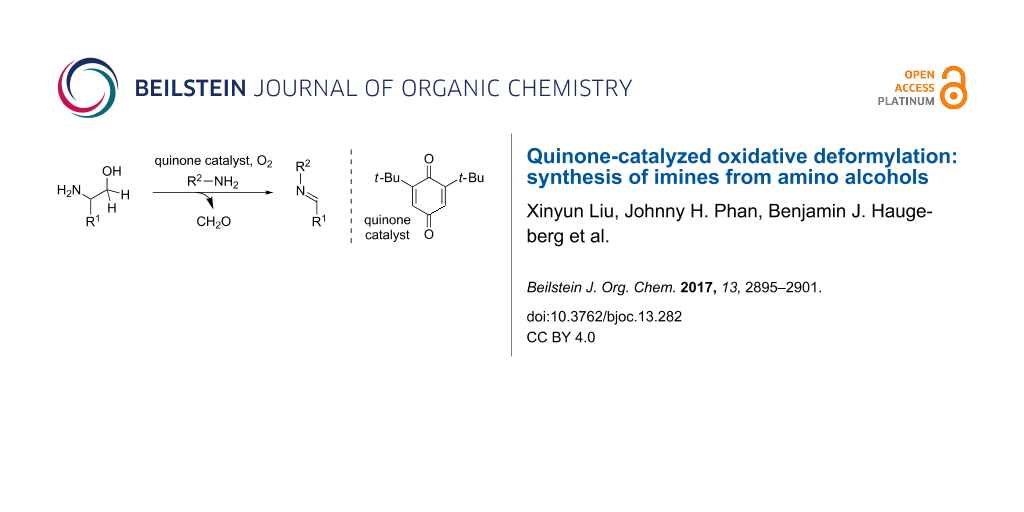
Abstract
A new method for imine synthesis by way of quinone-catalyzed oxidative deformylation of 1,2-amino alcohols is reported. A wide range of readily accessible amino alcohols and primary amines can be reacted to provide N-protected imine products. The methodology presented provides a novel organocatalytic approach for imine synthesis and demonstrates the synthetic versatility of quinone-catalyzed oxidative C–C bond cleavage.

Graphical Abstract
Introduction
Imines are extremely versatile intermediates in organic chemistry [1-3]. Consequently, many synthetic methods have been developed for the preparation of imines (Scheme 1). The condensation of an amine with an aldehyde or ketone is the oldest and most commonly employed method for imine synthesis [4]. More recently, the catalytic dehydrogenation of amines mediated by metal and organic catalysts has begun to emerge as an alternative approach for the preparation of imines [5,6]. The majority of these methods involve cleavage of a C−H bond at the α-position of an amine substrate [7-28]. Methods that deliver imines through amine α-C−C bond cleavage are far less common [29-32] despite the fact that these methods employ renewable resources, such as amino acids and their derivatives, as starting materials. In fact, only a few reports describing the oxidative deformylation of amino alcohols have been published [33-35], and in all of these reports stoichiometric oxidants, such as NaIO4 and Pb(OAc)4, must be employed to enable the desired transformations. Given that 1,2-amino alcohols are readily accessible from feedstock chemicals such as styrenes [36-38] and amino acids [39], the development of a new methodology to transform these materials into high-value imine products under catalytic conditions has the potential to be broadly useful. Herein, we report a new method that utilizes quinone catalysis to enable the synthesis of imines via oxidative deformylation of amino alcohols.
![[1860-5397-13-282-i1]](/bjoc/content/inline/1860-5397-13-282-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Established methods for the preparation of imines vs this work.
Scheme 1: Established methods for the preparation of imines vs this work.
Our group has recently reported the quinone-catalyzed decarboxylative homologation of α-amino acids [32], which demonstrated for the first time that quinone organocatalysts can be utilized to enable oxidative C–C bond cleavage to provide versatile imine intermediates. To further exploit the utility of this chemistry, we sought to develop a new method for the preparation of a wide range of imine products through the quinone-catalyzed deformylation of 1,2-amino alcohols. Such a transformation would not only facilitate rapid access to a variety of N-protected imines, but would also provide a novel approach for utilizing feedstock chemicals for the preparation of these valuable synthetic intermediates.
We envisioned a process wherein a 1,2-amino alcohol 1 would undergo condensation with an appropriate quinone catalyst 2 to deliver iminoquinone 3 (Scheme 2). Deformylation of 3 would generate N-arylimine 4. Subsequent transimination with amine 6 would provide the desired imine product 7 and a reduced form of the catalyst 5, which would be expected to undergo oxidative turnover through one of two possible mechanisms (i.e., 5 → 3 or 5 → 2).
![[1860-5397-13-282-i2]](/bjoc/content/inline/1860-5397-13-282-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed catalytic cycle for quinone-catalyzed deformylation.
Scheme 2: Proposed catalytic cycle for quinone-catalyzed deformylation.
Results and Discussion
With this plan in mind, we first explored the ability of several quinone catalysts to promote the deformylation of 2-phenylglycinol (1a) to deliver N-PMP imine 7a (Table 1). We selected quinone catalysts (2a−c) that have previously been utilized in amine oxidation reactions [21,32,40,41], and began with reaction conditions similar to those developed for our quinone-catalyzed oxidative decarboxylation chemistry [32]. To our delight, the desired deformylation product 7a was formed in 63% yield when catalyst 2a was employed (Table 1, entry 1). Quinone 2b failed to deliver imine 7a (Table 1, entry 2), but commercially available quinone 2c provided 7a in a promising 59% yield (Table 1, entry 3). Next, we examined the effect of base on the reaction using quinone 2c as the catalyst (Table 1, entries 4−7). Unfortunately, no improvement in reaction efficiency was observed when different bases were employed (Table 1, entries 4−6, 0−55% yield); however, exclusion of the base provided imine 7a in good yield (Table 1, entry 7, 85%). Decreasing the loading of catalyst 2c under these conditions reduced the yield of imine 7a (Table 1, entry 8, 64% yield), as did changing the identity of the catalyst (Table 1, entries 9 and 10, 62% and 0% respectively). Finally, we examined a range of solvents in an effort to further improve efficiency (Table 1, entries 11−17). No improvements in reaction efficiency were observed (Table 1, 0−72% yield), but it was noted that polar, protic solvents are critical in enabling the efficient deformylation of phenylglycinol.
Table 1: Optimization of quinone-catalyzed oxidative deformylation of phenylglycinol (1a).
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-282-i5.svg?max-width=637&scale=1.0)
|
||||
| Entry | Catalyst | Solvent | Base | Yield [%]a |
| 1 | 2a | EtOH | Et3N | 63 |
| 2 | 2b | EtOH | Et3N | 0 |
| 3 | 2c | EtOH | Et3N | 59 |
| 4 | 2c | EtOH | DABCO | 55 |
| 5 | 2c | EtOH | DBU | 0 |
| 6 | 2c | EtOH | K2CO3 | 17 |
| 7 | 2c | EtOH | none | 85 |
| 8b | 2c | EtOH | none | 64 |
| 9 | 2a | EtOH | none | 62 |
| 10 | 2b | EtOH | none | 0 |
| 11 | 2c | iPrOH | none | 72 |
| 12 | 2c | H2O | none | 47 |
| 13 | 2c | MeCN | none | 28 |
| 14 | 2c | DMSO | none | 13 |
| 15c | 2c | THF | none | 0 |
| 16 | 2c | PhMe | none | 11 |
| 17 | 2c | CHCl3 | none | 3 |
aDetermined by 1H NMR using benzyl ether as an internal standard.
b10 mol % quinone was used.
cReaction carried out at 50 °C.
With optimized conditions in hand, we next explored the scope of this methodology by employing a range of 1,2-amino alcohol substrates 1 (Table 2). As reported in Table 1, the reaction involving phenylglycinol gave the desired N-PMP imine (7a) in 85% yield (Table 2, entry 1). ortho-Substitution of the arene is reasonably well-tolerated, as 2-methylphenylglycinol (1b) and 2-chlorophenylglycinol (1c) delivered the corresponding imines in 68% yield (Table 2, entries 2 and 3). The meta-fluoro derivative provided imine 7d in 60% yield (Table 2, entry 4). Electronic effects were studied by examining a series of para-substituted phenylglycinol derivatives (Table 2, entries 5−9). Both electron-donating and electron-withdrawing substituents were tolerated, but no obvious trends in the reactivity patterns were observed (47−77% yield). Thiophenyl amino alcohol 1j was also subjected to the optimized conditions and the corresponding imine 7j was formed in 47% yield (Table 2, entry 10). Unfortunately, aliphatic 1,2-amino alcohols, such as valinol (1k), failed to undergo deformylation under the current conditions (Table 2, entry 11).
Table 2: Quinone-catalyzed oxidative deformylation of various amino alcohols.
![[Graphic 2]](/bjoc/content/inline/1860-5397-13-282-i6.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Amino alcohol 1 | Product 7 | Yield [%]a | ||
|
1
2 3 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-13-282-i7.svg?max-width=637&scale=1.0)
|
1a, R = H
1b, R = Me 1c, R = Cl |
![[Graphic 4]](/bjoc/content/inline/1860-5397-13-282-i8.svg?max-width=637&scale=1.0)
|
7a, R = H
7b, R = Me 7c, R = Cl |
85
68 68 |
| 4 |
![[Graphic 5]](/bjoc/content/inline/1860-5397-13-282-i9.svg?max-width=637&scale=1.0)
|
1d |
![[Graphic 6]](/bjoc/content/inline/1860-5397-13-282-i10.svg?max-width=637&scale=1.0)
|
7d | 60 |
|
5
6 7 8 9 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-13-282-i11.svg?max-width=637&scale=1.0)
|
1e, R = Me
1f, R = OMe 1g, R = Cl 1h, R = F 1i, R = CF3 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-13-282-i12.svg?max-width=637&scale=1.0)
|
7e, R = Me
7f, R = OMe 7g, R = Cl 7h, R = F 7i, R = CF3 |
68
77 66 54 47 |
| 10 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-13-282-i13.svg?max-width=637&scale=1.0)
|
1j |
![[Graphic 10]](/bjoc/content/inline/1860-5397-13-282-i14.svg?max-width=637&scale=1.0)
|
7j | 47 |
| 11 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-13-282-i15.svg?max-width=637&scale=1.0)
|
1k |
![[Graphic 12]](/bjoc/content/inline/1860-5397-13-282-i16.svg?max-width=637&scale=1.0)
|
7k | 0 |
aDetermined by 1H NMR using benzyl ether as an internal standard (average of two replicates).
Next, we investigated the use of various amine reaction partners 6 to access a variety of imine products 7 from phenylglycinol (1a, Table 3). The reaction with aniline (6l, Table 3, entry 2, 68% yield) showed reduced reaction efficiency compared to that with para-anisidine (6a, Table 3, entry 1, 85% yield). When para-fluoroaniline (6m) was employed as the reaction partner, imine 7m was produced in a 77% yield (Table 3, entry 3). α-Branched amines are effective reaction partners, providing the corresponding imines 7n−p in modest yields (Table 3, entries 4–6, 42−66% yield). From these results, it can be concluded that increasing the steric bulk at the α-position of the amine results in decreased reaction efficiency. Phenethylamine (6q) provided only a 17% yield of the corresponding imine (7q, Table 3, entry 7), potentially due to its increased nucleophilicity, which may result in inhibition of catalysis via condensation with quinone 2c. We also tested several electron deficient amides (6r−t) in these reactions (Table 3, entries 8−10). Unfortunately, only sulfinamide 6t provided the desired imine 7t (Table 3, entry 10, 22% yield). In all three cases, a significant amount of benzaldehyde was observed, indicating that electron deficient primary amides (such as 6r−t) are either incapable of promoting transimination, or the resulting imines (7r−t) are hydrolyzed under the current reaction conditions. Notably, imines 7n [42-45] and 7t [46-48] are useful imines for diastereoselective 1,2-addition reactions.
Table 3: Quinone-catalyzed oxidative deformylation using various amine reaction partners.
![[Graphic 13]](/bjoc/content/inline/1860-5397-13-282-i17.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Amine 6 | Product 7 | Yield [%]a | ||
|
1
2 3 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-13-282-i18.svg?max-width=637&scale=1.0)
|
6a, R = OMe
6l, R = H 6m, R = F |
![[Graphic 15]](/bjoc/content/inline/1860-5397-13-282-i19.svg?max-width=637&scale=1.0)
|
7a, R = OMe
7l, R = H 7m, R = F |
85
68 77 |
| 4 |
![[Graphic 16]](/bjoc/content/inline/1860-5397-13-282-i20.svg?max-width=637&scale=1.0)
|
6n |
![[Graphic 17]](/bjoc/content/inline/1860-5397-13-282-i21.svg?max-width=637&scale=1.0)
|
7n | 66 |
| 5 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-13-282-i22.svg?max-width=637&scale=1.0)
|
6o |
![[Graphic 19]](/bjoc/content/inline/1860-5397-13-282-i23.svg?max-width=637&scale=1.0)
|
7o | 42 |
| 6 |
![[Graphic 20]](/bjoc/content/inline/1860-5397-13-282-i24.svg?max-width=637&scale=1.0)
|
6p |
![[Graphic 21]](/bjoc/content/inline/1860-5397-13-282-i25.svg?max-width=637&scale=1.0)
|
7p | 56 |
| 7 |
![[Graphic 22]](/bjoc/content/inline/1860-5397-13-282-i26.svg?max-width=637&scale=1.0)
|
6q |
![[Graphic 23]](/bjoc/content/inline/1860-5397-13-282-i27.svg?max-width=637&scale=1.0)
|
7q | 17 |
| 8 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-13-282-i28.svg?max-width=637&scale=1.0)
|
6r |
![[Graphic 25]](/bjoc/content/inline/1860-5397-13-282-i29.svg?max-width=637&scale=1.0)
|
7r | 0 |
| 9 |
![[Graphic 26]](/bjoc/content/inline/1860-5397-13-282-i30.svg?max-width=637&scale=1.0)
|
6s |
![[Graphic 27]](/bjoc/content/inline/1860-5397-13-282-i31.svg?max-width=637&scale=1.0)
|
7s | 0 |
| 10 |
![[Graphic 28]](/bjoc/content/inline/1860-5397-13-282-i32.svg?max-width=637&scale=1.0)
|
6t |
![[Graphic 29]](/bjoc/content/inline/1860-5397-13-282-i33.svg?max-width=637&scale=1.0)
|
7t | 22 |
aDetermined by 1H NMR using benzyl ether as an internal standard (average of two replicates).
Following these substrate scope studies, we next examined the quinone-catalyzed C–C bond cleavage of analogous substrates (Scheme 3). First, we tested isomeric amino alcohol iso-1a, which provided imine 7a in a yield comparable to that observed when phenylglycinol was used as a substrate. Notably, the mechanism of this reaction likely involves initial formation of benzaldehyde, followed by condensation with para-anisidine, to deliver imine 7a. Vicinal diamine 8 was also a compatible substrate, delivering imine 7a in 63% yield. Finally, we subjected diol 9 to the optimal reaction conditions; no product was observed, indicating that condensation between the substrate and catalyst to form an iminoquinone intermediate is likely required for productive reactivity.
![[1860-5397-13-282-i3]](/bjoc/content/inline/1860-5397-13-282-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Studies of quinone-catalyzed C−C bond cleavage in related substrates.
Scheme 3: Studies of quinone-catalyzed C−C bond cleavage in related substrates.
To demonstrate the synthetic utility of this methodology, we performed a sequential oxidative deformylation/Mukaiyama−Mannich addition under our previously reported conditions for decarboxylative amino acid homologation (Scheme 4) [32]. In this reaction sequence, (thio)silyl ketene acetal 10 was united with 2-phenylglycinol and para-anisidine in a two-step, one-pot process to provide β-amino acid derivative 11 in a 60% yield. The overall reaction sequence provides a unique method for the production of the high-value β-amino acid derivatives [49,50] from 1,2-amino alcohols.
![[1860-5397-13-282-i4]](/bjoc/content/inline/1860-5397-13-282-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Sequential oxidative deformylation/Mukaiyama−Mannich addition using phenylglycinol.
Scheme 4: Sequential oxidative deformylation/Mukaiyama−Mannich addition using phenylglycinol.
Conclusion
In conclusion, we have developed a novel method for the synthesis of imines from 1,2-amino alcohols. This chemistry features an unprecedented application of quinone organocatalysis to enable oxidative deformylation under aerobic conditions. Future work will involve mechanistic studies and the development of new catalysts to expand the scope of this chemistry.
Supporting Information
| Supporting Information File 1: Experimental procedures, compound characterization data, and copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 1014.1 KB | Download |
Acknowledgements
Financial support from the NSF (EPS-0993806) and The University of Kansas is gratefully acknowledged. Additional support for this work was provided by the National Institutes of Health Graduate Training Program in Dynamic Aspects of Chemical Biology Grant T32 GM08545 from NIGMS (to B. J. H.). Support for NMR instrumentation was provided by NIH Shared Instrumentation Grants No. S10OD016369 and S10RR024664, and NSF Major Research Instrumentation Grant No. 0320648.
References
-
Kobayashi, S.; Ishitani, H. Chem. Rev. 1999, 99, 1069–1094. doi:10.1021/cr980414z
Return to citation in text: [1] -
Adams, J. P. J. Chem. Soc., Perkin Trans. 1 2000, 125–139. doi:10.1039/a808142e
Return to citation in text: [1] -
Kobayashi, S.; Mori, Y.; Fossey, J. S.; Salter, M. M. Chem. Rev. 2011, 111, 2626–2704. doi:10.1021/cr100204f
Return to citation in text: [1] -
Schiff, H. Justus Liebigs Ann. Chem. 1864, 131, 118–119. doi:10.1002/jlac.18641310113
Return to citation in text: [1] -
Patil, R. D.; Adimurthy, S. Asian J. Org. Chem. 2013, 2, 726–744. doi:10.1002/ajoc.201300012
Return to citation in text: [1] -
Chen, B.; Wang, L.; Gao, S. ACS Catal. 2015, 5, 5851–5876. doi:10.1021/acscatal.5b01479
Return to citation in text: [1] -
Éll, A. H.; Samec, J. S. M.; Brasse, C.; Bäckvall, J.-E. Chem. Commun. 2002, 1144–1145. doi:10.1039/b202117j
Return to citation in text: [1] -
Murahashi, S.-I.; Okano, Y.; Sato, H.; Nakae, T.; Komiya, N. Synlett 2007, 1675–1678. doi:10.1055/s-2007-984515
Return to citation in text: [1] -
Zhu, B.; Angelici, R. J. Chem. Commun. 2007, 21, 2157–2159. doi:10.1039/b700555e
Return to citation in text: [1] -
Jiang, G.; Chen, J.; Huang, J.-S.; Che, C.-M. Org. Lett. 2009, 11, 4568–4571. doi:10.1021/ol9018166
Return to citation in text: [1] -
Allen, J. M.; Lambert, T. H. J. Am. Chem. Soc. 2011, 133, 1260–1262. doi:10.1021/ja109617y
Return to citation in text: [1] -
Patil, R. D.; Adimurthy, S. Adv. Synth. Catal. 2011, 353, 1695–1700. doi:10.1002/adsc.201100100
Return to citation in text: [1] -
Prades, A.; Peris, E.; Albrecht, M. Organometallics 2011, 30, 1162–1167. doi:10.1021/om101145y
Return to citation in text: [1] -
Rueping, M.; Zhu, S.; Koenigs, R. M. Chem. Commun. 2011, 47, 12709–12711. doi:10.1039/c1cc15643h
Return to citation in text: [1] -
Rueping, M.; Zhu, S.; Koenigs, R. M. Chem. Commun. 2011, 47, 8679–8681. doi:10.1039/c1cc12907d
Return to citation in text: [1] -
Freeman, D. B.; Furst, L.; Condie, A. G.; Stephenson, C. R. J. Org. Lett. 2012, 14, 94–97. doi:10.1021/ol202883v
Return to citation in text: [1] -
Largeron, M.; Fleury, M.-B. Angew. Chem., Int. Ed. 2012, 51, 5409–5412. doi:10.1002/anie.201200587
Return to citation in text: [1] -
Park, J. H.; Ko, K. C.; Kim, E.; Park, N.; Ko, J. H.; Ryu, D. H.; Ahn, T. K.; Lee, J. Y.; Son, S. U. Org. Lett. 2012, 14, 5502–5505. doi:10.1021/ol302584y
Return to citation in text: [1] -
Rueping, M.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C.; Leonori, D.; Vila, C. Chem. – Eur. J. 2012, 18, 5170–5174. doi:10.1002/chem.201200050
Return to citation in text: [1] -
Rueping, M.; Zoller, J.; Fabry, D. C.; Poscharny, K.; Koenigs, R. M.; Weirich, T. E.; Mayer, J. Chem. – Eur. J. 2012, 18, 3478–3481. doi:10.1002/chem.201103242
Return to citation in text: [1] -
Wendlandt, A. E.; Stahl, S. S. Org. Lett. 2012, 14, 2850–2853. doi:10.1021/ol301095j
Return to citation in text: [1] [2] -
Zhu, S.; Rueping, M. Chem. Commun. 2012, 48, 11960–11962. doi:10.1039/c2cc36995h
Return to citation in text: [1] -
Zhang, E.; Tian, H.; Xu, S.; Yu, X.; Xu, Q. Org. Lett. 2013, 15, 2704–2707. doi:10.1021/ol4010118
Return to citation in text: [1] -
Bergonzini, G.; Schindler, C. S.; Wallentin, C.-J.; Jacobsen, E. N.; Stephenson, C. R. J. Chem. Sci. 2014, 5, 112–116. doi:10.1039/C3SC52265B
Return to citation in text: [1] -
Furukawa, S.; Suga, A.; Komatsu, T. Chem. Commun. 2014, 50, 3277–3280. doi:10.1039/C4CC00024B
Return to citation in text: [1] -
Wang, J.; Lu, S.; Cao, X.; Gu, H. Chem. Commun. 2014, 50, 5637–5640. doi:10.1039/c4cc01389a
Return to citation in text: [1] -
Wendlandt, A. E.; Stahl, S. S. J. Am. Chem. Soc. 2014, 136, 506–512. doi:10.1021/ja411692v
Return to citation in text: [1] -
Beatty, J. W.; Stephenson, C. R. J. Acc. Chem. Res. 2015, 48, 1474–1484. doi:10.1021/acs.accounts.5b00068
Return to citation in text: [1] -
Leonard, N. J.; Rebenstorf, M. A. J. Am. Chem. Soc. 1945, 67, 49–51. doi:10.1021/ja01217a016
Return to citation in text: [1] -
McGarvey, G. J.; Williams, J. M.; Hiner, R. N.; Matsubara, Y.; Oh, T. J. Am. Chem. Soc. 1986, 108, 4943–4952. doi:10.1021/ja00276a040
Return to citation in text: [1] -
Shimizu, M.; Makino, H. Tetrahedron Lett. 2001, 42, 8865–8868. doi:10.1016/S0040-4039(01)01945-1
Return to citation in text: [1] -
Haugeberg, B. J.; Phan, J. H.; Liu, X.; O'Connor, T. J.; Clift, M. D. Chem. Commun. 2017, 53, 3062–3065. doi:10.1039/C7CC00485K
Return to citation in text: [1] [2] [3] [4] [5] -
Burger, E. C.; Tunge, J. A. J. Am. Chem. Soc. 2006, 128, 10002–10003. doi:10.1021/ja063115x
Return to citation in text: [1] -
Zuo, Z.; MacMillan, D. W. C. J. Am. Chem. Soc. 2014, 136, 5257–5260. doi:10.1021/ja501621q
Return to citation in text: [1] -
Zuo, Z.; Cong, H.; Li, W.; Choi, J.; Fu, G. C.; MacMillan, D. W. C. J. Am. Chem. Soc. 2016, 138, 1832–1835. doi:10.1021/jacs.5b13211
Return to citation in text: [1] -
Reddy, K. L.; Dress, K. R.; Sharpless, K. B. Tetrahedron Lett. 1998, 39, 3667–3670. doi:10.1016/S0040-4039(98)00644-3
Return to citation in text: [1] -
Reddy, K. L.; Sharpless, K. B. J. Am. Chem. Soc. 1998, 120, 1207–1217. doi:10.1021/ja9728177
Return to citation in text: [1] -
Williamson, K. S.; Yoon, T. P. J. Am. Chem. Soc. 2010, 132, 4570–4571. doi:10.1021/ja1013536
Return to citation in text: [1] -
Nystrom, R. F.; Brown, W. G. J. Am. Chem. Soc. 1947, 69, 2548–2549. doi:10.1021/ja01202a082
Return to citation in text: [1] -
Corey, E. J.; Achiwa, K. J. Am. Chem. Soc. 1969, 91, 1429–1432. doi:10.1021/ja01034a027
Return to citation in text: [1] -
Leon, M. A.; Liu, X.; Phan, J. H.; Clift, M. D. Eur. J. Org. Chem. 2016, 4508–4515. doi:10.1002/ejoc.201600786
Return to citation in text: [1] -
Boga, C.; Savoia, D.; Umani-Ronchi, A. Tetrahedron: Asymmetry 1990, 1, 291–294. doi:10.1016/S0957-4166(00)86316-8
Return to citation in text: [1] -
Annunziata, R.; Benaglia, M.; Cinquini, M.; Cozzi, F.; Raimondi, L. Tetrahedron Lett. 1998, 39, 3333–3336. doi:10.1016/S0040-4039(98)00484-5
Return to citation in text: [1] -
Nancy; Ghosh, S.; Singh, N.; Nanda, G. K.; Venugopalan, P.; Bharatam, P. V.; Trehan, S. Chem. Commun. 2003, 1420–1421. doi:10.1039/b300478c
Return to citation in text: [1] -
Chen, J.; Pandey, R. K.; Cunico, R. F. Tetrahedron: Asymmetry 2005, 16, 941–947. doi:10.1016/j.tetasy.2005.01.025
Return to citation in text: [1] -
Ellman, J. A.; Owens, T. D.; Tang, T. P. Acc. Chem. Res. 2002, 35, 984–995. doi:10.1021/ar020066u
Return to citation in text: [1] -
Ellman, J. A. Pure Appl. Chem. 2003, 75, 39–46. doi:10.1351/pac200375010039
Return to citation in text: [1] -
Robak, M. T.; Herbage, M. A.; Ellman, J. A. Chem. Rev. 2010, 110, 3600–3740. doi:10.1021/cr900382t
Return to citation in text: [1] -
Cheng, R. P.; Gellman, S. H.; DeGrado, W. F. Chem. Rev. 2001, 101, 3219–3232. doi:10.1021/cr000045i
Return to citation in text: [1] -
Weiner, B.; Szymański, W.; Janssen, D. B.; Minnaard, A. J.; Feringa, B. L. Chem. Soc. Rev. 2010, 39, 1656–1691. doi:10.1039/b919599h
Return to citation in text: [1]
| 1. | Kobayashi, S.; Ishitani, H. Chem. Rev. 1999, 99, 1069–1094. doi:10.1021/cr980414z |
| 2. | Adams, J. P. J. Chem. Soc., Perkin Trans. 1 2000, 125–139. doi:10.1039/a808142e |
| 3. | Kobayashi, S.; Mori, Y.; Fossey, J. S.; Salter, M. M. Chem. Rev. 2011, 111, 2626–2704. doi:10.1021/cr100204f |
| 29. | Leonard, N. J.; Rebenstorf, M. A. J. Am. Chem. Soc. 1945, 67, 49–51. doi:10.1021/ja01217a016 |
| 30. | McGarvey, G. J.; Williams, J. M.; Hiner, R. N.; Matsubara, Y.; Oh, T. J. Am. Chem. Soc. 1986, 108, 4943–4952. doi:10.1021/ja00276a040 |
| 31. | Shimizu, M.; Makino, H. Tetrahedron Lett. 2001, 42, 8865–8868. doi:10.1016/S0040-4039(01)01945-1 |
| 32. | Haugeberg, B. J.; Phan, J. H.; Liu, X.; O'Connor, T. J.; Clift, M. D. Chem. Commun. 2017, 53, 3062–3065. doi:10.1039/C7CC00485K |
| 49. | Cheng, R. P.; Gellman, S. H.; DeGrado, W. F. Chem. Rev. 2001, 101, 3219–3232. doi:10.1021/cr000045i |
| 50. | Weiner, B.; Szymański, W.; Janssen, D. B.; Minnaard, A. J.; Feringa, B. L. Chem. Soc. Rev. 2010, 39, 1656–1691. doi:10.1039/b919599h |
| 7. | Éll, A. H.; Samec, J. S. M.; Brasse, C.; Bäckvall, J.-E. Chem. Commun. 2002, 1144–1145. doi:10.1039/b202117j |
| 8. | Murahashi, S.-I.; Okano, Y.; Sato, H.; Nakae, T.; Komiya, N. Synlett 2007, 1675–1678. doi:10.1055/s-2007-984515 |
| 9. | Zhu, B.; Angelici, R. J. Chem. Commun. 2007, 21, 2157–2159. doi:10.1039/b700555e |
| 10. | Jiang, G.; Chen, J.; Huang, J.-S.; Che, C.-M. Org. Lett. 2009, 11, 4568–4571. doi:10.1021/ol9018166 |
| 11. | Allen, J. M.; Lambert, T. H. J. Am. Chem. Soc. 2011, 133, 1260–1262. doi:10.1021/ja109617y |
| 12. | Patil, R. D.; Adimurthy, S. Adv. Synth. Catal. 2011, 353, 1695–1700. doi:10.1002/adsc.201100100 |
| 13. | Prades, A.; Peris, E.; Albrecht, M. Organometallics 2011, 30, 1162–1167. doi:10.1021/om101145y |
| 14. | Rueping, M.; Zhu, S.; Koenigs, R. M. Chem. Commun. 2011, 47, 12709–12711. doi:10.1039/c1cc15643h |
| 15. | Rueping, M.; Zhu, S.; Koenigs, R. M. Chem. Commun. 2011, 47, 8679–8681. doi:10.1039/c1cc12907d |
| 16. | Freeman, D. B.; Furst, L.; Condie, A. G.; Stephenson, C. R. J. Org. Lett. 2012, 14, 94–97. doi:10.1021/ol202883v |
| 17. | Largeron, M.; Fleury, M.-B. Angew. Chem., Int. Ed. 2012, 51, 5409–5412. doi:10.1002/anie.201200587 |
| 18. | Park, J. H.; Ko, K. C.; Kim, E.; Park, N.; Ko, J. H.; Ryu, D. H.; Ahn, T. K.; Lee, J. Y.; Son, S. U. Org. Lett. 2012, 14, 5502–5505. doi:10.1021/ol302584y |
| 19. | Rueping, M.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C.; Leonori, D.; Vila, C. Chem. – Eur. J. 2012, 18, 5170–5174. doi:10.1002/chem.201200050 |
| 20. | Rueping, M.; Zoller, J.; Fabry, D. C.; Poscharny, K.; Koenigs, R. M.; Weirich, T. E.; Mayer, J. Chem. – Eur. J. 2012, 18, 3478–3481. doi:10.1002/chem.201103242 |
| 21. | Wendlandt, A. E.; Stahl, S. S. Org. Lett. 2012, 14, 2850–2853. doi:10.1021/ol301095j |
| 22. | Zhu, S.; Rueping, M. Chem. Commun. 2012, 48, 11960–11962. doi:10.1039/c2cc36995h |
| 23. | Zhang, E.; Tian, H.; Xu, S.; Yu, X.; Xu, Q. Org. Lett. 2013, 15, 2704–2707. doi:10.1021/ol4010118 |
| 24. | Bergonzini, G.; Schindler, C. S.; Wallentin, C.-J.; Jacobsen, E. N.; Stephenson, C. R. J. Chem. Sci. 2014, 5, 112–116. doi:10.1039/C3SC52265B |
| 25. | Furukawa, S.; Suga, A.; Komatsu, T. Chem. Commun. 2014, 50, 3277–3280. doi:10.1039/C4CC00024B |
| 26. | Wang, J.; Lu, S.; Cao, X.; Gu, H. Chem. Commun. 2014, 50, 5637–5640. doi:10.1039/c4cc01389a |
| 27. | Wendlandt, A. E.; Stahl, S. S. J. Am. Chem. Soc. 2014, 136, 506–512. doi:10.1021/ja411692v |
| 28. | Beatty, J. W.; Stephenson, C. R. J. Acc. Chem. Res. 2015, 48, 1474–1484. doi:10.1021/acs.accounts.5b00068 |
| 5. | Patil, R. D.; Adimurthy, S. Asian J. Org. Chem. 2013, 2, 726–744. doi:10.1002/ajoc.201300012 |
| 6. | Chen, B.; Wang, L.; Gao, S. ACS Catal. 2015, 5, 5851–5876. doi:10.1021/acscatal.5b01479 |
| 46. | Ellman, J. A.; Owens, T. D.; Tang, T. P. Acc. Chem. Res. 2002, 35, 984–995. doi:10.1021/ar020066u |
| 47. | Ellman, J. A. Pure Appl. Chem. 2003, 75, 39–46. doi:10.1351/pac200375010039 |
| 48. | Robak, M. T.; Herbage, M. A.; Ellman, J. A. Chem. Rev. 2010, 110, 3600–3740. doi:10.1021/cr900382t |
| 4. | Schiff, H. Justus Liebigs Ann. Chem. 1864, 131, 118–119. doi:10.1002/jlac.18641310113 |
| 32. | Haugeberg, B. J.; Phan, J. H.; Liu, X.; O'Connor, T. J.; Clift, M. D. Chem. Commun. 2017, 53, 3062–3065. doi:10.1039/C7CC00485K |
| 32. | Haugeberg, B. J.; Phan, J. H.; Liu, X.; O'Connor, T. J.; Clift, M. D. Chem. Commun. 2017, 53, 3062–3065. doi:10.1039/C7CC00485K |
| 32. | Haugeberg, B. J.; Phan, J. H.; Liu, X.; O'Connor, T. J.; Clift, M. D. Chem. Commun. 2017, 53, 3062–3065. doi:10.1039/C7CC00485K |
| 39. | Nystrom, R. F.; Brown, W. G. J. Am. Chem. Soc. 1947, 69, 2548–2549. doi:10.1021/ja01202a082 |
| 42. | Boga, C.; Savoia, D.; Umani-Ronchi, A. Tetrahedron: Asymmetry 1990, 1, 291–294. doi:10.1016/S0957-4166(00)86316-8 |
| 43. | Annunziata, R.; Benaglia, M.; Cinquini, M.; Cozzi, F.; Raimondi, L. Tetrahedron Lett. 1998, 39, 3333–3336. doi:10.1016/S0040-4039(98)00484-5 |
| 44. | Nancy; Ghosh, S.; Singh, N.; Nanda, G. K.; Venugopalan, P.; Bharatam, P. V.; Trehan, S. Chem. Commun. 2003, 1420–1421. doi:10.1039/b300478c |
| 45. | Chen, J.; Pandey, R. K.; Cunico, R. F. Tetrahedron: Asymmetry 2005, 16, 941–947. doi:10.1016/j.tetasy.2005.01.025 |
| 36. | Reddy, K. L.; Dress, K. R.; Sharpless, K. B. Tetrahedron Lett. 1998, 39, 3667–3670. doi:10.1016/S0040-4039(98)00644-3 |
| 37. | Reddy, K. L.; Sharpless, K. B. J. Am. Chem. Soc. 1998, 120, 1207–1217. doi:10.1021/ja9728177 |
| 38. | Williamson, K. S.; Yoon, T. P. J. Am. Chem. Soc. 2010, 132, 4570–4571. doi:10.1021/ja1013536 |
| 33. | Burger, E. C.; Tunge, J. A. J. Am. Chem. Soc. 2006, 128, 10002–10003. doi:10.1021/ja063115x |
| 34. | Zuo, Z.; MacMillan, D. W. C. J. Am. Chem. Soc. 2014, 136, 5257–5260. doi:10.1021/ja501621q |
| 35. | Zuo, Z.; Cong, H.; Li, W.; Choi, J.; Fu, G. C.; MacMillan, D. W. C. J. Am. Chem. Soc. 2016, 138, 1832–1835. doi:10.1021/jacs.5b13211 |
| 21. | Wendlandt, A. E.; Stahl, S. S. Org. Lett. 2012, 14, 2850–2853. doi:10.1021/ol301095j |
| 32. | Haugeberg, B. J.; Phan, J. H.; Liu, X.; O'Connor, T. J.; Clift, M. D. Chem. Commun. 2017, 53, 3062–3065. doi:10.1039/C7CC00485K |
| 40. | Corey, E. J.; Achiwa, K. J. Am. Chem. Soc. 1969, 91, 1429–1432. doi:10.1021/ja01034a027 |
| 41. | Leon, M. A.; Liu, X.; Phan, J. H.; Clift, M. D. Eur. J. Org. Chem. 2016, 4508–4515. doi:10.1002/ejoc.201600786 |
© 2017 Liu et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)