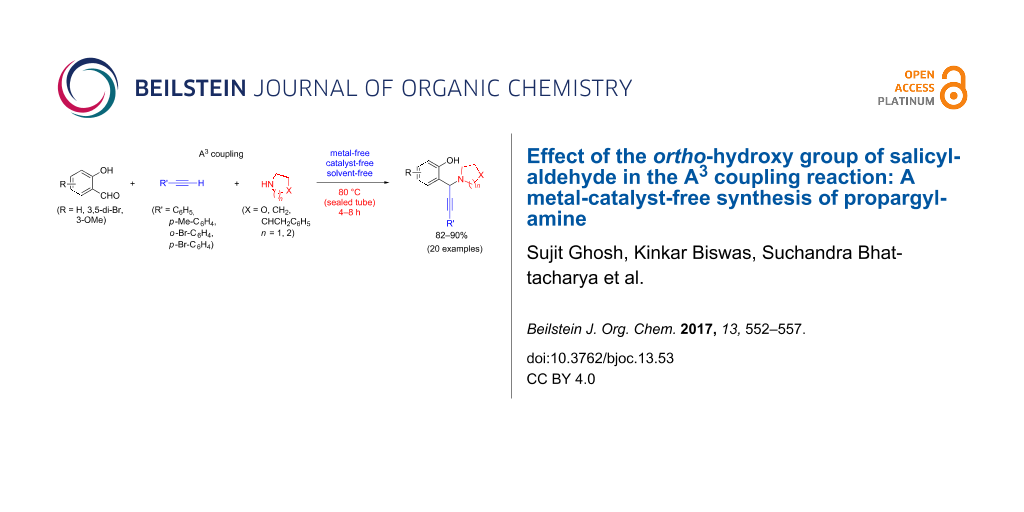
Abstract
The synthesis of propargylamines via A3 coupling mostly under metal-catalyzed procedures is well known. This work invented an unprecedented effect of salicylaldehyde, one of the A3 coupling partners, which could lead to the formation of propargylamine, an important pharmaceutical building block, in the absence of any metal catalyst and under mild conditions. The role of the hydroxy group in ortho position of salicylaldehyde has been explored, which presumably activates the Csp–H bond of the terminal alkyne leading to the formation of propargylamines in good to excellent yields, thus negating the function of the metal catalyst. This observation is hitherto unknown, tested for a variety of salicylaldehyde, amine and acetylene, established as a general protocol, and is believed to be of interest for synthetic chemists from green chemistry.

Graphical Abstract
Introduction
Propargylamines are important synthetic intermediates for the preparation of natural products [1], potential therapeutic agents [2], oxotremorine analogues [3] and multifunctional amino derivatives [4,5]. Compounds like resagiline or selegiline (structures 1 and 2, Scheme 1) bearing a propargylamine moiety, are familiar as potent selective, irreversible monoamine oxidase (MAO) type B inhibitors [6] often used for the treatment of neuropsychiatric disorders such as Alzheimer’s and Parkinson diseases. These alkynylamines are also important building blocks for the synthesis of N-bearing compounds such as β-lactams [7,8], pyrroles [9], pyrrolidines [10], pyrrolophanes [11], 3-aminobenzofurans [12], aminoindolizines [13], 2-aminoimidazoles [14], oxazolidinones [15], and quinolines [16] (Scheme 1).
![[1860-5397-13-53-i1]](/bjoc/content/inline/1860-5397-13-53-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Representative examples of bioactive compounds bearing a propargylamine moiety and synthesis of various N-heterocycles from propargylamine-containing intermediates.
Scheme 1: Representative examples of bioactive compounds bearing a propargylamine moiety and synthesis of var...
Because of diverse applications of propargylamine, several methods are developed among which the three–component reaction of aldehyde, amine and terminal alkyne, commonly known as A3 coupling reaction, remains the most common and straightforward method for the synthesis of propargylamine. The A3 coupling reaction is reported under transition-metal-catalyzed conditions using copper [12,17-19], gold [17,20,21], silver [17,22], zinc [17,23], nickel [24], iron [25], mercury [26], cobalt [27], iridium [28], ruthenium [29], indium [30] etc. Other methods towards the synthesis of propargylamine include: alkynylation of imine [31-33], enamine [34], and Csp³–H bonds adjacent to N-atoms [35,36]. In the A3 coupling, the role of the metal catalyst is believed to activate the terminal acetylene primarily, which then undergoes a nucleophilic addition to the iminium electrophile generated from the aldehyde and the amine. Among different transition metals, copper metal has been mostly explored as the catalyst to activate the terminal acetylene, though there is a possibility for the Glaser coupling of the terminal alkyne as the byproduct [37]. Interestingly, hitherto there is no report for the A3 coupling reaction in the absence of a metal catalyst except of one example in a three-component reaction using an alkynylcarboxylic acid instead of a terminal alkyne [38]. In this case, activation of the Csp–COOH occurs via decarboxylation followed by the coupling with an iminium electrophile to produce the propargylamine. Although the strategy is interesting, functionalized acetylene carboxylic acids are difficultly accessible and the reaction is less 'atom economic'. Therefore, the development of a metal-free and straightforward greener protocol for the preparation of propargylamine is highly desirable. A schematic presentation of various metal-catalyzed protocols including the present work is outlined in Scheme 2.
![[1860-5397-13-53-i2]](/bjoc/content/inline/1860-5397-13-53-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Various metal-catalyzed methods for the synthesis of propargylamine.
Scheme 2: Various metal-catalyzed methods for the synthesis of propargylamine.
Results and Discussion
In connection with our interest in developing new organic reaction methodologies as well as to synthesize propargylamine, we performed one reaction of salicylaldehyde, morpholine and phenylacetylene at 80 °C in the presence of a Cu(I) dithiane-based complex as the catalyst that gave rise to the formation of 3a in good yield (89%, Table 1, entry 1). To check the role of our Cu(I) catalyst, we performed the same reaction under similar conditions in the absence of the copper catalyst (Table 1, entry 2). To our surprise, the same product spot was noticed on TLC plate and further work-up and purification afforded the product in comparable isolated yield. We became interested about our findings and wanted to explore the reaction further in the absence of a metal catalyst. We did the same experiment with o-anisaldehyde and 2-chlorobenzaldehyde without using any copper catalyst, and interestingly, there was no desired product formed in these two reactions (Table 1, entries 3 and 4). Conducting the same experiment with p-hydroxybenzaldehyde (Table 1, entry 5) or o-hydroxyacetopheneone (Table 1, entry 6) also did not generate the corresponding A3 coupled product.
Table 1: Optimization of metal-catalyst-free A3 coupling reaction.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-53-i4.svg?max-width=637&scale=1.0)
|
||||||
| Entry | R1 | R2 | Temperature (°C) | Time (h) | Solvent | Yield (%)a |
|---|---|---|---|---|---|---|
| 1b | H | 2-OH | 80 | 4 | neat | 89 |
| 2 | H | 2-OH | 80 | 4 | neat | 90 |
| 3 | H | 2-OMe | 80 | 4 | neat | no reaction |
| 4 | H | 2-Cl | 80 | 4 | neat | no reaction |
| 5 | H | 4-OH | 80 | 4 | neat | no reaction |
| 6 | Me | 2-OH | 80 | 4 | neat | no reaction |
| 7 | H | 2-OH | rt | 24 | neat | traces (<10%) |
| 8 | H | 2-OH | 60 | 10 | neat | 64 |
| 9c | H | 2-OH | 80 | 4 | MeCN | 85 |
| 10c | H | 2-OH | 80 | 4 | toluene | 88 |
| 11c | H | 2-OH | 80 | 4 | EtOH | 47 |
| 12d | H | 2-OH | 80 | 10 | neat | no A3 product, only imine formation |
| 13e | H | 2-OH | 80 | 10 | neat | no A3 product, only imine formation |
aYield of product after purification by column chromatography; busing a mixture of CuI and 1,3-dithiane ligand [1-(3-(p-tolylthio)propylthio)-4-methylbenzene] in 1:2 ratios (1 mol %); c2 mL of solvent was taken; dcyclohexylamine (1 mmol) was used instead of morpholine; ebenzylamine (1 mmol) was used instead of morpholine.
The above observations made us curious about any specific role of the hydroxy group in the ortho position of salicylaldehyde in A3 coupling. A search in the literature revealed vast examples of A3 coupling with different benzaldehydes. However, only few research groups [12,19-24] reported their experiments with salicylaldehyde only in the presence of metal catalysts (e.g., Cu, Au, Ag, Zn and Ni). We therefore explored further to optimize the reaction conditions in the absence of Cu catalysts at varying temperatures and solvents. In the optimization process (Table 1), we found that the reaction was very slow at room temperature even after 24 h (<10%; Table 1, entry 7), and raising the reaction temperature to 60 °C afforded an improved yield of the product (64%; Table 1, entry 8). We carried out the reaction in solvents like acetonitrile (Table 1, entry 9), toluene (Table 1, entry 10) and ethanol (Table 1, entry 11). Although high yields were achieved in the first two solvents (85–88%), the use of ethanol resulted in a low yield (47%). Thus the A3 coupling of salicylaldehyde, phenylacetylene and morpholine can be achieved under solvent-free conditions as well as in solvents like toluene or acetonitrile with the formation of the coupled product in 85–90% isolated yields. We also conducted similar reactions with cyclohexylamine and benzylamine instead of morpholine. In both cases, we ended up with the formation of corresponding imines (Table 1, entries 12 and 13).
After the successful optimization, further extension of the reaction protocol was made by varying the other two components viz. the secondary cyclic amine and the terminal alkyne. The results are not only encouraging but constitute a hitherto unknown general protocol for the preparation of propargylamines under metal-catalyst-free A3 coupling of salicylaldehyde as the aldehyde component (Figure 1). Changing the phenylacetylene to other substituted arylacetylenes like p-tolylacetylene, o-bromophenylacetylene, p-bromophenylacetylene or switching from morpholine to other amines like piperidine, 4-benzylpiperidine and pyrrolidine also react smoothly to afford the corresponding propargylamine (the A3 coupled product) in 82–90% isolated yields (Figure 1, 3a–j). Substituted salicylaldehyde like 3,5-dibromosalicylaldehyde or o-vanillin also reacted easily when treated with morpholine and phenylacetylene, p-tolylacetylene or o- and p-bromophenylacetylene as the other coupling components at 80–90 °C (Figure 1, 3k–p). Varying the secondary amine component with 4-benzylpiperidine, piperidine also worked efficiently to produce the corresponding propargylamine derivatives (3q–t). All products were characterized by FTIR, 1H and 13C NMR spectral data. Scaling up the reaction to gram-scale operation using phenylacetylene, morpholine and salicylaldehyde (5 mmol each) also afforded the desired propargylamine cleanly in 86% yield.
![[1860-5397-13-53-1]](/bjoc/content/figures/1860-5397-13-53-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Synthesis of various propargylamines from various salicylaldehydes under metal-catalyst-free conditions.
Figure 1: Synthesis of various propargylamines from various salicylaldehydes under metal-catalyst-free condit...
A mixture of salicylaldehyde or its derivative (1 mmol), amine (1 mmol) and alkyne (1 mmol) was stirred at 80 °C (90 °C for dibromo derivative of salicylaldehyde) in a sealed tube for 4–8 h (4 h for 3a–l and 8 h for 3m–t). Isolated yields after purification by column chromatography are given.
Mechanism
As regard to the mechanism of the reaction, it is generally believed that the metal catalyst activates the terminal alkyne so as to generate metal acetylide species, which then undergoes nucleophilic addition at the electrophilic iminium ion 4, eventually providing the A3 coupled product (Scheme 3). In the absence of the metal catalyst, the sp carbon (C–H) of the alkyne is less nucleophilic in nature. However, after the formation of the iminium ion 4 (as it happens in all such cases), the hydroxy group in the ortho position may undergo deprotonation forming an unstable o-quinonoid intermediate 5, which presumably activates the sp carbon (C–H) of the alkyne more nucleophilic via H-bond formation to 5, and finally to form the more stable A3 coupled product 3. Although we are not sure about the exact routes to the formation of 3, other results indirectly corroborate the proposition. For example, the inability of the o-methoxybenzaldehyde, p-hydroxybenzaldehyde and o–hydroxyacetophenone to undergo a similar reaction under metal−free conditions could be explained by this mechanism. The p-hydroxybenzaldehyde might give rise to the corresponding p-quinonoid species but not activates the sp carbon (C–H) of the alkyne. Again, in the case of o-hydroxyacetophenone, the corresponding iminium salt might form with difficulty as well as the iminium carbon would be less elctrophilic. Furthermore, the reaction does not proceed well in a protic solvent like ethanol. This suggests that the hydroxy group of salicylaldehyde can also make the hydrogen bonding with protic ethanol solvent and thereby reducing the possibility of activating the sp carbon (C–H) of the alkyne. Furthermore, reactions with primary amines like cyclohexylamine and benzylamine produce only the imine and no A3-coupled product, signifying that imines are less efficient than iminium species to initiate further reaction with terminal alkyne.
![[1860-5397-13-53-i3]](/bjoc/content/inline/1860-5397-13-53-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Plausible mechanism for the metal-free A3 coupling from salicylaldehyde.
Scheme 3: Plausible mechanism for the metal-free A3 coupling from salicylaldehyde.
Conclusion
In conclusion, the present study demonstrates an unusual but added role of the hydroxy group of salicylaldehyde, which paves the way to develop metal-free as well as solvent-free A3 coupling reactions leading to the formation of propargylamine – the useful synthetic intermediate and an important unit of many bio-active compounds. The reaction conditions are straightforward and products are obtained in good to excellent yields. The metal-free approach also offers the advantages of avoiding any possible byproduct arising out from the Glaser coupling of terminal alkynes as well as contamination with metal species. The present protocol supersedes the only metal-catalyst-free approach from acetylene carboxylic acids that are difficultly accessible and with low atom economy. Thus the present reaction from easily available A3 components leading to the formation of propargylamine under metal-catalyst-free and solvent-free conditions could attract the interest of synthetic and medicinal chemists.
Supporting Information
| Supporting Information File 1: Experimental procedure, characterization data and scan copies of 1H and 13C NMR spectra (3a–t). | ||
| Format: PDF | Size: 3.4 MB | Download |
References
-
Su, J.; Liu, H.; Hua, R. Int. J. Mol. Sci. 2015, 16, 3599–3608. doi:10.3390/ijms16023599
Return to citation in text: [1] -
Zlotos, D. P.; Blender, W.; Holzgrabe, U. Expert Opin. Ther. Pat. 1990, 9, 1029.
Return to citation in text: [1] -
Nilson, B. M.; Vargas, H. M.; Ringdahl, B.; Hacksell, U. J. Med. Chem. 1992, 35, 285–294. doi:10.1021/jm00080a013
Return to citation in text: [1] -
Konishi, M.; Ohkuma, H.; Tsuno, T.; Oki, T.; VanDuyne, G. D.; Clardy, J. J. Am. Chem. Soc. 1990, 112, 3715–3716. doi:10.1021/ja00165a097
Return to citation in text: [1] -
Huffman, M. A.; Yasuda, N.; DeCamp, E. A.; Grabowski, E. J. J. J. Org. Chem. 1995, 60, 1590–1594. doi:10.1021/jo00111a016
Return to citation in text: [1] -
Yu, P. H.; Davis, B. A.; Boulton, A. A. J. Med. Chem. 1992, 35, 3705–3713. doi:10.1021/jm00098a017
Return to citation in text: [1] -
Ma, S.; Wu, B.; Jiang, X. J. Org. Chem. 2005, 70, 2588–2593. doi:10.1021/jo0480996
Return to citation in text: [1] -
Matsuda, I.; Sakakibara, J.; Nagashima, H. Tetrahedron Lett. 1991, 32, 7431–7434. doi:10.1016/0040-4039(91)80126-q
Return to citation in text: [1] -
Yamamoto, Y.; Hayashi, H.; Saigoku, T.; Nishiyama, H. J. Am. Chem. Soc. 2005, 127, 10804–10805. doi:10.1021/ja053408a
Return to citation in text: [1] -
Harvey, D. F.; Sigano, D. M. J. Org. Chem. 1996, 61, 2268–2272. doi:10.1021/jo9519930
Return to citation in text: [1] -
Fürstner, A.; Szillat, H.; Stelzer, F. J. Am. Chem. Soc. 2000, 122, 6785–6786. doi:10.1021/ja001034
Return to citation in text: [1] -
Li, H.; Liu, J.; Yan, B.; Li, Y. Tetrahedron Lett. 2009, 50, 2353–2357. doi:10.1016/j.tetlet.2009.02.204
Return to citation in text: [1] [2] [3] -
Yan, B.; Liu, Y. Org. Lett. 2007, 9, 4323–4326. doi:10.1021/ol701886e
Return to citation in text: [1] -
Ermolat’ev, D. S.; Bariwal, J. B.; Steenackers, H. P. L.; De Keersmaecker, S. C. J.; Van der Eycken, E. V. Angew. Chem. 2010, 122, 9655–9658. doi:10.1002/ange.201004256
Return to citation in text: [1] -
Lee, E.-S.; Yeom, H.-S.; Hwang, J.-H.; Shin, S. Eur. J. Org. Chem. 2007, 3503–3507. doi:10.1002/ejoc.200700210
Return to citation in text: [1] -
Xiao, F.; Chen, Y.; Liu, Y.; Wang, J. Tetrahedron 2008, 64, 2755–2761. doi:10.1016/j.tet.2008.01.046
Return to citation in text: [1] -
Peshkov, V. A.; Pereshivko, O. P.; Van der Eycken, E. V. Chem. Soc. Rev. 2012, 41, 3790–3807. doi:10.1039/c2cs15356d
Return to citation in text: [1] [2] [3] [4] -
Kerim, M. D.; El Kaim, L. Synlett 2016, 27, 1572–1576. doi:10.1055/s-0035-1561396
Return to citation in text: [1] -
Aliaga, M. J.; Ramon, D. J.; Yus, M. Org. Biomol. Chem. 2010, 8, 43–46. doi:10.1039/b917923b
Return to citation in text: [1] [2] -
Villaverde, G.; Corma, A.; Iglesias, M.; Sánchez, F. ACS Catal. 2012, 2, 399–406. doi:10.1021/cs200601w
Return to citation in text: [1] [2] -
Borah, B. J.; Borah, S. J.; Saikia, K.; Dutta, D. K. Catal. Sci. Technol. 2014, 4, 4001–4009. doi:10.1039/C4CY00666F
Return to citation in text: [1] [2] -
Trose, M.; Dell’Acqua, M.; Pedrazzini, T.; Pirovano, V.; Gallo, E.; Rossi, E.; Caselli, A.; Abbiati, G. J. Org. Chem. 2014, 79, 7311–7320. doi:10.1021/jo500981r
Return to citation in text: [1] [2] -
Mukhopadhyay, C.; Rana, S. Catal. Commun. 2009, 11, 285–289. doi:10.1016/j.catcom.2009.10.016
Return to citation in text: [1] [2] -
Namitharan, K.; Pitchumani, K. Eur. J. Org. Chem. 2010, 411–415. doi:10.1002/ejoc.200901084
Return to citation in text: [1] [2] -
Huo, X.; Liu, J.; Wang, B.; Zhang, H.; Yang, Z.; She, X.; Xi, P. J. Mater. Chem. A 2013, 1, 651–656. doi:10.1039/c2ta00485b
Return to citation in text: [1] -
Li, P.-H.; Wang, L. Chin. J. Chem. 2005, 23, 1076–1080. doi:10.1002/cjoc.200591076
Return to citation in text: [1] -
Chen, W.-W.; Bi, H.-P.; Li, C.-J. Synlett 2010, 475–479. doi:10.1055/s-0029-1219173
Return to citation in text: [1] -
Sakaguchi, S.; Mizuta, T.; Furuwan, M.; Kubo, T.; Ishii, Y. Chem. Commun. 2004, 1638–1639. doi:10.1039/b404430d
Return to citation in text: [1] -
Bonfield, E. R.; Li, C.-J. Org. Biomol. Chem. 2007, 5, 435–437. doi:10.1039/B613596J
Return to citation in text: [1] -
Lima da Silva, T.; Scheibler Rambo, R.; da Silveira Rampon, D.; Schwartz Radatz, C.; Benvenutti, E. V.; Russowsky, D.; Schneider, P. H. J. Mol. Catal. A 2015, 399, 71–78. doi:10.1016/j.molcata.2015.01.021
Return to citation in text: [1] -
Wei, C.; Li, C.-J. J. Am. Chem. Soc. 2002, 124, 5638–5639. doi:10.1021/ja026007t
Return to citation in text: [1] -
Zani, L.; Alesi, S.; Cozzi, P. G.; Bolm, C. J. Org. Chem. 2006, 71, 1558–1562. doi:10.1021/jo052273o
Return to citation in text: [1] -
Lu, Y.; Johnstone, T. C.; Arndtsen, B. A. J. Am. Chem. Soc. 2009, 131, 11284–11285. doi:10.1021/ja904185b
Return to citation in text: [1] -
Koradin, C.; Polborn, K.; Knochel, P. Angew. Chem., Int. Ed. 2002, 41, 2535–2538. doi:10.1002/1521-3773(20020715)41:14<2535::AID-ANIE2535>3.0.CO;2-M
Return to citation in text: [1] -
Li, Z.; Li, C.-J. J. Am. Chem. Soc. 2004, 126, 11810–11811. doi:10.1021/ja0460763
Return to citation in text: [1] -
Xu, X.; Li, X. Org. Lett. 2009, 11, 1027–1029. doi:10.1021/ol802974b
Return to citation in text: [1] -
Albaladejo, M. J.; Alonso, F.; Moglie, Y.; Yus, M. Eur. J. Org. Chem. 2012, 3093–3104. doi:10.1002/ejoc.201200090
Return to citation in text: [1] -
Park, K.; Heo, Y.; Lee, S. Org. Lett. 2013, 15, 3322–3325. doi:10.1021/ol401358t
Return to citation in text: [1]
| 35. | Li, Z.; Li, C.-J. J. Am. Chem. Soc. 2004, 126, 11810–11811. doi:10.1021/ja0460763 |
| 36. | Xu, X.; Li, X. Org. Lett. 2009, 11, 1027–1029. doi:10.1021/ol802974b |
| 37. | Albaladejo, M. J.; Alonso, F.; Moglie, Y.; Yus, M. Eur. J. Org. Chem. 2012, 3093–3104. doi:10.1002/ejoc.201200090 |
| 38. | Park, K.; Heo, Y.; Lee, S. Org. Lett. 2013, 15, 3322–3325. doi:10.1021/ol401358t |
| 1. | Su, J.; Liu, H.; Hua, R. Int. J. Mol. Sci. 2015, 16, 3599–3608. doi:10.3390/ijms16023599 |
| 6. | Yu, P. H.; Davis, B. A.; Boulton, A. A. J. Med. Chem. 1992, 35, 3705–3713. doi:10.1021/jm00098a017 |
| 12. | Li, H.; Liu, J.; Yan, B.; Li, Y. Tetrahedron Lett. 2009, 50, 2353–2357. doi:10.1016/j.tetlet.2009.02.204 |
| 17. | Peshkov, V. A.; Pereshivko, O. P.; Van der Eycken, E. V. Chem. Soc. Rev. 2012, 41, 3790–3807. doi:10.1039/c2cs15356d |
| 18. | Kerim, M. D.; El Kaim, L. Synlett 2016, 27, 1572–1576. doi:10.1055/s-0035-1561396 |
| 19. | Aliaga, M. J.; Ramon, D. J.; Yus, M. Org. Biomol. Chem. 2010, 8, 43–46. doi:10.1039/b917923b |
| 4. | Konishi, M.; Ohkuma, H.; Tsuno, T.; Oki, T.; VanDuyne, G. D.; Clardy, J. J. Am. Chem. Soc. 1990, 112, 3715–3716. doi:10.1021/ja00165a097 |
| 5. | Huffman, M. A.; Yasuda, N.; DeCamp, E. A.; Grabowski, E. J. J. J. Org. Chem. 1995, 60, 1590–1594. doi:10.1021/jo00111a016 |
| 17. | Peshkov, V. A.; Pereshivko, O. P.; Van der Eycken, E. V. Chem. Soc. Rev. 2012, 41, 3790–3807. doi:10.1039/c2cs15356d |
| 20. | Villaverde, G.; Corma, A.; Iglesias, M.; Sánchez, F. ACS Catal. 2012, 2, 399–406. doi:10.1021/cs200601w |
| 21. | Borah, B. J.; Borah, S. J.; Saikia, K.; Dutta, D. K. Catal. Sci. Technol. 2014, 4, 4001–4009. doi:10.1039/C4CY00666F |
| 3. | Nilson, B. M.; Vargas, H. M.; Ringdahl, B.; Hacksell, U. J. Med. Chem. 1992, 35, 285–294. doi:10.1021/jm00080a013 |
| 15. | Lee, E.-S.; Yeom, H.-S.; Hwang, J.-H.; Shin, S. Eur. J. Org. Chem. 2007, 3503–3507. doi:10.1002/ejoc.200700210 |
| 2. | Zlotos, D. P.; Blender, W.; Holzgrabe, U. Expert Opin. Ther. Pat. 1990, 9, 1029. |
| 16. | Xiao, F.; Chen, Y.; Liu, Y.; Wang, J. Tetrahedron 2008, 64, 2755–2761. doi:10.1016/j.tet.2008.01.046 |
| 11. | Fürstner, A.; Szillat, H.; Stelzer, F. J. Am. Chem. Soc. 2000, 122, 6785–6786. doi:10.1021/ja001034 |
| 10. | Harvey, D. F.; Sigano, D. M. J. Org. Chem. 1996, 61, 2268–2272. doi:10.1021/jo9519930 |
| 14. | Ermolat’ev, D. S.; Bariwal, J. B.; Steenackers, H. P. L.; De Keersmaecker, S. C. J.; Van der Eycken, E. V. Angew. Chem. 2010, 122, 9655–9658. doi:10.1002/ange.201004256 |
| 9. | Yamamoto, Y.; Hayashi, H.; Saigoku, T.; Nishiyama, H. J. Am. Chem. Soc. 2005, 127, 10804–10805. doi:10.1021/ja053408a |
| 12. | Li, H.; Liu, J.; Yan, B.; Li, Y. Tetrahedron Lett. 2009, 50, 2353–2357. doi:10.1016/j.tetlet.2009.02.204 |
| 19. | Aliaga, M. J.; Ramon, D. J.; Yus, M. Org. Biomol. Chem. 2010, 8, 43–46. doi:10.1039/b917923b |
| 20. | Villaverde, G.; Corma, A.; Iglesias, M.; Sánchez, F. ACS Catal. 2012, 2, 399–406. doi:10.1021/cs200601w |
| 21. | Borah, B. J.; Borah, S. J.; Saikia, K.; Dutta, D. K. Catal. Sci. Technol. 2014, 4, 4001–4009. doi:10.1039/C4CY00666F |
| 22. | Trose, M.; Dell’Acqua, M.; Pedrazzini, T.; Pirovano, V.; Gallo, E.; Rossi, E.; Caselli, A.; Abbiati, G. J. Org. Chem. 2014, 79, 7311–7320. doi:10.1021/jo500981r |
| 23. | Mukhopadhyay, C.; Rana, S. Catal. Commun. 2009, 11, 285–289. doi:10.1016/j.catcom.2009.10.016 |
| 24. | Namitharan, K.; Pitchumani, K. Eur. J. Org. Chem. 2010, 411–415. doi:10.1002/ejoc.200901084 |
| 7. | Ma, S.; Wu, B.; Jiang, X. J. Org. Chem. 2005, 70, 2588–2593. doi:10.1021/jo0480996 |
| 8. | Matsuda, I.; Sakakibara, J.; Nagashima, H. Tetrahedron Lett. 1991, 32, 7431–7434. doi:10.1016/0040-4039(91)80126-q |
| 12. | Li, H.; Liu, J.; Yan, B.; Li, Y. Tetrahedron Lett. 2009, 50, 2353–2357. doi:10.1016/j.tetlet.2009.02.204 |
| 24. | Namitharan, K.; Pitchumani, K. Eur. J. Org. Chem. 2010, 411–415. doi:10.1002/ejoc.200901084 |
| 17. | Peshkov, V. A.; Pereshivko, O. P.; Van der Eycken, E. V. Chem. Soc. Rev. 2012, 41, 3790–3807. doi:10.1039/c2cs15356d |
| 22. | Trose, M.; Dell’Acqua, M.; Pedrazzini, T.; Pirovano, V.; Gallo, E.; Rossi, E.; Caselli, A.; Abbiati, G. J. Org. Chem. 2014, 79, 7311–7320. doi:10.1021/jo500981r |
| 17. | Peshkov, V. A.; Pereshivko, O. P.; Van der Eycken, E. V. Chem. Soc. Rev. 2012, 41, 3790–3807. doi:10.1039/c2cs15356d |
| 23. | Mukhopadhyay, C.; Rana, S. Catal. Commun. 2009, 11, 285–289. doi:10.1016/j.catcom.2009.10.016 |
| 31. | Wei, C.; Li, C.-J. J. Am. Chem. Soc. 2002, 124, 5638–5639. doi:10.1021/ja026007t |
| 32. | Zani, L.; Alesi, S.; Cozzi, P. G.; Bolm, C. J. Org. Chem. 2006, 71, 1558–1562. doi:10.1021/jo052273o |
| 33. | Lu, Y.; Johnstone, T. C.; Arndtsen, B. A. J. Am. Chem. Soc. 2009, 131, 11284–11285. doi:10.1021/ja904185b |
| 34. | Koradin, C.; Polborn, K.; Knochel, P. Angew. Chem., Int. Ed. 2002, 41, 2535–2538. doi:10.1002/1521-3773(20020715)41:14<2535::AID-ANIE2535>3.0.CO;2-M |
| 29. | Bonfield, E. R.; Li, C.-J. Org. Biomol. Chem. 2007, 5, 435–437. doi:10.1039/B613596J |
| 30. | Lima da Silva, T.; Scheibler Rambo, R.; da Silveira Rampon, D.; Schwartz Radatz, C.; Benvenutti, E. V.; Russowsky, D.; Schneider, P. H. J. Mol. Catal. A 2015, 399, 71–78. doi:10.1016/j.molcata.2015.01.021 |
| 27. | Chen, W.-W.; Bi, H.-P.; Li, C.-J. Synlett 2010, 475–479. doi:10.1055/s-0029-1219173 |
| 28. | Sakaguchi, S.; Mizuta, T.; Furuwan, M.; Kubo, T.; Ishii, Y. Chem. Commun. 2004, 1638–1639. doi:10.1039/b404430d |
| 25. | Huo, X.; Liu, J.; Wang, B.; Zhang, H.; Yang, Z.; She, X.; Xi, P. J. Mater. Chem. A 2013, 1, 651–656. doi:10.1039/c2ta00485b |
| 26. | Li, P.-H.; Wang, L. Chin. J. Chem. 2005, 23, 1076–1080. doi:10.1002/cjoc.200591076 |
© 2017 Ghosh et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)