. Chem. 2019, 17, 703–711. doi:10.1039/c8ob02864h
Return to citation in text: [1] -
Hill, J.; Wicks, C.; Pounder, A.; Tam, W. Tetrahedron Lett. 2019, 60, 150990. doi:10.1016/j.tetlet.2019.150990
Return to citation in text: [1] [2] [3] [4] -
Edmunds, M.; Menard, M. L.; Tam, W. Synth. Commun. 2015, 45, 458–466. doi:10.1080/00397911.2014.965330
Return to citation in text: [1] -
Pounder, A.; Bishop, F.; Chen, L. D.; Tam, W. Eur. J. Org. Chem. 2021, 1901–1908. doi:10.1002/ejoc.202100093
Return to citation in text: [1] -
Nagamoto, M.; Nishimura, T. Chem. Commun. 2015, 51, 13791–13794. doi:10.1039/c5cc05432j
Return to citation in text: [1] [2] [3] -
Tanaka, K.; Tanaka, M.; Suemune, H. Tetrahedron Lett. 2005, 46, 6053–6056. doi:10.1016/j.tetlet.2005.07.004
Return to citation in text: [1] -
Stemmler, R. T.; Bolm, C. Adv. Synth. Catal. 2007, 349, 1185–1198. doi:10.1002/adsc.200600583
Return to citation in text: [1] -
Lough, A. J.; Ho, A.; Tam, W. IUCrData 2020, 5, x200265. doi:10.1107/s2414314620002655
Return to citation in text: [1] -
Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, 2016.
Return to citation in text: [1] -
Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] -
Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/b508541a
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] -
Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104. doi:10.1063/1.3382344
Return to citation in text: [1] -
Miertuš, S.; Scrocco, E.; Tomasi, J. Chem. Phys. 1981, 55, 117–129. doi:10.1016/0301-0104(81)85090-2
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] -
CYLview, 1.0b; Université de Sherbrooke, 2009, http://www.cylview.org.
Return to citation in text: [1] -
Hyatt, I. F. D.; Anderson, H. K.; Morehead, A. T., Jr.; Sargent, A. L. Organometallics 2008, 27, 135–147. doi:10.1021/om700842d
Return to citation in text: [1] [2] -
von Delius, M.; Le, C. M.; Dong, V. M. J. Am. Chem. Soc. 2012, 134, 15022–15032. doi:10.1021/ja305593y
Return to citation in text: [1] -
Wang, M.; Zhang, X.; Chen, Z.; Tang, Y.; Lei, M. Sci. China: Chem. 2014, 57, 1264–1275. doi:10.1007/s11426-014-5102-2
Return to citation in text: [1] -
Chaplin, A. B.; Hooper, J. F.; Weller, A. S.; Willis, M. C. J. Am. Chem. Soc. 2012, 134, 4885–4897. doi:10.1021/ja211649a
Return to citation in text: [1] -
Pawley, R. J.; Huertos, M. A.; Lloyd-Jones, G. C.; Weller, A. S.; Willis, M. C. Organometallics 2012, 31, 5650–5659. doi:10.1021/om300647n
Return to citation in text: [1] [2]
| 67. | Lough, A. J.; Ho, A.; Tam, W. IUCrData 2020, 5, x200265. doi:10.1107/s2414314620002655 |
| 69. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 74. | Hyatt, I. F. D.; Anderson, H. K.; Morehead, A. T., Jr.; Sargent, A. L. Organometallics 2008, 27, 135–147. doi:10.1021/om700842d |
| 75. | von Delius, M.; Le, C. M.; Dong, V. M. J. Am. Chem. Soc. 2012, 134, 15022–15032. doi:10.1021/ja305593y |
| 76. | Wang, M.; Zhang, X.; Chen, Z.; Tang, Y.; Lei, M. Sci. China: Chem. 2014, 57, 1264–1275. doi:10.1007/s11426-014-5102-2 |
| 77. | Chaplin, A. B.; Hooper, J. F.; Weller, A. S.; Willis, M. C. J. Am. Chem. Soc. 2012, 134, 4885–4897. doi:10.1021/ja211649a |
| 78. | Pawley, R. J.; Huertos, M. A.; Lloyd-Jones, G. C.; Weller, A. S.; Willis, M. C. Organometallics 2012, 31, 5650–5659. doi:10.1021/om300647n |
| 70. | Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/b508541a |
| 72. | Miertuš, S.; Scrocco, E.; Tomasi, J. Chem. Phys. 1981, 55, 117–129. doi:10.1016/0301-0104(81)85090-2 |
| 72. | Miertuš, S.; Scrocco, E.; Tomasi, J. Chem. Phys. 1981, 55, 117–129. doi:10.1016/0301-0104(81)85090-2 |
| 69. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 70. | Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/b508541a |
| 71. | Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. J. Chem. Phys. 2010, 132, 154104. doi:10.1063/1.3382344 |
| 69. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 70. | Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/b508541a |
| 72. | Miertuš, S.; Scrocco, E.; Tomasi, J. Chem. Phys. 1981, 55, 117–129. doi:10.1016/0301-0104(81)85090-2 |
| 70. | Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/b508541a |
| 72. | Miertuš, S.; Scrocco, E.; Tomasi, J. Chem. Phys. 1981, 55, 117–129. doi:10.1016/0301-0104(81)85090-2 |
| 78. | Pawley, R. J.; Huertos, M. A.; Lloyd-Jones, G. C.; Weller, A. S.; Willis, M. C. Organometallics 2012, 31, 5650–5659. doi:10.1021/om300647n |
| 69. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 72. | Miertuš, S.; Scrocco, E.; Tomasi, J. Chem. Phys. 1981, 55, 117–129. doi:10.1016/0301-0104(81)85090-2 |
| 74. | Hyatt, I. F. D.; Anderson, H. K.; Morehead, A. T., Jr.; Sargent, A. L. Organometallics 2008, 27, 135–147. doi:10.1021/om700842d |
| 69. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 70. | Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/b508541a |
| 70. | Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/b508541a |
| 72. | Miertuš, S.; Scrocco, E.; Tomasi, J. Chem. Phys. 1981, 55, 117–129. doi:10.1016/0301-0104(81)85090-2 |
| 69. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 1. | Nicolaou, K. C. Proc. R. Soc. A 2014, 470, 20130690. doi:10.1098/rspa.2013.0690 |
| 18. | Villeneuve, K.; Riddell, N.; Jordan, R. W.; Tsui, G. C.; Tam, W. Org. Lett. 2004, 6, 4543–4546. doi:10.1021/ol048111g |
| 19. | Riddell, N.; Tam, W. J. Org. Chem. 2006, 71, 1934–1937. doi:10.1021/jo052295a |
| 20. | Villeneuve, K.; Riddell, N.; Tam, W. Tetrahedron 2006, 62, 3823–3836. doi:10.1016/j.tet.2005.11.081 |
| 21. | Qin, H.; Chen, J.; Li, K.; He, Z.; Zhou, Y.; Fan, B. Chem. – Asian J. 2018, 13, 2431–2434. doi:10.1002/asia.201800492 |
| 22. | Yang, Q.; Yu, L.; Xu, J.; Li, S.; Liu, S.; Chen, H.; Zhou, Y.; Wang, L.; Fan, B. Tetrahedron: Asymmetry 2014, 25, 957–961. doi:10.1016/j.tetasy.2014.06.007 |
| 23. | Chao, K. C.; Rayabarapu, D. K.; Wang, C.-C.; Cheng, C.-H. J. Org. Chem. 2001, 66, 8804–8810. doi:10.1021/jo010609y |
| 59. | Allen, A.; Le Marquand, P.; Burton, R.; Villeneuve, K.; Tam, W. J. Org. Chem. 2007, 72, 7849–7857. doi:10.1021/jo7012884 |
| 69. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 14. | Rayabarapu, D. K.; Cheng, C.-H. Acc. Chem. Res. 2007, 40, 971–983. doi:10.1021/ar600021z |
| 15. | Lautens, M.; Fagnou, K.; Hiebert, S. Acc. Chem. Res. 2003, 36, 48–58. doi:10.1021/ar010112a |
| 16. | Boutin, R.; Koh, S.; Tam, W. Curr. Org. Synth. 2019, 16, 460–484. doi:10.2174/1570179416666181122094643 |
| 17. | Vivek Kumar, S.; Yen, A.; Lautens, M.; Guiry, P. J. Chem. Soc. Rev. 2021, 50, 3013–3093. doi:10.1039/d0cs00702a |
| 60. | Deng, Y.; Yang, W.; Yao, Y.; Yang, X.; Zuo, X.; Yang, D. Org. Biomol. Chem. 2019, 17, 703–711. doi:10.1039/c8ob02864h |
| 6. | Kundu, K.; McCullagh, J. V.; Morehead, A. T. J. Am. Chem. Soc. 2005, 127, 16042–16043. doi:10.1021/ja0564416 |
| 7. | Colby, D. A.; Bergman, R. G.; Ellman, J. A. Chem. Rev. 2010, 110, 624–655. doi:10.1021/cr900005n |
| 8. | Leung, J. C.; Krische, M. J. Chem. Sci. 2012, 3, 2202–2209. doi:10.1039/c2sc20350b |
| 9. | Willis, M. C. Chem. Rev. 2010, 110, 725–748. doi:10.1021/cr900096x |
| 10. | Newton, C. G.; Wang, S.-G.; Oliveira, C. C.; Cramer, N. Chem. Rev. 2017, 117, 8908–8976. doi:10.1021/acs.chemrev.6b00692 |
| 11. | Zeng, X. Chem. Rev. 2013, 113, 6864–6900. doi:10.1021/cr400082n |
| 12. | Yang, L.; Huang, H. Chem. Rev. 2015, 115, 3468–3517. doi:10.1021/cr500610p |
| 13. | Murphy, S. K.; Dong, V. M. Chem. Commun. 2014, 50, 13645–13649. doi:10.1039/c4cc02276a |
| 57. | Madan, S.; Cheng, C.-H. J. Org. Chem. 2006, 71, 8312–8315. doi:10.1021/jo061477h |
| 70. | Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/b508541a |
| 2. | Negishi, E.-i. Angew. Chem., Int. Ed. 2011, 50, 6738–6764. doi:10.1002/anie.201101380 |
| 3. | Ruiz-Castillo, P.; Buchwald, S. L. Chem. Rev. 2016, 116, 12564–12649. doi:10.1021/acs.chemrev.6b00512 |
| 4. | Suzuki, A. Angew. Chem., Int. Ed. 2011, 50, 6722–6737. doi:10.1002/anie.201101379 |
| 5. | Biffis, A.; Centomo, P.; Del Zotto, A.; Zecca, M. Chem. Rev. 2018, 118, 2249–2295. doi:10.1021/acs.chemrev.7b00443 |
| 58. | Pounder, A.; Ho, A.; Macleod, M.; Tam, W. Curr. Org. Synth. 2021, 18, 446–474. doi:10.2174/1570179417666210105121115 |
| 72. | Miertuš, S.; Scrocco, E.; Tomasi, J. Chem. Phys. 1981, 55, 117–129. doi:10.1016/0301-0104(81)85090-2 |
| 39. | Koh, S.; Pounder, A.; Brown, E.; Tam, W. Eur. J. Org. Chem. 2020, 4558–4562. doi:10.1002/ejoc.202000672 |
| 40. | Koh, S.; Pounder, A.; Brown, E.; Tam, W. Org. Lett. 2020, 22, 3433–3437. doi:10.1021/acs.orglett.0c00900 |
| 41. | Cheng, H.; Yang, D. J. Org. Chem. 2012, 77, 9756–9765. doi:10.1021/jo3018507 |
| 42. | Yang, X.; Yang, W.; Yao, Y.; Deng, Y.; Yang, D. Org. Chem. Front. 2019, 6, 1151–1156. doi:10.1039/c8qo01403e |
| 43. | Long, Y.; Li, X.; Pan, X.; Ding, D.; Xu, X.; Zuo, X.; Yang, D.; Wang, S.; Li, C. Catal. Lett. 2014, 144, 419–433. doi:10.1007/s10562-013-1136-x |
| 44. | Yang, D.; Long, Y.; Zhang, J.; Zeng, H.; Wang, S.; Li, C. Organometallics 2010, 29, 3477–3480. doi:10.1021/om100384q |
| 45. | Chen, J.; Zou, L.; Zeng, C.; Zhou, Y.; Fan, B. Org. Lett. 2018, 20, 6859–6862. doi:10.1021/acs.orglett.8b02980 |
| 46. | Jack, K.; Fatila, E.; Hillis, C.; Tam, W. Synth. Commun. 2013, 43, 1181–1187. doi:10.1080/00397911.2011.626140 |
| 47. | Li, L.-P.; Rayabarapu, D. K.; Nandi, M.; Cheng, C.-H. Org. Lett. 2003, 5, 1621–1624. doi:10.1021/ol034251z |
| 48. | Tan, Y.; Yao, Y.; Yang, W.; Lin, Q.; Huang, G.; Tan, M.; Chen, S.; Chen, D.; Yang, D. Adv. Synth. Catal. 2020, 362, 139–145. doi:10.1002/adsc.201901152 |
| 49. | Shen, G.; Khan, R.; Yang, F.; Yang, Y.; Pu, D.; Gao, Y.; Zhan, Y.; Luo, Y.; Fan, B. Asian J. Org. Chem. 2019, 8, 97–102. doi:10.1002/ajoc.201800569 |
| 50. | Li, Y.; Yang, W.; Cheng, G.; Yang, D. J. Org. Chem. 2016, 81, 4744–4750. doi:10.1021/acs.joc.6b00667 |
| 51. | Millet, R.; Gremaud, L.; Bernardez, T.; Palais, L.; Alexakis, A. Synthesis 2009, 2101–2112. doi:10.1055/s-0029-1216838 |
| 52. | Yang, D.; Liang, N. Org. Biomol. Chem. 2014, 12, 2080–2086. doi:10.1039/c3ob42199f |
| 53. | Yang, D.; Hu, P.; Long, Y.; Wu, Y.; Zeng, H.; Wang, H.; Zuo, X. Beilstein J. Org. Chem. 2009, 5, No. 53. doi:10.3762/bjoc.5.53 |
| 55. | Fleming, M. J.; McManus, H. A.; Rudolph, A.; Chan, W. H.; Ruiz, J.; Dockendorff, C.; Lautens, M. Chem. – Eur. J. 2008, 14, 2112–2124. doi:10.1002/chem.200701775 |
| 72. | Miertuš, S.; Scrocco, E.; Tomasi, J. Chem. Phys. 1981, 55, 117–129. doi:10.1016/0301-0104(81)85090-2 |
| 32. | Yang, J.; Sekiguchi, Y.; Yoshikai, N. ACS Catal. 2019, 9, 5638–5644. doi:10.1021/acscatal.9b00655 |
| 33. | Sawano, T.; Ou, K.; Nishimura, T.; Hayashi, T. Chem. Commun. 2012, 48, 6106–6108. doi:10.1039/c2cc31880f |
| 34. | Chen, W.; Yang, W.; Wu, R.; Yang, D. Green Chem. 2018, 20, 2512–2518. doi:10.1039/c7gc03772d |
| 35. | Lin, Q.; Yang, W.; Yao, Y.; Chen, S.; Tan, Y.; Chen, D.; Yang, D. Org. Lett. 2019, 21, 7244–7247. doi:10.1021/acs.orglett.9b02452 |
| 36. | Sakae, R.; Hirano, K.; Satoh, T.; Miura, M. Angew. Chem., Int. Ed. 2015, 54, 613–617. doi:10.1002/anie.201409104 |
| 37. | Lee, H.; Lee, B. Y.; Yun, J. Org. Lett. 2015, 17, 764–766. doi:10.1021/ol503598w |
| 38. | Prakash, S.; Chang, Y.-C.; Cheng, C.-H. Chem. – Asian J. 2018, 13, 1664–1668. doi:10.1002/asia.201800433 |
| 56. | Lautens, M.; Rovis, T. Tetrahedron 1999, 55, 8967–8976. doi:10.1016/s0040-4020(99)00456-1 |
| 69. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 28. | Ballantine, M.; Menard, M. L.; Tam, W. J. Org. Chem. 2009, 74, 7570–7573. doi:10.1021/jo901504n |
| 29. | Yen, A.; Choo, K.-L.; Yazdi, S. K.; Franke, P. T.; Webster, R.; Franzoni, I.; Loh, C. C. J.; Poblador-Bahamonde, A. I.; Lautens, M. Angew. Chem. 2017, 129, 6404–6408. doi:10.1002/ange.201700632 |
| 30. | Villeneuve, K.; Tam, W. J. Am. Chem. Soc. 2006, 128, 3514–3515. doi:10.1021/ja058621l |
| 31. | Peng, F.; Fan, B.; Shao, Z.; Pu, X.; Li, P.; Zhang, H. Synthesis 2008, 3043–3046. doi:10.1055/s-2008-1067268 |
| 69. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 24. | Shih, H.-T.; Shih, H.-H.; Cheng, C.-H. Org. Lett. 2001, 3, 811–814. doi:10.1021/ol0069204 |
| 25. | Jack, K.; Tam, W. J. Org. Chem. 2013, 78, 3416–3420. doi:10.1021/jo400104q |
| 26. | Huang, D.-J.; Cheng, C.-H. J. Organomet. Chem. 1995, 490, C1–C7. doi:10.1016/0022-328x(94)05335-9 |
| 27. | Nishimura, T.; Kawamoto, T.; Sasaki, K.; Tsurumaki, E.; Hayashi, T. J. Am. Chem. Soc. 2007, 129, 1492–1493. doi:10.1021/ja068488c |
| 70. | Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/b508541a |
| 62. | Edmunds, M.; Menard, M. L.; Tam, W. Synth. Commun. 2015, 45, 458–466. doi:10.1080/00397911.2014.965330 |
| 63. | Pounder, A.; Bishop, F.; Chen, L. D.; Tam, W. Eur. J. Org. Chem. 2021, 1901–1908. doi:10.1002/ejoc.202100093 |
| 61. | Hill, J.; Wicks, C.; Pounder, A.; Tam, W. Tetrahedron Lett. 2019, 60, 150990. doi:10.1016/j.tetlet.2019.150990 |
| 61. | Hill, J.; Wicks, C.; Pounder, A.; Tam, W. Tetrahedron Lett. 2019, 60, 150990. doi:10.1016/j.tetlet.2019.150990 |
| 69. | Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215–241. doi:10.1007/s00214-007-0310-x |
| 70. | Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/b508541a |
| 70. | Weigend, F.; Ahlrichs, R. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. doi:10.1039/b508541a |
| 72. | Miertuš, S.; Scrocco, E.; Tomasi, J. Chem. Phys. 1981, 55, 117–129. doi:10.1016/0301-0104(81)85090-2 |
| 65. | Tanaka, K.; Tanaka, M.; Suemune, H. Tetrahedron Lett. 2005, 46, 6053–6056. doi:10.1016/j.tetlet.2005.07.004 |
| 66. | Stemmler, R. T.; Bolm, C. Adv. Synth. Catal. 2007, 349, 1185–1198. doi:10.1002/adsc.200600583 |
| 61. | Hill, J.; Wicks, C.; Pounder, A.; Tam, W. Tetrahedron Lett. 2019, 60, 150990. doi:10.1016/j.tetlet.2019.150990 |
| 61. | Hill, J.; Wicks, C.; Pounder, A.; Tam, W. Tetrahedron Lett. 2019, 60, 150990. doi:10.1016/j.tetlet.2019.150990 |
| 64. | Nagamoto, M.; Nishimura, T. Chem. Commun. 2015, 51, 13791–13794. doi:10.1039/c5cc05432j |
| 64. | Nagamoto, M.; Nishimura, T. Chem. Commun. 2015, 51, 13791–13794. doi:10.1039/c5cc05432j |
| 64. | Nagamoto, M.; Nishimura, T. Chem. Commun. 2015, 51, 13791–13794. doi:10.1039/c5cc05432j |
| 72. | Miertuš, S.; Scrocco, E.; Tomasi, J. Chem. Phys. 1981, 55, 117–129. doi:10.1016/0301-0104(81)85090-2 |
| 58. | Pounder, A.; Ho, A.; Macleod, M.; Tam, W. Curr. Org. Synth. 2021, 18, 446–474. doi:10.2174/1570179417666210105121115 |
© 2022 Ho et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.